
Abstract
Background
Anti–N-methyl-D-aspartate receptor immunoglobulin G antibodies directed against the GluN1 subunit are considered highly specific for anti-N-methyl-D-aspartate receptor encephalitis, a severe clinical syndrome characterized by seizures, psychiatric symptoms, orofacial dyskinesia and autonomic dysfunction.
Case presentation
Here we report a 33 year old Caucasian male patient with clinically definite multiple sclerosis who was found to be positive for anti-N-methyl-D-aspartate receptor antibodies. Rituximab therapy was initiated. On the 18 months follow-up visit the patient was found to be clinically stable, without typical signs of anti-N-methyl-D-aspartate receptor encephalitis.
Conclusion
Our findings add to the growing evidence for a possible association between anti-N-methyl-D-aspartate receptor encephalitis and demyelinating diseases.
Similar content being viewed by others


Background
Patients with immunoglobulin (Ig) G antibodies to the GluN1 subunit of the N-methyl-D-aspartate receptor (NMDAR) develop a characteristic clinical syndrome that was termed anti-NMDA receptor encephalitis [1]. These patients may present with acute behavioural changes, dyskinesia or seizures followed by a decrease in consciousness, psychosis, central hypoventilation and autonomic dysregulation. Here we report a male patient who had signs and symptoms highly suggestive of multiple sclerosis (MS) and met the McDonald criteria [2] and yet was found to be seropositive for NMDAR IgG antibodies. Given the high specificity of this antibody, the question arises whether NMDAR encephalitis may mimic MS or whether NMDAR IgG is a coincidental finding either without clinical significance or indicative of a clinically silent (prodromal) state of the disease. Nowadays diagnostic opportunities may challenge the clinician’s appraisal and impose once more the question: treat the patient or his lab results – or both?
Case presentation
This 33 year-old Caucasian male patient presented to our emergency department with mild left-sided hemiparesis that had developed within days. Six years prior, he had been treated for subacute-onset diplopia at another hospital. At that time, MRI demonstrated several demyelinating periventricular lesions with one gadolinium enhancing lesion. Cerebrospinal fluid (CSF) analysis revealed oligoclonal bands. Following a glucocorticoid pulse therapy complete remission was reached. He was lost to follow up and no further MRI scans or neurological examinations were performed since he remained asymptomatic and felt to be in good health.
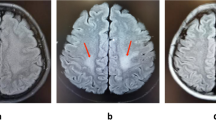
Cranial MRI on admission demonstrated multiple T2 hyperintense lesions (periventricular, juxtacortical, infratentorial), one of them with gadolinium enhancement (Figure 1). Upon comparison to the MRI six years before, an obvious increase of the lesion load was noted. Visually evoked potentials showed prolonged latencies in both eyes. CSF analysis revealed a borderline pleocytosis (5 cells/μl, 95% lymphocytes). The IgG index (1.3) indicated intrathecal IgG synthesis. A polyspecific intrathecal immunoglobulin synthesis against rubella, varicella and herpes simplex virus but not measles (MRZH reaction) was detected. Oligoclonal bands were found to be positive in serum and cerebrospinal fluid with additional bands in the CSF. Blood work and urine testing were unremarkable without evidence of chronic infection (syphilis, borreliosis, HIV, HBV and HCV), vitamin B12 deficiency or systemic autoimmune disease (ANA, ENA, ANCA, RF, dsDNA and anti-phospholipid antibodies, ACE). The patient was diagnosed with relapsing remitting MS according to the revised McDonald criteria [2] and received a glucocorticoid pulse (5 days, methylprednisolone 1 g/day i.v.) followed by complete recovery. 3 weeks later he returned complaining of new-onset paroxysmal tingling and cramping in his left hand and was found to have tonic spasms that responded well to another course of glucocorticoids and intermittent low dose carbamazepine therapy. Given the higher incidence of tonic spasms in neuromyelitis optica compared to MS [3] we decided to test for aquaporin-4 (AQ4) autoantibodies before initiation of immunomodulatory therapy. Serum samples were sent to an accredited commercial laboratory with long-standing experience (Stöcker Laboratories, Euroimmun AG, Lübeck) that employs a biochip to test for AQ4 autoantibodies in a cell based assay. This biochip consists of a mosaic of fixed human embryonal kidney 239 cells each expressing different recombinant antigens (AQ4, Glu1 NMDAR, AMPAR, GABA-bR, LGI, CASPR2, Amphiphysin, GAD, Hu, Ri, Yo, Tr, MAG, Myelin, Ma/Ta, Glycine receptor) in addition to frozen sections of rat hippocampus and cerebellum. The patient turned out to be AQ4 autoantibody negative but surprisingly IgG directed against the NR1 subunit of the NMDAR (titre 1:100) was detected and confirmed by a typical staining pattern on rat brain. Control testing with an independent serum sample yielded the same result. A third serum sample was sent to a second laboratory (A. Vincent, Imunology Laboratory, Churchill Hospital, Oxford, GB) and confirmed the results and titres.

