
Abstract
Background
Borrelia burgdorferi sensu lato (sl), the causative agent of Lyme borreliosis, is transmitted by ticks of the genus Ixodes as vector. For identification of Borrelia infections in ticks a TaqMan™ minor groove binder (MGB) probe-based quantitative real time PCR (qPCR) was established targeting the 5S-23S intergenic spacer. Extension to a duplex qPCR included an Ixodes spp. positive control to verify successful DNA isolation. Besides qPCR, an osp A-specific conventional PCR for species-specific identification of B. spielmanii was established. Afterwards 1000 I. ricinus flagged in the city of Hanover, Germany, were investigated for B. burgdorferi sl infections followed by species identification. Furthermore, I. hexagonus ticks were investigated to proof applicability of the PCRs.
Results
Quantitative real time PCR (qPCR) identifying B. burgdorferi sl in ticks was able to detect 1-10 copies per reaction. B. spielmanii osp A-specific conventional PCR was also highly specific and showed no cross reactions with the other tested Borrelia species. From 1000 hanoveranian ticks 24.3% were positive compared to only 7.4% positives by dark-field microscopy. Related to tick stage 1.7% larvae, 18.1% nymphs, and 34.6% adults were positive. The most frequent species was B. garinii, followed by B. afzelii, B. spielmanii, B. valaisiana and B. burgdorferi sensu stricto (ss). 70.6% of I. ricinus were mono-infected, whereas 28.0% and 1.4% were infected with two and three Borrelia species, respectively. From 232 I. hexagonus collected from hedgehogs in different sites of Germany, qPCR detected 5.7% to be infected with B. burgdorferi sl, which were identified as B. afzelii, B. garinii and B. spielmanii.
Conclusions
The evaluated qPCR to detect B. burgdorferi sl in Ixodes spp. is highly specific and sensitive. As a duplex qPCR including detection of Ixodes spp. DNA it is the first DNA based technique incorporating a control for successful DNA isolation from the vector tick. Establishment of a B. spielmanii specific conventional PCR filled the gap in PCR identification of principal European Borrelia genospecies. Practical application showed that all European pathogenic Borrelia spp. were present in I. ricinus flagged in recreational areas of the city of Hanover and confirmed I. hexagonus as reservoir for pathogenic Borrelia spp.
Similar content being viewed by others


Background
Ticks of the genus Ixodes are transmission vectors for Borrelia burgdorferi sensu lato (sl), the causative agent of Lyme borreliosis (LB). In Central Europe, the sheep or forest tick I. ricinus is the main vector for B. burgdorferi sl with a distribution area between the 66th and 39th degree of latitude and from Portugal to Russia [1]. Besides this classical vector, the hedgehog tick I. hexoganus, the fox tick I. canisuga, and the sea bird tick I. uriae contribute to the circulation of B. burgdorferi sl in Europe [2–4]. The B. burgdorferi sl-complex comprises worldwide today at least 15 spirochete species, namely B. burgdorferi sensu stricto (ss), Borrelia afzelii, Borrelia andersonii, Borrelia bissettii, Borrelia californiensis, Borrelia garinii, Borrelia japonica, Borrelia lusitaniae, Borrelia sinica, Borrelia spielmanii, Borrelia tanukii, Borrelia turdi, Borrelia valaisiana as well as the recently described Borrelia carolinensis[5] and Borrelia americana[6]. Furthermore, B. bavariensis sp. nov. [7] formerly included in the B. garinii species (OspA serotype 4) and B. yangtze sp. nov. [8] were proposed as new species. From these, B. burgdorferi ss, B. afzelii, B. bissettii, B. garinii, B. lusitaniae, B. spielmanii, B. valaisiana and B. bavariensis sp. nov. are recognized in Central Europe. While B. burgdorferi ss, B. afzelii, B. garinii, and B. spielmanii are confirmed as causative organisms of Lyme disease, the pathogenic potential of the remaining species is still unclear. However, the presence of B. bissettii, B. lusitaniae, and B. valaisiana in human patients [9–13] provides evidence that these species might also cause LB. While B. burgdorferi ss is the most common species in the United States, in Germany the pathogens B. garinii and B. afzelii are more frequent than B. burgdorferi ss [14–17].
In Europe, the overall mean prevalence of Borrelia spirochetes in ticks is 13.7% with the highest I. ricinus infection rates in Central Europe and a significant increase in the infection rate of adult ticks from Western to Eastern Europe [18]. In Germany, approximately 1% of larvae, 4 to 18% of nymphs and 10 to 35% of adult ticks are infected with B. burgdorferi sl (reviewed in [19]). Thereby prevalence variations not only depend on the region, but also on the examination methods known to exhibit unequal sensitivity and specificity values. In both cases, PCR techniques are certainly superior to other techniques used to detect B. burgdorferi sl.
Here we present the establishment of a highly sensitive and specific quantitative real time PCR (qPCR) based on TaqMan™ minor groove binder (MGB)-probes to detect Borrelia burgdorferi sl in ticks. The inference on individual Borrelia burden was assured by targeting a single copy region, the 5S-23S intergenic spacer [20]. A further aim was to establish a species-specific conventional PCR to discriminate B. spielmanii from other European genospecies. When applying the established PCR methods in a study on ticks flagged in the city of Hanover we found that all assured human pathogenic Borrelia species distributed in Europe were present in the collected hanoveranian ticks.
Results
Borrelia burgdorferi sl genus-specific quantitative real time PCR
Genomic DNA amplification of the 5S-23S intergenic spacer region with the genus-specific primer/TaqMan™-MGB-probe combination resulted for each tested Borrelia species (B. burgdorferi ss, B. afzelii, B. garinii, B. valaisiana and B. spielmanii) in bands of 67 bp which were verified by sequencing.
