
Abstract
Background
Trisomy 1q and monosomy 3p deriving from a t(1;3) is an infrequent event. The clinical characteristics of trisomy 1q41-qter have been described but there is not a delineation of the syndrome. The 3p25.3-pter monosomy syndrome (MIM 613792) characteristics include low birth weight, microcephaly, psychomotor and growth retardation and abnormal facies.
Case presentation
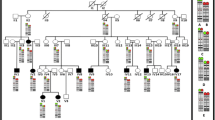
A 2 years 8 months Mexican mestizo male patient was evaluated due to a trisomy 1q and monosomy 3p derived from a familial t(1;3)(q41;q26.3). Four female carriers of the balanced translocation and one relative that may have been similarly affected as the proband were identified. The implicated chromosomal regions were defined by microarray analysis, the patient had a trisomy 1q41-qter of 30.3 Mb in extension comprising about 240 protein coding genes and a monosomy 3p26.3-pter of 1.7 Mb including only the genes CNTN6 (MIM 607220) and CHL1 (MIM 607416), which have been implicated in dendrite development. Their contribution to the phenotype, regarding the definition of trisomy 1q41-qter and monosomy 3p26.3-pter syndromes are discussed.
Conclusion
We propose that a trisomy 1q41-qter syndrome should be considered in particular when the following characteristics are present: postnatal growth delay, macrocephaly, wide fontanelle, triangular facies, frontal bossing, thick eye brows, down slanting palpebral fissures, hypertelorism, flat nasal bridge, hypoplasic nostrils, long filtrum, high palate, microretrognathia, ear abnormalities, neural abnormalities (in particular ventricular dilatation), psychomotor developmental delay and mental retardation. Our patient showed most of these clinical characteristics with exception of macrocephaly, possibly due to a compensatory effect by haploinsufficiency of the two genes lost from 3p. The identification of carriers has important implications for genetic counseling as the risk of a new born with either a der(3) or der(1) resulting from an adjacent-1 segregation is of 25% for each of them, as the products of adjacent-2 or 3:1 segregations are not expected to be viable.
Similar content being viewed by others
Background
There are 14 patients reported with trisomy 1q and monosomy 3p deriving from a t(1;3) which per se is an infrequent event (Table 1) [1–7]. The clinical characteristics of distal trisomy 1q syndrome have been described in several cases but a precise characterization of the syndrome has not been achieved (Table 2) [8–21]. In this regard, only 9 cases with a pure trisomy have been reported [8, 11, 13, 14, 16–18, 20], some of them correspond to small interstitial duplications [14, 16–18]; and in 5 of them, a translocation with the short arm of an acrocentric chromosome is implicated [8, 11, 13, 20]. Other cases are derived from an unbalanced translocation that have a small monosomic segment from another chromosome [9, 10, 12, 15, 19, 21], additionally the proximal break point varies between 1q41 [9, 11, 15, 17, 20, 21] and 1q42 [8–12, 14, 18, 19], but only two of them have been studied by genomic methodologies [19, 21] (Table 2).
Several cases of 3p25.3-pter monosomy syndrome (MIM 613792) have been delineated (Table 3) [22–29]. The clinical manifestations of monosomy 3p syndrome include low birth weight, microcephaly, trigonocephaly, hypotonia, psychomotor and growth retardation, among others (Table 3). Although intellectual deficits are almost invariably associated with cytogenetically visible 3p deletions, patients with infrequent 3p25-p26 or terminal deletions display normal intelligence or mild abnormalities [26, 27, 30]. A critical region has been identified for monosomy 3p and several genes have been proposed as responsible for the phenotypic features [24, 25, 30], however none critical region or candidate genes have been identified for terminal trisomy 1q syndrome [12, 20, 21, 31]. We report the case of a patient with a trisomy 1q and monosomy 3p derived from a familial t(1;3)(q41;q26.3). The chromosomal regions involved were defined by high-density microarray techniques and their effects in the phenotype regarding the definition of the syndromes are discussed.