Magnetic resonance imaging. MR FLAIR imaging demonstrates multiple hyperintense lesions suggestive of multiple sclerosis (A-C, E). One of the lesions (arrow) was found to be Gadolinium (Gd) enhancing on T1 scans (D, F).
Further diagnostic tests were performed: Electroencephalography showed a normal alpha-rhythm without evidence for epileptic activity or slowing. Neuropsychological evaluation (Wechsler Memory Scale Revised Edition; Digit span forward/backward; Spatial span backward/forward; Logical memory I, Go/Nogo, Divided attention version I/auditive-visual) demonstrated normal cognitive functions. A psychiatric exploration as well as thorax/adomen CT and spinal cord MRI were unremarkable. A B cell depleting therapy with rituximab was started and well tolerated. A complete B cell depletion was confirmed by immune cell phenotyping. Since then (18 months) the patient has been monitored at short intervals and repeatedly been assessed for seizures, cognitive decline or behavioral changes as well as symptoms of MS relapse but was considered clinically stable. In addition MRI demonstrated paraclinical stability 12 months after the onset of symptoms. NMDAR Ab testing demonstrated persistence at an unchanged titre of 1:100.
Discussion
Here we describe a patient with clinically definite MS and the presence of NMADR serum antibodies. He was carefully evaluated for seizures, dyskinesia, psychiatric or neuropsychological symptoms which were all negative. Thus, the clinical line-up found was best compatible with relapsing remitting MS and not considered indicative of NMDAR encephalitis.
In contrast to IgA and IgM antibodies directed against the NMDA receptor, NMDAR IgG serum antibodies are in general considered highly specific for NMDAR encephalitis. However, depending on the diagnostic test, false positive IgG serum results have been reported in up to 3% of healthy controls [4, 5]. In our patient, a positive cell based assay [6], was confirmed by a typical staining pattern on rat brain tissue, and in another laboratory. According to previously published studies CSF adds to the sensitivity [7] and the specificity [4] of NMDAR antibody tests. Unfortunately CSF was not available to confirm intrathecal synthesis and the patient declined to undergo a second lumbar puncture which limits the significance of our findings. Also unfortunately, we were unable to obtain high quality images of the IHC/IF stainings done by Labor Stöcker which would have enhanced our report.
Monosymptomatic disease courses of NMDAR encephalitis (“forms frustes”) have been reported. Accordingly, NMDAR IgG Ab were recently described at low frequency (1.7%) in a cohort of epilepsy patients who did not display other features of encephalitis [8]. Similarily, patients with isolated psychiatric symptoms at onset or during relapse were reported [9]. In addition persistence of NMDAR IgG after an initial episode of encephalitis as well as relapses following long clinically silent periods have been reported [10]. Therefore, NMDAR serum IgG may as well precede the clinical onset of NMDAR encephalitis.
A correlation of serum titers with disease activity is a subject of ongoing debate, but a recent study show that higher antibody titers are associated with poor outcome or the presence of teratoma [7]. Our patient had a rather low serum titer of 1:100 (normal <1:10) and tumor screening remained negative.
Anecdotal evidence points towards a possible association of NMDAR encephalitis with demyelinating disease: NMDAR IgG have been reported in children with demyelinating syndromes (ADEM, ON) [11] and extensive myelitis [12]. In addition two cases of patients that initially presented with typical symptoms of NMDAR encephalitis and developed a disease closely mimicking neuromyelitis optica have been reported [13, 14] and a range of demyelinating conditions were reported in case series in adults and children [15, 16].
In contrast a study testing NMDAR IgG antibodies in a small cohort of MS patients as a disease control failed to detect the autoantibody [6]. Herpes encephalitis has been reported as a possible trigger of NMDA receptor antibody formation and consecutive encephalitis [17–19]. The mechanisms remain unknown. However, both molecular mimicry and breakdown of immunologic tolerance towards the NMDAR released by neuronal damage in an inflamed environment have been discussed [17, 18]. Whether tissue damage and inflammation in MS may also trigger the formation of this antibody remains speculative.