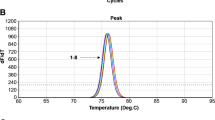
When testing specificity, no amplification products were observed by using DNA of related spirochetes or laboratory bred I. ricinus larvae. Regarding sensitivity, qPCR detected between 1 to10 IGS copies in the individual runs when using serial plasmid standard dilutions as template. This detection limit was also reached for the B. spielmanii- and B. lusitaniae-IGS plasmid standard showing one mismatch with the reverse primer. Since the DNA elution volume from homogenised ticks is 100 μl and 2 μl of them are used as template in qPCR, this correlates with a burden of ≥50-500 Borrelia individuals per tick. In this context spiking experiments revealed no negative impact of tick material on qPCR detection.
Duplex quantitative real time PCR targeting ITS2 of Ixodes spp. as positive control
By testing the designed primer/TaqMan™-MGB-probe combination targeting the ITS2 sequence of Ixodes spp. all isolated tick DNAs (280 I. ricinus, 8 I. hexagonus, and 8 I. canisuga) resulted in positive qPCR signals. Based on serially diluted plasmid standards, the qPCR was able to detect 1 ITS2-copy. By using the ABsolute™ Blue QPCR low Rox Mix in the duplex qPCR reaction setup there was no obvious effect on B. burgdorferi sl detection whereas this detection failed when the Brilliant® QPCR Master Mix was used.
Partial ITS2 sequencing of eight I. ricinus individuals showed 100% sequence identity with the primer-probe combination. For one I. hexagonus (accession no. GQ330526), a substitution (C→T) was identified within the probe sequence. Of I. canisuga, all individuals possessed a substitution (C→T) within the probe sequence, and one individual (accession no. GQ330529) had a second substitution (T→C). In the reverse primer sequence, an insertion (A) was observed in three individuals (accession nos. GQ330528, GQ330529, and GQ330534). Additionally, one of them showed a substitution (C→T, accession no. GQ330529). The sequence of the forward primer could not be considered since this primer was used to generate the partial I. canisuga ITS2 sequences.
Evaluation of the species-specific conventional PCR
Specificity examination of the newly designed B. spielmanii specific osp A primers as well as the published rpo B primers for B. burgdorferi ss, B. afzelii, B. garinii, and B. valaisiana[21] using DNA of the different Borrelia spp. as well as from related spirochetes (Treponema sp. and Leptospira spp.) resulted in bands with the corresponding Borrelia spp. DNA only, no cross reactions were observed. B. spielmanii osp A amplification resulted in a PCR product measuring 151 bp in length and was confirmed by sequencing. Sequence alignment revealed an osp A forward primer identity of 82% and 69% with B. bissettii and B. lusitaniae, respectively. From the last 7 bases on the primer's 3' end at most 3 bases were identical, but not the very last one. Concerning the reverse osp A primer sequence identities were 85% and 76% with the above mentioned Borrelia spp. From the last 7 bases on the primer's 3' end at most 4 bases were identical, but not the last two ones. Further B. spielmanii osp A-specific primer evaluation proved negative PCR results when using DNA of 100 laboratory bred Ixodes larvae as template. No inhibitory effect was noticed by amplification of B. spielmanii DNA in presence of DNA of the other Borrelia species.
Analysis of Borrelia infections and species distribution in ticks of the city of Hanover
Morphological determination revealed that the 1000 ticks collected in recreation areas of the city of Hanover were all belonging to the species I. ricinus. Stage distribution was as follows: 60 larvae, 507 nymphs, and 433 adults (229 males and 204 females). Of these, 74 were spirochete positive in dark-field microscopy (DFM), but only 54 of them were confirmed by genus-specific quantitative real time PCR (qPCR). In all, 243 ticks gave a positive qPCR signal whereas in 8 ticks the results were questionable. Table 1 shows the DFM and qPCR results in more detail. With the qPCR positive and questionable ticks species-specific conventional PCR runs were performed. In 212 out of the 243 qPCR positive ticks and in 6 out of the 8 questionable ticks the B. burgdorferi sl species differentiation was successful. From the 218 Borrelia species differentiated ticks, 154 ticks (70.6%) were mono-infected, 61 (28.0%) were infected with two and 3 (1.4%) were infected with three species of the Borrelia burgdorferi sl-complex. Results considering the stage distribution of the I. ricinus ticks are listed in Table 2. The most frequent mono-infection was with B. afzelii closely followed by B. garinii whereas B. burgdorferi ss caused the least common mono-infection. Frequent double-infections were the combinations B. garinii/B. spielmanii, B. garinii/B. valaisiana, B. afzelii/B. garinii and B. afzelii/B. spielmanii. The combinations B. afzelii/B. burgdorferi ss/B. spielmanii and B. garinii/B. spielmanii/B. valaisiana were found in triple infections. Detailed species distribution results and combinations are shown in Table 3.
Analysis of Borrelia infections in ticks collected from hedgehogs
The 238 ticks collected from hedgehogs were represented by 92 nymphs and 146 adult females. All were morphologically identified as I. hexagonus. Genus-specific qPCR was successfully performed with 232 individuals of which 12 (5.7%) were positive for B. burgdorferi sl. These 12 positive ticks were composed of 2 nymphs and 10 adult females resulting in 2.17% positive nymphs and 6.85% positive adult females.
With species-specific conventional PCR in 4 ticks the Borrelia species could be determined. Two ticks were infected with B. afzelii, one with B. spielmanii, and one was double infected with B. afzelii and B. garinii.