Case presentation
Clinical report
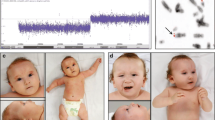
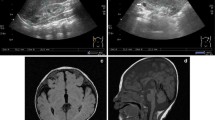
The proband is a 2 years 8 months Mexican mestizo male (Figure 1, IV.1), first known at 4 months of age due to dysmorphic features and mental development arrest. He is the only child of a young, apparently healthy and unrelated couple. He had two maternal uncles who died during childhood and presented congenital diseases, one of them also had dysmorphic features. The pregnancy was 38 weeks long and was complicated by a threat of miscarriage in the first trimester. His weight was 2,880gr (P10), height 51 cm (P50), and Apgar score 8/9. He could sit without support at 12 months of age; however so far he has not achieved speech, cannot walk and does not control sphincters. He suffered from esophageal reflux at 5 months of age, pneumonia at 8 months and was treated for dacryostenosis at 12 months. At present he has a weight of 11 kg (<P5), height 84 cm (<P5) and head circumference of 48 cm (P25). He has brachicephaly, triangular facies, horizontal palpebral fissures, micrognathia and several dysmorphic features, low set and retroposition of the ears, widely spaced nipples and hypotrophic limbs (Figure 2 and Tables 1, 2 and 3). Heart and renal malformations were discarded. The MRI showed ventriculomegaly and a subarachnoid cyst.

Pedigree of the family.

Facial characteristics of the proband are shown. A) Frontal view: triangular face and horizontal palpebral fissures. B) Lateral view: micrognathia, and posteriorly rotated low set ears.
Cytogenetic analysis
The GTG banding demonstrated additional material on 3p26.3 in the proband (Figure 3A), the karyotype of the mother (Figure 1, III.6) revealed a balanced translocation between chromosomes 1 and 3 (Figure 3B), also present in other three female family members (Figure 1) with a chromosomal complement 46,XX,t(1;3)(q41;p26.3). FISH analyses using subtelomeric probes (ToTelVysion™, Vysis Abbott, Inc. Abbott Park, IL, USA), mixture 1:1p (CEB108/T7, green), 1q (D1S3738, orange) and mixture 3: 3p (D3S4559, green), 3q (D3S4560, orange) confirmed the presence of the derivative chromosome 3 in the proband and showed the balanced translocation in his mother. Additionaly, mixture 1 includes Xp/Yp (yellow) and centromeric X (aqua) probes, while mixture 3 contains probes 22q (yellow) and BCR (aqua) (Figure 3C and D). The final chromosomal formula for the patient was 46,XY,der(3)t(1;3)(q41;p26.3)mat.ish der(3)t(1;3)(D1S3738+,D3S4559-,D3S4560+) and 46,XX,t(1;3)(q41;p26.3)mat.ish t(1;3)(CEB108/T7+,D1S3738-,D3S4559+;D1S3738+, D3S4559-,D3S4560+) for his mother.

Cytogenetic analysis. A. Partial karyotype of the proband showing additional material in 3p26.3. B. Partial karyotype of mother’s patient (individual III.6) showing a balanced t(1;3)(q41;p26.3). C. FISH analysis in patient’s metaphase showing a der(3) with both subtelomeric probes 1q and 3q in orange (the arrow indicates the der(3) and normal chromosomes 1 and 3 are indicated). D. FISH analysis in a metaphase of individual III.6 showing a balanced translocation, the der(1) with subtelomeric 1p and 3p probes both in green and the der(3) with 1q and 3q probes in orange are indicated with arrows, the normal chromosomes 1 and 3 are also indicated.
Microarray analysis
Genomic DNA was extracted from a peripheral blood sample. Copy-number analysis was performed using the Cytoscan HD array kit (Affymetrix Inc., Santa Clara, CA, USA). Data was analyzed with Chromosome Analysis Suite 2.0 (Affymetrix Inc.), using 50 markers and 200 kb resolution. Joining algorithm was applied for copy-number altered segments interrupted by <200 kb normal state data. Mapping was based on the human genome assembly Feb 2009 (GRCh 37/hg19). The analysis on the proband revealed a chromosome 1q41-qter duplication (pos.218,920,024-249,224,684) and a chromosome 3pter-p26.3 deletion (pos.61,891-1,774,215) (Figure 4). In addition, a 640 kb duplication was detected on chromosome 14q32.33 (pos.106,072,250-106,712,665), this alteration corresponds to a highly represented CNV region, according to the Database of Genomic Variants [32, 33].

Microarray analysis. A. The 30.3 Mb duplication in 1q41-qter and B. The 1.71 Mb deletion in 3p26.3-pter identified on the proband by microarray mapping. The solid bars at the top of each panel indicate the copy-number altered regions and chromosome bands are annotated at the bottom. The chromosome scheme at the left of each panel is not to scale.