Conclusion
The growing availability of chip technologies that perform simultaneous testing of multiple antibodies independent of the clinical syndrome and with limited confirmatory tests poses the risk of false-positive results and challenges clinicians and laboratory physicians alike who have to decide whether these autoantibodies may indicate an evolving autoimmune process or represent a clinical non-significant epiphenomenon. Further research is warranted to clarify a possible link between demyelinating diseases and NMDAR encephalitis.
In general, diagnosis of any autoimmune disease necessitates the combination of a compatible clinical picture together with conformed laboratory findings and treatment should not be based on laboratory findings alone.
Consent
Written informed consent was obtained from the patient for publication of this Case Report and any accompanying images. A copy of the written consent is available for review by the Editor-in-Chief of this journal.
Abbreviations
- ACE:
-
Angiotensin-converting-enzyme
- ANA:
-
Antinuclear antibodies
- ENA:
-
Extractable nuclear antigens
- HBV:
-
Hepatitis B virus
- HCV:
-
Hepatitis C virus
- IgG:
-
Immunoglobulin G
- MS:
-
Multiple sclerosis
- NMDAR:
-
N-methyl-D-aspartate receptor
- RF:
-
Rheumatoid factor.
References
Dalmau J, Gleichman AJ, Hughes EG, Rossi JE, Peng X, Lai M, Dessain SK, Rosenfeld MR, Balice-Gordon R, Lynch DR: Anti-NMDA-receptor encephalitis: case series and analysis of the effects of antibodies. Lancet Neurol. 2008, 7: 1091-1098. 10.1016/S1474-4422(08)70224-2.
Polman CH, Reingold SC, Banwell B, Clanet M, Cohen JA, Filippi M, Fujihara K, Havrdova E, Hutchinson M, Kappos L, Lublin FD, Montalban X, O'Connor P, Sandberg-Wollheim M, Thompson AJ, Waubant E, Weinshenker B, Wolinsky JS: Diagnostic criteria for multiple sclerosis: 2010 revisions to the McDonald criteria. Ann Neurol. 2011, 69: 292-302. 10.1002/ana.22366.
Kim SM, Go MJ, Sung JJ, Park KS, Lee KW: Painful tonic spasm in neuromyelitis optica: incidence, diagnostic utility, and clinical characteristics. Arch Neurol. 2012, 69: 1026-1031.
Viaccoz A, Desestret V, Ducray F, Picard G, Cavillon G, Rogemond V, Antoine JC, Delattre JY, Honnorat J: Clinical specificities of adult male patients with NMDA receptor antibodies encephalitis. Neurology. 2014, 82: 556-563. 10.1212/WNL.0000000000000126.
Dahm L, Ott C, Steiner J, Stepniak B, Teegen B, Saschenbrecker S, Hammer C, Borowski K, Begemann M, Lemke S, Rentzsch K, Probst C, Martens H, Wienands J, Spalletta G, Weissenborn K, Stocker W, Ehrenreich H: Seroprevalence of autoantibodies against brain antigens in health and disease. Ann Neurol. 2014, 76: 82-94. 10.1002/ana.24189.
Suh-Lailam BB, Haven TR, Copple SS, Knapp D, Jaskowski TD, Tebo AE: Anti-NMDA-receptor antibody encephalitis: performance evaluation and laboratory experience with the anti-NMDA-receptor IgG assay. Clin Chim Acta. 2013, 421: 1-6.
Gresa-Arribas N, Titulaer MJ, Torrents A, Aguilar E, McCracken L, Leypoldt F, Gleichman AJ, Balice-Gordon R, Rosenfeld MR, Lynch D, Graus F, Dalmau J: Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study. Lancet Neurol. 2013
Brenner T, Sills GJ, Hart Y, Howell S, Waters P, Brodie MJ, Vincent A, Lang B: Prevalence of neurologic autoantibodies in cohorts of patients with new and established epilepsy. Epilepsia. 2013, 54: 1028-1035. 10.1111/epi.12127.
Kayser MS, Titulaer MJ, Gresa-Arribas N, Dalmau J: Frequency and characteristics of isolated psychiatric episodes in anti-N-methyl-d-aspartate receptor encephalitis. JAMA Neurol. 2013, 70: 1133-1139. 10.1001/jamaneurol.2013.3216.