Discussion
Spirochetes of the B. burgdorferi sl-complex can be detected by dark-field and phase-contrast microscopy, Giemsa-stained smears, direct and indirect immunofluorescence, cultivation, and PCR methods. Detection of Borrelia infections in ticks is commonly done by dark-field microscopy (DFM) or PCR methods. However, DFM is less specific than PCR because all spirochete bacteria are diagnosed as positive, not exclusively Borrelia spp. Concerning sensitivity PCR is superior to DFM since successful DNA analysis does not require living organisms whereas DFM and also cultivation are dependent on living bacteria. But sensitivity is also varying among different PCR methods of which quantitative real time PCR (qPCR) is superior to conventional PCR. The established genus-specific B. burgdorferi sl-qPCR is based on probes enhancing specificity through a third oligonucleotide that has to match the target sequence. The designed TaqMan™ minor groove binder (MGB)-probe has an advantage over a conventional TaqMan™ probe regarding mismatch discrimination [22] and the use of a non-fluorescent quencher (NFQ) results in more precise fluorescence signal detection compared to dye quenchers. In conclusion, qPCR utilizing TaqMan™ MGB-probe is the most sensitive and specific detection system. The chosen target sequence, the 5S-23S intergenic spacer (IGS), represents a single copy region within the B. burgdorferi sl genome [20]. Thus, a reliable quantification of the Borrelia burden per tick or the like is given. Consistent with the sensitivity of a LightCycler hybridization probe based qPCR targeting the osp A gene of B. burgdorferi ss, B. afzelii, and B. garinii described by Rauter et al. [23], the detection limit of the established qPCR targeting all European genospecies was 1-10 copies. This corresponds to a detection limit of ≥50-500 Borrelia spirochetes per tick taken the volumes of eluted genomic DNA and template into account. To exclude that negative qPCR results follow from failed DNA isolations, a duplex qPCR was established targeting the Ixodes ITS2 sequence alongside the B. burgdorferi sl IGS sequence. The designed primer/probe combination detected the considered vector species I. ricinus, I. hexagonus and I. canisuga, which feed on humans and domestic mammals besides their wild-living hosts. However, I. hexagonus and I. canisuga are found less frequently parasitizing non wildlife species due to their nest adapted life cycle [24]. Noteworthy, duplex qPCR Borrelia detection was successful using the Thermo Fisher ABsolute™ Blue QPCR low Rox Mix but failed with the Stratagene Brilliant® QPCR Master Mix.
From 1000 I. ricinus collected in recreation areas of the city of Hanover (Lower Saxony, Northern Germany) 24.3% were positive for B. burgdorferi sl considering all developmental tick stages. Similar infection rates (22%) were found in Southern Germany [17], whereas in Central Germany (Thuringia) only 11.1% were positive [14]. Interestingly, both authors observed with 9.0 and 8.6% almost identical Borrelia infections in nymphs. In contrast, in the present study the number of infected nymphs was with 18.1% nearly doubled. Also a higher percentage of adults were positive in the present study (34.6%) than in Central Germany (21.0%) and Southern Germany (29.7%). With respect to larvae, the number of positives is comparable to these of Southern Germany (1.7% and 1.5%; no data for Central Germany). Infections in larvae can either result from transovarial transmission [25, 26], or, since this transmission is rather inefficient, from a blood meal that was interrupted and thus too short for nymph development.
In contrast to 24.3% positives in qPCR, DFM revealed only 7.4% positive ticks depicting the lower sensitivity of this method due to dependency on living spirochetes. This necessity of living bacteria and thus ticks for examination restricts also operational capacity, which is another disadvantage of the DFM technique. Furthermore, as mentioned above, DFM has a low specificity by reason that it is not possible to differentiate between Borrelia spp. and other spirochetes. This became apparent in 20 ticks that were spirochete positive in DFM but negative in genus-specific B. burgdorferi sl qPCR.
The most prevalent genospecies in the present study was B. garinii closely followed by B. afzelii. This is consistent with the findings of Hildebrandt et al. [14] and Fingerle et al. [17] whereas Rauter et al. [23] and Maetzel et al. [16] found B. afzelii more frequent than B. garinii. Interestingly, B. burgdorferi ss, the main causative genospecies of LB in the United States, was the third most common genospecies in Southern and Central Germany [14, 17] but the least frequent in the presented study site in Northern Germany. These prevalence results could potentially be influenced by cross reactions with B. bavariensis sp. nov. since this species was not included in species-specific rpo B PCR specificity tests by Lee et al. [21]. For B. lusitaniae and B. bissettii as further species of the B. burgdorferi sl complex occurring in Central Europe the authors excluded cross reactions by amplification experiments. The present study revealed that the same is true for B. spielmanii. Regarding the B. spielmanii osp A specific primer pair, the present study negated cross reactions with B. burgdorferi ss, B. afzelii, B. garinii and B. valaisiana by PCR experiments. Also for B. bissettii and B. lusitaniae osp A primer sequence comparison gave strong indication that cross reactions can not occur whereas for B. bavariensis sp. nov. no sequence data where available. Consequently, amplification of this Borrelia species by B. spielmanii osp A primers could potentially occur.