Discussion
The patient described in this report has a trisomy 1q41-qter of 30.3 Mb in extension (comprising about 240 protein coding genes) and has a monosomy 3p26.3-pter of only 1.7 Mb. These break points are similar to those found in at least 6 reported cases with der(3)t(1;3) (Table 1) [5–7]. It is interesting to note that 14 patients have been described with trisomy 1q and monosomy 3p, but only one of these cases has been studied by genomic techniques (aCGH) [7]. The case reported here has been addressed by high-density microarray analysis, and revealed different break points and chromosomal segment sizes than the previously published. We consider that our patient shows a phenotype that resembles trisomy 1q41-qter syndrome, as he has brachicephaly, triangular face, temporal indentation, hypertelorism, anteverted nostrils, low set and malformed ears, micrognathia and narrow palate, all of which are features of distal trisomy 1q syndrome [34]. Despite this, we consider that the small deleted region at chromosome 3p could be modifying the phenotype of our patient. For example, head circumference data reported for trisomy 1q shows that most of the patients (82%) have macrocephaly (Table 2) while pure monosomy 3p patients have microcephaly more frequently (53-70%) (Table 3). In this regard, our patient displayed normal head circumference, and considering the specific chromosomal regions implicated in this phenotype and therefore the genes within these loci (Table 2), it seems that macrocephaly is associated to a duplicated region close to or in 1q43 [13, 14, 16–19]. Recently, 1q43 has been proposed as a candidate region for microcephaly, in particular AKT3 has been considered as a candidate gene for this feature in patients with a microdeletion of 1q43-q44 [35]. On the other hand, according to the described patients with monosomy 3p, both proximal and distal deletions to 3p26 show microcephaly and so far a critical region has not been identified [24–26, 29]. Although this characteristic in neither of the two pure syndromes is always present, it is possible that a compensatory effect of both implicated regions could be taking place in our patient and, as observed in other patients with a der(3) of a t(1;3) (Table 1), he has not macro or microcephaly but he has brachicephaly. However, these malformations may be multifactorial in their etiology.
The specific effect of each of the chromosomal regions regarding a particular clinical characteristic cannot be extended to all the features in the patient reported here or in the previously described patients, as this analysis is further complicated by: 1) the precise loci involved, 2) the size of the chromosomal fragments implicated in both the translocations and the pure trisomy 1q and monosomy 3p, and 3) the number and function of the genes present. As a result, most of the alterations of the diverse cases are privative of a given patient. When considering the first eight cases reported with a der(3)t(1;3) (Table 1) [1–4], 50% have break points with a longer chromosome fragment implicated than in our patient. In fact, the patients with a more similar phenotype to our patient are the ones reported by Sunaga et al., [5] which have break points in 1q42 and 3p26. However, not even in these cases the similarity is complete as these patients show the brachicephaly and abnormalities of the neural central system (that similar to our case, one of them is ventricular dilatation). All these patients show a triangular face and temporal indentation, but they do not show hirsutism, while some other characteristics are not constant among the three cases (Table 1) [5]. Another interesting consideration is that these patients do not have congenital heart disease and all of them have mental retardation. A critical region for congenital heart disease has been designated to a 0.65 Mb region in 3p25 and four candidate genes have been proposed: SLC6A1 (MIM 137165), HRH1 (MIM 600167), ATG7 (MIM 608760) and CRELD1 (MIM 607170) [24, 25, 30]. This region is not implicated in our case (Tables 1 and 3) and this may explain why even though both chromosomal abnormalities involve heart disease, our case does not show this characteristic. Only a third of the patients with terminal trisomy 1q have congenital heart disease and apparently, there is not a critical region for this malformation (Table 2) [8, 9, 11, 13, 16, 17].