Hansen HC, Klingbeil C, Dalmau J, Li W, Weissbrich B, Wandinger KP: Persistent intrathecal antibody synthesis 15 years after recovering from anti-N-methyl-D-aspartate receptor encephalitis. JAMA Neurol. 2013, 70: 117-119. 10.1001/jamaneurol.2013.585.
Hacohen Y, Absoud M, Woodhall M, Cummins C, De Goede CG, Hemingway C, Jardine PE, Kneen R, Pike MG, Whitehouse WP, Wassmer E, Waters P, Vincent A, Lim M: Autoantibody biomarkers in childhood-acquired demyelinating syndromes: results from a national surveillance cohort. J Neurol Neurosurg Psychiatry. 2014, 85: 456-461. 10.1136/jnnp-2013-306411.
Outteryck O, Baille G, Hodel J, Giroux M, Lacour A, Honnorat J, Zephir H, Vermersch P: Extensive myelitis associated with anti-NMDA receptor antibodies. BMC Neurol. 2013, 13: 211-10.1186/1471-2377-13-211.
Kruer MC, Koch TK, Bourdette DN, Chabas D, Waubant E, Mueller S, Moscarello MA, Dalmau J, Woltjer RL, Adamus G: NMDA receptor encephalitis mimicking seronegative neuromyelitis optica. Neurology. 2010, 74: 1473-1475. 10.1212/WNL.0b013e3181dc1a7f.
Zoccarato M, Saddi MV, Serra G, Pelizza MF, Rosellini I, Peddone L, Ticca A, Giometto B, Zuliani L: Aquaporin-4 antibody neuromyelitis optica following anti-NMDA receptor encephalitis. J Neurol. 2013, 260: 3185-3187. 10.1007/s00415-013-7182-x.
Titulaer MJ, Hoftberger R, Iizuka T, Leypoldt F, McCracken L, Cellucci T, Benson LA, Shu H, Irioka T, Hirano M, Singh G, Calvo AC, Kaida K, Morales PS, Wirtz PW, Yamamoto T, Reindl M, Rosenfeld MR, Graus F, Saiz A, Dalmau J: Overlapping demyelinating syndromes and anti-N-methyl-D-aspartate receptor encephalitis. Ann Neurol. 2014, 75: 411-428. 10.1002/ana.24117.
Hacohen Y, Absoud M, Hemingway C, Jacobson L, Lin JP, Pike M, Pullaperuma S, Siddiqui A, Wassmer E, Waters P, Irani SR, Buckley C, Vincent A, Lim M: NMDA receptor antibodies associated with distinct white matter syndromes. Neurol Neuroimmunol Neuroinflamm. 2014, 1: 1-11. 10.4103/2347-8659.135565.
Leypoldt F, Titulaer MJ, Aguilar E, Walther J, Bonstrup M, Havemeister S, Teegen B, Lutgehetmann M, Rosenkranz M, Magnus T, Dalmau J: Herpes simplex virus-1 encephalitis can trigger anti-NMDA receptor encephalitis: case report. Neurology. 2013, 81: 1637-1639. 10.1212/WNL.0b013e3182a9f531.
Hacohen Y, Deiva K, Pettingill P, Waters P, Siddiqui A, Chretien P, Menson E, Lin JP, Tardieu M, Vincent A, Lim MJ: N-methyl-D-aspartate receptor antibodies in post-herpes simplex virus encephalitis neurological relapse. Mov Disord. 2014, 29: 90-96. 10.1002/mds.25626.
Desena A, Graves D, Warnack W, Greenberg BM: Herpes simplex encephalitis as a potential cause of anti-N-methyl-d-aspartate receptor antibody encephalitis: report of 2 cases. JAMA Neurol. 2014, 71: 344-346. 10.1001/jamaneurol.2013.4580.
Acknowledgments
Authors have no acknowledgment to declare.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AW, BK, SS, DHL and RA analyzed and interpreted patient data and contributed to the writing of the manuscript. AV performed laboratory analyses and critically reviewed the manuscript. PG performed MR imaging and critically reviewed the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Waschbisch, A., Kallmünzer, B., Schwab, S. et al. Demyelinating disease and anti-N-methyl-D-aspartate receptor immunoglobulin G antibodies: a case report. BMC Res Notes 7, 948 (2014). https://doi.org/10.1186/1756-0500-7-948
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1756-0500-7-948