In 33 out of the 251 positive/questionable ticks the Borrelia species could not be determined. One explanation is that the target sequence copy number was to low to produce visible bands in gel electrophoresis. Furthermore, B. lusitaniae, B. bissettii and B. bavariensis sp. nov. were not included in conventional species-specific PCR and thus could be considered as not determined species. However, B. lusitaniae is perpetuated by lizards and thus distributed mainly in Mediterranean countries [27]. In Germany, Fingerle et al. [17] found only 1 from 475 positive ticks collected at different study sites in Southern Germany to be infected with B. lusitaniae. The authors also identified B. bissettii in a human cerebrospinal fluid from Germany. Otherwise, this species is mainly reported in human patients from Slovenia [28] and Czech Republic [12, 13]. In Czech Republic, 0.5% ticks were found to be infected with B. bissettii and 0.8% with B. lusitaniae[29]. The recently proposed new species B. bavariensis sp. nov. was so far described by Margos et al. [7] in human samples from Bavaria (Germany), Villach (Austria), and Slovenia as well as by Gern et al. [30] in xenodiagnostic ticks fed on 2 Apodemus sylvaticus mice from the Staatswald in Switzerland [31]. Thus, a small proportion of the unidentified tick infections may be due to B. bissettii, B. lusitaniae or and B. bavariensis sp. nov., but in most of them PCR failed most likely due to insufficient template amounts.
Infections with more than one genospecies were observed in nearly one third of the positive ticks. Because adult ticks had one more blood meal than nymphs, it is not surprising that the number of multiple infected adults (32.1%) is doubled compared to nymphs (15.6%). The combination B. afzelii and B. spielmanii as well as B. garinii and B. valaisiana made the most common double infections. For the latter ones birds are competent reservoirs [32, 33]. Thus, bird-feeding ticks often are infected with both species. Kipp et al. [33] found that the blackbird and the song trush play an important role in the cycle of these two genospecies and at least blackbirds are very frequent in recreational or human inhabited areas. The combination B. garinii/B. valaisiana was also identified by Maetzel et al. [16] as the most common tick double infections. Interestingly, B. spielmanii was observed two times more in multiple infections (30 infected ticks, 12% of all infected ticks) than as single infection (15 infected ticks, 6% of all infected ticks).
Besides I ricinus, I. hexagonus ticks collected from hedgehogs were examined for Borrelia infection resulting in 5.7% positives. Identified Borrelia species were B. afzelii, B. garinii and B. spielmanii. Former studies revealed 2.6 and 2.7% positive I. hexagonus using DFM and direct immunofluorescence, respectively [2, 34]. In a conventional PCR based study, 11.5% of I. hexagonus collected from hedgehogs were infected with B. burgdorferi sl [35]. Nest adaptation of I. hexagonus suggests that the ticks in the present study were infected by hedgehogs, for which reservoir competency for the B. burgdorferi sl-complex was shown [25, 36].
Conclusions
With the TaqMan™ MGB-probe based qPCR a sensitive, specific, and rapid tool to detect spirochetes of the B. burgdorferi sl-complex was established. Enhancement to a duplex qPCR also targeting the 16S region of tested common ixodid ticks provides a positive control of successful isolation of DNA from the ticks besides the diagnostic purpose, assuring proper results not only for epidemiological studies but also for statements of diagnostic laboratories processing transmitted ticks from private parties. The designed B. spielmanii specific conventional PCR filled the gap to identify the most common European pathogenic Borrelia species by DNA amplification since Lee et al. [21] described PCR protocols for B. burgdorferi ss, B. afzelii, B. garinii and B. valaisiana only.
We have found that all pathogenic species of the B. burgdorferi sl-complex were present in ticks collected in recreational areas in the city of Hanover and B. afzelii, B. garinii and B. spielmanii were identified in I. hexagonus ticks collected from hedgehogs confirming their role as reservoirs for pathogenic species of the B. burgdorferi sl-complex.
Methods
Spirochete cultures for subsequent DNA isolation
Spirochetes were cultured to obtain genomic DNA for establishing the genus- and species-specific Borrelia PCR and to determine sensitivity and specificity. Furthermore, the DNA of different species of the Borrelia burgdorferi sl-complex served as species-specific PCR positive controls. The following organisms were cultured: B. burgdorferi ss (strain B31 and LW2), B. afzelii (strain NE632 and ZQ1), B. garinii (strain 1B29 and A87SB), B. valaisiana (strain VS116), B. spielmanii, Treponema sokranskii, Leptospira grippothyphosa, and L. interrogans. These spirochetes were cultivated in sterile filtered modified Barbour-Stoenner-Kelly medium (BSKH medium, Sigma-Aldrich, Taufkirchen, Germany) at 37°C in 50 ml culture flasks (Sarstedt, Nürmbrecht, Germany). Cultures were monitored twice a week with the dark-field microscope, and refilled with 15 ml of fresh medium. After 14 days the cultures were centrifuged at 8000 × g for 10 min and the resulting pellets diluted with 50 μl of sterile filtered PBS. Genomic DNA was isolated using the QIAamp® DNA Blood Mini Kit (Quiagen, Hilden), according to the manufacturer's instructions, and eluted in 100 μl of deionized H2O.
Genus-specific quantitative real time PCR to detect Borrelia burgdorferi sl
For genus-specific detection of the B. burgdorferi sl-complex, a TaqMan™ minor groove binder (MGB)-probe and corresponding primers were designed using the Primer Express™ software (Applied Biosystems, Darmstadt, Germany). As target sequence the 5S-23S intergenic spacer (IGS) was chosen and the design based on species recognized in Europe, whereby diversity was considered in sequence selection. Selected sequences were (GenBank accession numbers and origin in brackets): B. burgdorferi ss (AF497981, Czech Republic; DQ393308, France; AY583237, Russia), B. afzelii (AF497984, Czech Republic; DQ111066, France; DQ020300, Russia), B. garinii (AF497993, Czech Republic; AY163784; Latvia; AY163784, Russia), B. valaisiana (AF497988, Czech Republic; AF497989, Czech Republic; U78150, The Netherlands), and B. lusitaniae (AB091801, Turkey; AB091799, Turkey; DQ111065, France).