Microarray analysis on our patient demonstrated that two genes, CNTN6 (MIM 607220) and CHL1 (MIM 607416) (which has been previously implicated in neuronal development associated to dendrite migration and mental retardation), are located within the deleted region and might implicate an haploinsufficiency mechanism, as loss of these genes has been related to mild mental deficit or do not present symptoms at all [26, 27]. Studies about CNTN4 (MIM 607280) indicate that it belongs to the same family as CNTN6. In a patient described by Fernandez et al., [22], CNTN4 was disrupted by a balanced translocation, while the patient displayed severe clinical manifestations associated to monosomy 3p (in particular the facial features without triangular face or microcephaly) and mental retardation. Assuming that even if no chromosomal material was gain or lost in the case referred, CNTN4 is an important gene for brain development. Therefore, it has been considered that CNTN4 corresponds to the critical region or is one critical gene for the monosomy 3p clinical characteristics. In our patient, CNTN4 is not included in the deleted region as it is further centromeric, however CNTN6 is deleted and, considering that it belongs to the same gene family, its effect should not be underestimated. In addition, studies by Mercati et al., [36] have shown that CNTN6 is implicated in the same pathway as CHL1, i.e. the integrity of the neuronal development. CHL1 gene participates in brain development, interacting with a tyrosin phosphatase protein, regulating its activity in the complex signaling of the neuronal dendritic migration. Therefore, its deletion may have an important role in the mental retardation observed in our patient, and although its importance is difficult to assess, as in the patients described by Pohjola et al., [26] and Couco et al., [27] in whom these genes are lost, some of them have a borderline development, learning disabilities or are apparently normal. It is possible that in our patient, due to the presence of the trisomy 1q41-qter, the haploinsufficiency of both genes could have a synergic effect that in turn compromises the psychomotor development.
Analysis of the phenotypic impact that the chromosome 1q trisomy region may have on our patient is complicated by the fact that over 240 genes may be involved, and in general terms the additive effect of these genes may be related to the facial, cranial and cognitive abnormalities (Table 2). Although several cases with similar chromosomal abnormalities have been described, the contribution of specific regions in the development of the characteristics of the syndrome is not clear, due to: 1) the high number of genes implicated, 2) the variability of the break points and 3) the fact that not all of the cases correspond to pure terminal trisomies. Therefore, a trisomy 1q41-qter syndrome should be considered in particular when the following characteristics are present: postnatal growth delay, macrocephaly, wide fontanelle, triangular face, frontal bossing, thick eye brows, down slanting palpebral fissures, hypertelorism, flat nasal bridge, hypoplasic nostrils, long filtrum, high palate, microretrognathia, ear abnormalities, neural abnormalities (in particular ventricular dilatation), psychomotor delay and mental retardation (Table 2).
In the 1q41-q42 region are the loci for 5S rRNA and this situation may predispose to chromosomal rearrangements, in particular with the short arms of acrocentric chromosomes during the transcription of ribosomal genes [37]. This is illustrated in Table 1, as in 5 out of 9 cases with pure 1q trisomy, this region is implicated [8, 11, 13, 20]. Furthermore, recently a new case of trisomy 1q41-qter has been described in a patient who also has a partial trisomy 9pter-9q21.32, derived from a 3:1 segregation of a maternal balanced translocation, and it has been suggested that the break-point is associated with the presence of a fragile site (FRA1H) localized to 1q41-q42.1 [38].The chromosomal analysis of our family revealed 4 female carriers of the balanced translocation (Figure 1) and this has an important implication for genetic counseling. The pedigree shows 5 cases of spontaneous abortions, which might have resulted from an adjacent-1 segregation. There is also an uncle of the proband (Figure 1, III.11) that was described with psychomotor delay and dysmorphic features, and who died in infancy of unknown causes. We may suppose that this person had a der(3)t(1;3) resulting of adjacent-1 segregation, as with the proband. When considering the segregation of the chromosomes implicated in the translocation, the risk of a newborn with either a der(3) or der(1) resulting from an adjacent-1 segregation is of 25% for each of them, as the products of adjacent-2 or 3:1 segregations are not expected to be viable.
Conclusion
The case described here gives new insights into the complex definition of distal trisomy 1q and monosomy 3p26.3 syndromes. Even though in this case the trisomy affects such a large quantity of genes, it is possible to evaluate the clinical characteristics in order to understand their participation in the pathogenesis of the syndrome and has very important implications for the genetic assessment.
Consent
Written informed consent from the patient’s mother for publication of this Case report and any accompanying images. A copy of the written consent is available for review by the Editor of this journal.
Author’s contribution
AC carried out the general analysis of the case and drafted the manuscript. CGD, CIGM and VFMB provided the clinical evaluation and VFMB also drafted the manuscript. ABMJ, JVM, MQP carried out the cytogenetics analysis. KNM and LGL carried out the FISH studies. FFR and JB carried out the microarray analysis. SKE participated to the critical reading of the manuscript. All authors read and approved the final manuscript.
References
Yunis E, Egel H, Zúñiga R, Ramírez E, Torres de Caballero OM, Leibovici M: “de novo” trisomy 1q32→1qter and monosomy 3p25→3pter. Hum Genet. 1997, 36: 113-116.
Cook PJ, Fear CN, Povey S: A 1q translocation family segregating for peptidase C. Cytogenet Cell Genet. 1978, 22: 375-377. 10.1159/000130975.