Designed primer sequences and corresponding TaqMan™-MGB-probe were also checked for sequence matching with B. spielmanii (AF497994, Czech Republic; AM160603, Germany; AM183337, France) and B. bissettii (FJ431140, Czech Republic; FJ431142, Czech Republic; EF015627 USA) after sequence availability in the National Center for Biotechnology Information (NCBI) database. Primer and probe nucleotide sequences are listed in Table 4. The probe was purchased from Applied Biosystems (Darmstadt, Germany) and primers from Invitrogen (Karlsruhe, Germany).
Performance of primers and probe in quantitative real time PCR (qPCR) was tested with isolated DNA from the above mentioned Borrelia cultures. The reaction was set up using the Brilliant® QPCR Master Mix (Stratagene, Heidelberg, Germany): 8.49 μl deionized H2O, 12.50 μl Brilliant buffer (containing SureStart®Taq Polymerase), 0.75 μl forward and reverse primer (100 μM each), respectively, 0.13 μl probe (50 μM), 0.38 μl diluted ROX as reference dye (1:500 dilution), and 2 μl template. Thermal cycling conditions were: 10 min at 94°C followed by 40 cycles of 20 sec at 94°C, 60 sec at 56°C and 45 sec at 72°C. Experiments and data analysis were performed using the Mx3005 Multiplex Quantitative PCR System (Stratagene, Heidelberg, Germany). Amplification products were analyzed via gel electrophoresis (2% agarose gels). The bands were cut out, ligated into pCR® 4-TOPO® vector followed by transformation of Escherichia coli One Shot® TOP 10 cells (TOPO TA Cloning® Kit for Sequencing; Invitrogen, Karlsruhe, Germany). Plasmid DNA was obtained using the NucleoSpin® Plasmid Kit (Macherey-Nagel, Dueren, Germany) following the manufacturer's recommendations and sequenced at the SEQLAB Sequence laboratories (Göttingen, Germany). Sequences were verified by using BLAST searching the non-redundant NCBI database http://www.ncbi.nlm.nih.gov/BLAST/.
The plasmid containing the verified sequence of B. afzelii strain NE632 served as plasmid standard in prospective qPCR runs to ensure effective PCR amplification and detection of the fluorescence signal as well as to compile standard curves for determination of the IGS copy number and thus for determination of the Borrelia burden in individual ticks. For these standards, 10-fold serial plasmid dilutions ranging from 100 to 106 copies per reaction were prepared.
While the primer/probe combination perfectly matched the IGS sequence of B. burgdorferi ss, B. afzelii, B. garinii, B. bissettii, and B. valaisiana, there was one mismatch in position 8 of the reverse primer sequence (C instead of T) with B. spielmanii and B. lusitaniae. To examine whether this mismatch has an impact on detection, qPCR runs were performed using a plasmid standard containing the targeted IGS-sequence of B. spielmanii and B. lusitaniae as template.
Specificity and sensitivity of the genus-specific quantitative real time PCR
To test specificity, isolated DNA of cultured related spirochetes (Treponema sp. and Leptospira spp.) was checked for cross reactivity in qPCR runs. Besides this control, genomic DNA of 100 laboratory bred I. ricinus larvae was also used as negative control. These ticks were processed in the same manner as field-collected ticks described below.
Sensitivity testing was conducted by amplification of the plasmid standard serially diluted from 100 to 106 copies. Furthermore, DNA of different dilutions from cultured B. afzelii spirochetes (strain NE632) as well as spiked negative laboratory bred larvae, nymphs, and adults of I. ricinus were amplified to investigate the effect of tick material on the sensitivity of the qPCR.
Duplex genus-specific quantitative real time PCR including a DNA isolation positive control
For verification of successful genomic DNA isolation, a duplex quantitative real time PCR was established targeting the ITS2-sequence of Ixodes-ticks besides the IGS-sequence of the B. burgdorferi sl-complex. The positive control primer/TaqMan™-MGB-probe combination was designed based on the ITS2-sequence of I. ricinus (GenBank accession number D88884) using the AlleleID® 7 software (version 7.01., Premier Biosoft International, Palo Alto, USA). The nucleotide sequences of the primers and corresponding TaqMan™-MGB-probe are listed in Table 4. The probe was purchased from Applied Biosystems (Darmstadt, Germany) and primers from Invitrogen (Karlsruhe, Germany). To ensure detection of the three most common mammalian Ixodes spp. feeding on mammals in Central Europe (I. ricinus, I. hexagonus and I. canisuga), the primer-probe combination was tested on genomic DNA of the following individuals: 280 I. ricinus (9 larvae, 134 nymphs, 71 adult males, and 66 adult females), 8 I. hexagonus (adult females), and 8 I. canisuga (1 larvae and 7 adult females).
To confirm qPCR signal detection, the genomic DNA of each eight individuals of the three Ixodes spp. was amplified with different primer combinations covering different lengths of the Ixodes ITS2-sequence. Used forward primers were: 5'-TTC TTT TGG CGT GGA TGT TGT TCG-3', 5'-TGC GTC GTA GCC TTC-3', and 5'-CTT CTT GCT CGA AGG AGA G-3'. Used reverse primers were: 5'-GCA TCG CTT TCG ATT CGA CAA AAA-3' and 5'-GGG GGT TGT CTC GCC TGA TGT G-3'. Nucleotide data obtained by sequencing the PCR products cloned into pCR® 4-TOPO® have been submitted to the GenBank database under accession numbers GQ330512 - GQ330535. The sequence deposited under the acc. no. GQ330512 (I. ricinus amplified with the primer combination 5'-TTC TTT TGG CGT GGA TGT TGT TCG-3' and 5'-GGG GGT TGT CTC GCC TGA TGT G-3') represented the positive control plasmid standard in subsequent qPCR runs.
The set up for the genus-specific qPCR was adapted insofar as for the volume of Ixodes ITS2-specific primers and probe the amount of deionized H2O was reduced. Besides the used Brilliant® QPCR Master Mix (Stratagene, Heidelberg, Germany), the ABsolute™ Blue QPCR low Rox Mix (Thermo Fisher Scientific, Hamburg, Germany) was tested for qPCR setup. The qPCR temperature profile was retained unchanged.
Species-specific conventional PCR to analyse B. burgdorferi sl species distribution
Conventional species-specific PCRs can be used to differentiate species of the B. burgdorferi sl-complex. It was performed for each collected tick (described below) positive in genus-specific qPCR. The species B. burgdorferi ss, B. afzelii, B. garinii and B. valaisiana were detected with previously described specific primers targeting the RNA polymerase subunit B (rpo B) gene [21]. To detect B. spielmanii, outer surface protein A (osp A) gene specific primers were designed using the Lasergene PrimerSelect program (DNASTAR, version 5.06; GATC Biotech, Konstanz, Germany) as primer design software. The following osp A sequences from European B. spielmanii isolates were used for primer design: Accession nos. DQ133517 (France), AF102057 (The Netherlands), and EU545183 (Turkey). The nucleotide sequences of all species-specific primers supplied by Invitrogen (Karlsruhe, Germany) are listed in Table 4. Specificity of the designed osp A primers as well as the published rpo B primers was tested with DNA from B. spielmanii, B. burgdorferi ss, B. afzelii, B. garinii, and B. valaisiana, and other related spirochetes (Treponema spp. and Leptospira spp.) cultured as described above. For B. bissettii and B. lusitaniae specificity was checked via sequence alignment (Align Plus 5, vs. 5.04; Scientific & Educational software, Cary, USA) based on acc nos. DQ393323 and EF457558, respectively. Sequence information for B. bavariensis sp. nov. osp A were not available in the databases. Furthermore, specificity was tested on 100 laboratory bred I. ricinus larvae and additionally via Nucleotide BLAST http://blast.ncbi.nlm.nih.gov/Blast.cgi. The PCR product resulting from B. spielmanii DNA amplification was cloned and sequenced for verification.
PCR setup for the conventional species-specific PCR was as follows: 3 μl template was added to 14.2 μl deionized H2O, 2.5 μl 10× buffer, 2.5 μl 10× CoralLoad PCR buffer, 0.5 μl MgCl2 (50 mM), 0.5 μl deoxyribonucleotide triphosphates (10 mM each), 0.8 μl gene-specific forward and reverse primer (10 μM each), respectively, and 0.2 μl Taq Polymerase (5 U/μl; Qiagen, Hilden, Germany). PCR cycling (40 cycles) was performed using for B. burgdorferi ss, B. afzelii, B. garinii and B. valaisiana the annealing temperatures described by Lee et al. [21] and 57°C for B. spielmanii. The following temperature profile was used: Initial denaturing at 94°C for 4 min, denaturing at 94°C for 30 sec, annealing for 30 sec, extending primers at 72°C for 30 sec and final elongation for 4 min.
Analysis of B. burgdorferi sl infections in ticks of the city of Hanover
In the city of Hanover, the capital of the federal state Lower Saxony, 1000 ticks were collected in recreation areas using the flagging method for subsequent investigation of infection with the B. burgdorferi sl-complex. Furthermore, the dark-field microscopy (DFM) technique should be compared to the established qPCR method.
To perform the DFM analysis, every selected tick was placed in an individual tube and mechanically homogenized with single-use polystyrene homogenizers (Roth, Karlsruhe, Germany) in 50 μl sterile filtered phosphate buffered saline (PBS; pH 7.2). From the homogenized solution, 10 μl were placed on a microscope slide and entirely examined for spirochetes using a DFM microscope (Zeiss, Jena, Germany). If one spirochete was found, the sample was determined as positive. After DFM analysis, the vials containing the ticks' homogenized solution were stored at -75°C until DNA isolation as described above.
When performing genus-specific qPCR, the micro titre plates contained duplicates of each tick sample as well as serially diluted plasmid standards to generate standard curves and a no-template control. Each tick positive in genus-specific qPCR was examined with species-specific conventional PCR to obtain information about the species distribution.
Analysis of Borrelia infections in ticks collected from hedgehogs
Besides hanoverian ticks, 238 ticks collected from hedgehogs in 23 different sites of the federal states Schleswig-Holstein, North-Rhine Westphalia, Saxony, Baden-Württemberg, Bavaria, and Berlin were analysed for infection with B. burgdorferi sl by genus-specific quantitative real time PCR and positive ticks were analysed for species distribution via conventional PCR.
References
Gern L, Humair F: Ecology of Borrelia burgdorferi sensu lato in Europe. Lyme borreliosis: biology, epidemiology and control. Edited by: Gray JS, Kahl O, Lane RS, Stanek G. 2002, United Kingdom: CAB International, 49-174.
Liebisch A, Olbrich S, Brand A, Liebisch G, Mourettou-Kunitz M: Natürliche Infektionen der Zeckenart Ixodes hexagonus mit Borrelien (Borrelia burgdorferi). Tierärztl Umschau. 1989, 44: 809-810.