Schinzel A: Duplication-deletion with partial trisomy 1q and partial monosomy 3p resulting from a maternal reciprocal translocation rcp(1;3)(q32;p25). J Med Genet. 1981, 18: 64-68. 10.1136/jmg.18.1.64.
McCarthy GT, Fear CN, Berry AC: Three children with partial trisomy 1q and partial monosomy 3p. J Med Genet. 1986, 23: 466-467. 10.1136/jmg.23.5.466.
Sunaga Y, Ohtsuka T, Nagashima K, Kuroume T: Two siblings with partial trisomy 1(q42.3-ter). Brain Dev. 1993, 15: 119-124. 10.1016/0387-7604(93)90048-D.
Kozma C, Slavotinek AM, Meck JM: Segregation of a t(1;3) translocation in multiple affected family members with both types of adjacent-1 segregants. Am J Med Genet. 2004, 124A: 118-128. 10.1002/ajmg.a.20332.
Li C, Mahajan V, Wang JC, Paes B: Monosomy 3pter-p25.3 and trisomy 1q42.13-qter in a boy with profound growth and developmental restriction, multiple congenital anomalies, and early death. Pediatr Neonatol. 2013, 54: 202-206. 10.1016/j.pedneo.2013.01.009.
Chia NL, Bousfield LR, Poon CC, Trudinger BJ: Trisomy (1q)(q42-qter): confirmation of a syndrome. Clin Genet. 1988, 34: 224-229.
Rasmussen SA, Frias JL, Lafer CZ, Eunpu DL, Zackai EH: Partial duplication 1q: report of four patients and review of the literature. Am J Med Genet. 1990, 36: 137-143. 10.1002/ajmg.1320360203.
Kennerknecht I, Barbi G, Rodens K: Dup(1q)(q42-qter) syndrome: case report and review of literature. Am J Med Genet. 1993, 47: 1157-1160. 10.1002/ajmg.1320470805.
Verschuuren-Bemelmans CC, Leegte B, Hodenius TM, Cobben JM: Trisomy 1q42→qter in a sister and brother: further delineation of the “trisomy 1q42→qter syndrome”. Am J Med Genet. 1995, 58: 83-86. 10.1002/ajmg.1320580116.
Concolino D, Cinti R, Ferraro L, Moricca MT, Strisciuglio P: Partial trisomy 1 (q42 →qter): a new case with a mild phenotype. J Med Genet. 1998, 35: 75-77. 10.1136/jmg.35.1.75.
Villa N, Sala E, Colombo D, Dell’Orto M, Dalprá L: Monosomy and trisomy 1q44-qter in two sisters originating from a half cryptic 1q;15p translocation. J Med Genet. 2000, 37: 612-615. 10.1136/jmg.37.8.612.
De Brasi D, Rossi E, Giglio S, D’Agostino A, Titomanlio L, Farina V, Andria G, Sebastio G: Inv dup del (1)(pter– > q44::q44– > q42:) with the classical phenotype of trisomy 1q42-qter. Am J Med Genet. 2001, 104: 127-130. 10.1002/ajmg.1589.
Emberger W, Petek E, Kroisel PM, Zierler H, Wagner K: Clinical and molecular cytogenetic characterization of two patients with partial trisomy 1q41-qter: further delineation of partial trisomy 1q syndrome. Am J Med Genet. 2001, 104: 312-318. 10.1002/ajmg.10096.
Morava E, Jackson KE, Tsien F, Marble MR: Trisomy 1q43 syndrome: a consistent phenotype with macrocephaly, characteristic face, developmental delay and cardiac anomalies. Genet Couns. 2004, 15: 449-453.
Polityko A, Starke H, Rumyantseva N, Claussen U, Liehr T, Raskin S: Three cases with rare interstitial rearrangements of chromosome 1 characterized by multicolor banding. Cytogenet Genome Res. 2005, 111: 171-174. 10.1159/000086388.
Coccé MC, Villa O, Obregon MG, Salido M, Barreiro C, Solé F, Gallego MS: Duplication dup(1)(q41q44) defined by fluorescence in situ hybridization: delineation of the “trisomy 1q42→qter syndrome”. Cytogenet Genome Res. 2007, 118: 84-86. 10.1159/000106446.
Percesepe A, Lugli L, Pierluigi M, Cavani S, Malacame M, Roversi MF, Ferrati F, Forabosco A: Pure segmental trisomy 1q42-qter in a boy with a severe phenotype. Am J Med Genet. 2007, 143A: 2339-2342. 10.1002/ajmg.a.31890.