Estrada-Pena A, Oteo JA, Estrada-Pena R, Gortazar C, Osacar JJ, Moreno JA, Castella J: Borrelia burgdorferi sensu lato in ticks (Acari: Ixodidae) from two different foci in Spain. Exp Appl Acarol. 1995, 19: 173-180. 10.1007/BF00046289.
Olsen B, Jaenson TG, Noppa L, Bunikis J, Bergstrom S: A Lyme borreliosis cycle in seabirds and Ixodes uriae ticks. Nature. 1993, 362: 340-342. 10.1038/362340a0.
Rudenko N, Golovchenko M, Grubhoffer L, Oliver JH: Borrelia carolinensis sp. nov., a new (14th) member of the Borrelia burgdorferi sensu lato complex from the southeastern region of the United States. J Clin Microbiol. 2009, 47: 134-141. 10.1128/JCM.01183-08.
Rudenko N, Golovchenko M, Lin T, Gao L, Grubhoffer L, Oliver JH: Delineation of a new species of the Borrelia burgdorferi sensu lato complex, Borrelia americana sp. nov. J Clin Microbiol. 2009, 47: 3875-3880. 10.1128/JCM.01050-09.
Margos G, Vollmer SA, Cornet M, Garnier M, Fingerle V, Wilske B, Bormane A, Vitorino L, Collares-Pereira M, Drancourt M, Kurtenbach K: A new Borrelia species defined by multilocus sequence analysis of housekeeping genes. Exp Appl Acarol. 2009, 75: 5410-5416.
Chu CY, Liu W, Jiang BG, Wang DM, Jiang WJ, Zhao QM, Zhang PH, Wang ZX, Tang GP, Yang H, Cao WC: Novel genospecies of Borrelia burgdorferi sensu lato from rodents and ticks in southwestern China. J Clin Microbiol. 2008, 46: 3130-3133. 10.1128/JCM.01195-08.
Ryffel K, Peter O, Rutti B, Suard A, Dayer E: Scored antibody reactivity determined by immunoblotting shows an association between clinical manifestations and presence of Borrelia burgdorferi sensu stricto, B. garinii, B. afzelii, and B. valaisiana in humans. J Clin Microbiol. 1999, 37: 4086-4092.
Collares-Pereira M, Couceiro S, Franca I, Kurtenbach K, Schafer SM, Vitorino L, Goncalves L, Baptista S, Vieira ML, Cunha C: First isolation of Borrelia lusitaniae from a human patient. J Clin Microbiol. 2004, 42: 1316-1318. 10.1128/JCM.42.3.1316-1318.2004.
da Franca I, Santos L, Mesquita T, Collares-Pereira M, Baptista S, Vieira L, Viana I, Vale E, Prates C: Lyme borreliosis in Portugal caused by Borrelia lusitaniae? Clinical report on the first patient with a positive skin isolate. Wien Klin Wochenschr. 2005, 117: 429-432. 10.1007/s00508-005-0386-z.
Rudenko N, Golovchenko M, Mokracek A, Piskunova N, Ruzek D, Mallatova N, Grubhoffer L: Detection of Borrelia bissettii in cardiac valve tissue of a patient with endocarditis and aortic valve stenosis in the Czech Republic. J Clin Microbiol. 2008, 46: 3540-3543. 10.1128/JCM.01032-08.
Rudenko N, Golovchenko M, Ruzek D, Piskunova N, Mallatova N, Grubhoffer L: Molecular detection of Borrelia bissettii DNA in serum samples from patients in the Czech Republic with suspected borreliosis. FEMS Microbiol Lett. 2009, 292: 274-281. 10.1111/j.1574-6968.2009.01498.x.
Hildebrandt A, Schmidt KH, Wilske B, Dorn W, Straube E, Fingerle V: Prevalence of four species of Borrelia burgdorferi sensu lato and coinfection with Anaplasma phagocytophila in Ixodes ricinus ticks in central Germany. Eur J Clin Microbiol Infect Dis. 2003, 22: 364-367. 10.1007/s10096-003-0926-2.
Kampen H, Rotzel DC, Kurtenbach K, Maier WA, Seitz HM: Substantial rise in the prevalence of Lyme borreliosis spirochetes in a region of western Germany over a 10-year period. Exp Appl Acarol. 2004, 70: 1576-1582.
Maetzel D, Maier WA, Kampen H: Borrelia burgdorferi infection prevalences in questing Ixodes ricinus ticks (Acari: Ixodidae) in urban and suburban Bonn, western Germany. Parasitol Res. 2005, 95: 5-12. 10.1007/s00436-004-1240-3.
Fingerle V, Schulte-Spechtel UC, Ruzic-Sabljic E, Leonhard S, Hofmann H, Weber K, Pfister K, Strle F, Wilske B: Epidemiological aspects and molecular characterization of Borrelia burgdorferi s.l. from southern Germany with special respect to the new species Borrelia spielmanii sp. nov. Int J Med Microbiol. 2008, 298: 279-290. 10.1016/j.ijmm.2007.05.002.
Rauter C, Hartung T: Prevalence of Borrelia burgdorferi sensu lato genospecies in Ixodes ricinus ticks in Europe: a metaanalysis. Exp Appl Acarol. 2005, 71: 7203-7216.
Hubalek Z, Halouzka J: Prevalence rates of Borrelia burgdorferi sensu lato in host-seeking Ixodes ricinus ticks in Europe. Parasitol Res. 1998, 84: 167-172.
Schwartz JJ, Gazumyan A, Schwartz I: rRNA gene organization in the Lyme disease spirochete, Borrelia burgdorferi. J Bacteriol. 1992, 174: 3757-3765.
Lee SH, Kim BJ, Kim JH, Park KH, Kim SJ, Kook YH: Differentiation of Borrelia burgdorferi sensu lato on the basis of RNA polymerase gene (rpoB) sequences. J Clin Microbiol. 2000, 38: 2557-2562.
Yao Y, Nellaker C, Karlsson H: Evaluation of minor groove binding probe and Taqman probe PCR assays: Influence of mismatches and template complexity on quantification. Mol Cell Probes. 2006, 20: 311-316.
Rauter C, Oehme R, Diterich I, Engele M, Hartung T: Distribution of clinically relevant Borrelia genospecies in ticks assessed by a novel, single-run, real-time PCR. J Clin Microbiol. 2002, 40: 36-43. 10.1128/JCM.40.1.36-43.2002.
Liebisch A, Walter G: Ticks of domestic and wild animals in Germany: on the occurrence and biology of the hedgehog tick (Ixodes hexgonus) and the fox tick (Ixodes canisuga). Dtsch Tierarztl Wochenschr. 1986, 93: 447-450.
Gern L, Rouvinez E, Toutoungi LN, Godfroid E: Transmission cycles of Borrelia burgdorferi sensu lato involving Ixodes ricinus and/or I. hexagonus ticks and the European hedgehog, Erinaceus europaeus, in suburban and urban areas in Switzerland. Folia Parasitol (Praha). 1997, 44: 309-314.
Nefedova V: Studies on the transovarial transmission of Borrelia burgdorferi sensu lato in the taiga tick Ixodes persulcatus. Folia Parasitol (Praha). 2004, 51: 67-71.
Richter D, Matuschka FR: Perpetuation of the Lyme disease spirochete Borrelia lusitaniae by lizards. Exp Appl Acarol. 2006, 72: 4627-4632.
Picken RN, Cheng Y, Strle F, Picken MM: Patient isolates of Borrelia burgdorferi sensu lato with genotypic and phenotypic similarities of strain 25015. J Infect Dis. 1996, 174: 1112-1115.
Hulinska D, Votypka J, Kriz B, Holinkova N, Novakova J, Hulinsky V: Phenotypic and genotypic analysis of Borrelia spp. isolated from Ixodes ricinus ticks by using electrophoretic chips and real-time polymerase chain reaction. Folia Microbiol (Praha). 2007, 52: 315-324. 10.1007/BF02932085.
Gern L, Douet V, López Z, Rais O, Morán Cadenas F: Diversity of Borrelia genospecies in Ixodes ricinus ticks in a Lyme borreliosis endemic area in Switzerland identified by using new probes for reverse line blotting. Ticks Tick Borne Dis. 2010, 1: 23-29. 10.1016/j.ttbdis.2009.11.001.
Huegli D, Hu CM, Humair PF, Wilske B, Gern L: Apodemus species mice are reservoir hosts of Borrelia garinii OspA serotype 4 in Switzerland. J Clin Microbiol. 2002, 40: 4735-4737. 10.1128/JCM.40.12.4735-4737.2002.
Kurtenbach K, De Michelis S, Sewell HS, Etti S, Schafer SM, Hails R, Collares-Pereira M, Santos-Reis M, Hanincova K, Labuda M, Bormane A, Donaghy M: Distinct combinations of Borrelia burgdorferi sensu lato genospecies found in individual questing ticks from Europe. Exp Appl Acarol. 2001, 67: 4926-4929.
Kipp S, Goedecke A, Dorn W, Wilske B, Fingerle V: Role of birds in Thuringia, Germany, in the natural cycle of Borrelia burgdorferi sensu lato, the Lyme disease spirochaete. Int J Med Microbiol. 2006, 296 (Suppl 40): 125-128. 10.1016/j.ijmm.2006.01.001.
Finkbeiner-Weber B: The role of the hedgehog and its ectoparasites for the epidemiology of Lyme-borreliosis. Thesis. 1996, Tierärztliche Hochschule Hannover, Institute for Parasitology, 146-[http://elib.tiho-hannover.de/dissertations/96finkbeiner-weber-b.pdf]
Hubbard MJ, Baker AS, Cann KJ: Distribution of Borrelia burgdorferi s.l. spirochaete DNA in British ticks (Argasidae and Ixodidae) since the 19th century, assessed by PCR. Med Vet Entomol. 1998, 12: 89-97. 10.1046/j.1365-2915.1998.00088.x.
Gray JS, Kahl O, Janetzki-Mittman C, Stein J, Guy E: Acquisition of Borrelia burgdorferi by Ixodes ricinus ticks fed on the European hedgehog, Erinaceus europaeus L. Exp Appl Acarol. 1994, 18: 485-491. 10.1007/BF00051470.
Acknowledgements
The authors want to thank Gabriele Liebisch (Zecklab, Burgwedel) for kindly providing Ixodes canisuga ticks, Dania Richter and Annette Moter (Charite, Berlin) for kindly providing Borrelia spielmanii and Treponema sokranskii, respectively, and the Innovative Veterinary Diagnostics Laboratory (Hanover) for kindly providing Leptospira grippothyphosa and L. interrogans.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
VMM collected the ticks, carried out the molecular work and participated in analysis. EE carried out duplex quantitative real time PCR and participated in analysis. TS and CE conceived and designed the study and participated in analysis. CS drafted the manuscript, analysed and interpreted the data, and participated in the design of the study. All authors read and approved the final manuscript.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Strube, C., Montenegro, V.M., Epe, C. et al. Establishment of a minor groove binder-probe based quantitative real time PCR to detect Borrelia burgdorferi sensu lato and differentiation of Borrelia spielmanii by osp A-specific conventional PCR. Parasites Vectors 3, 69 (2010). https://doi.org/10.1186/1756-3305-3-69
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-3305-3-69