Kulikowski LD, Bellucco FT, Nogueira SI, Christofolini DM, Smith M de A, de Mello CB, Brunoni D, Melaragno MI: Pure duplication 1q41-qter: further delineation of trisomy 1q syndromes. Am J Med Genet. 2008, 146A: 2663-2667. 10.1002/ajmg.a.32510.
Shin YB, Nam SO, Seo EJ, Kim HH, Chang CL, Lee EY, Son HC, Hwan SH: Partial trisomy 1q41 syndrome delineated by whole genomic array comparative genome hybridization. J Korean Med Sci. 2008, 23: 1097-1101. 10.3346/jkms.2008.23.6.1097.
Fernandez T, Morgan T, Davis N, Klin A, Morris A, Farhi A, Lifton RP, State MW: Disruption of Contactin 4 (CNTN4) results in development delay and other features of 3p deletion syndrome. Am J Med Genet. 2004, 74: 1286-1293.
Fernandez TV, García-González IJ, Mason CE, Hernández-Zaragoza G, Ledezma-Rodríguez VC, Anguiano-Alvarez VM, E’Vega R, Gutiérrez-Angulo M, Maya ML, García-Bejarano HE, González-Cruz M, Barrios S, Atorga R, López-Cardona MG, Armendariz-Borunda J, State MW, Dávalos NO: Molecular characterization of a patient with 3p deletion syndrome and a review of the literature. Am J Med Genet. 2008, 146A: 2746-2752. 10.1002/ajmg.a.32533.
Gunnarsson C, Foyn Bruun C: Molecular characterization and clinical features of a patient with an interstitial deletion of 3p25.3-p26.1. Am J Med Genet. 2010, 152A: 3110-3114. 10.1002/ajmg.a.33353.
Shuib S, McMullan D, Rattenberry E, Barber RM, Rahman F, Zatyka M, Chapman C, Macdonald F, Lati F, Davison V, Maher ER: Microarray based analysis of 3p25-p26 deletions (3p- syndrome). Am J Med Genet. 2009, 149A: 2099-2105. 10.1002/ajmg.a.32824.
Pohjola P, de Leeuw N, Penttinen M, Kääriälnen H: Terminal 3p deletions in two families—correlation between molecular karyotype and phenotype. Am J Med Genet. 2010, 152A: 441-446. 10.1002/ajmg.a.33215.
Cuoco C, Ronchetto P, Gimelli S, Béna F, Divizia MT, Lerone M, Miravelli-Badenier M, Mascaretti M, Gimelli G: Microarray based analysis of an inherited terminal 3p26.3 deletion, containing only the CHL1 gene, from a normal father to his two affected children. Orphanet J Rare Dis. 2011, 6: 12-10.1186/1750-1172-6-12. doi:10.1186/1750-1172-6-12
Chen CP, Su YN, Chen CY, Su JW, Chern SR, Town DD, Wang W: Pure partial monosomy 3p(3p25.3-pter): prenatal diagnosis and array comparative genomic hybridization characterization. Taiwan J Obstet Gynecol. 2012, 51: 435-439. 10.1016/j.tjog.2012.07.022.
Peltekova IT, Macdonald A, Armour CM: Microdeletion on 3p25 in a patient with features of 3p deletion syndrome. Am J Med Genet. 2012, 158A: 2583-2586. 10.1002/ajmg.a.35559.
Malmgren H, Sahién S, Wide K, Lundvall M, Biennow E: Distal 3p deletion syndrome: detailed molecular cytogenetic and clinical characterization of three small distal deletions and review. Am J Med Genet. 2007, 143A: 2143-2149. 10.1002/ajmg.a.31902.
Michels VV, Berseth CL, O’Brien JF, Dewald G: Duplication of part of chromosome 1q: clinical report and review of literature. Am J Med Genet. 1984, 18: 125-134. 10.1002/ajmg.1320180116.
Database of Genomic Variants. [http://projects.tcag.ca/variation/]
Zhang J, Feuk L, Duggan GE, Khaja R, Scherer SW: Development of bioinformatics resources for display and analysis of copy number and other structural variants in the human genome. Cytogenet Genome Res. 2006, 115: 205-214. 10.1159/000095916.
Leisti J, Aula P: Partial trisomy 1 (q42 leads to ter). Clin Genet. 1980, 18: 371-378.
Ballif BC, Rosenfeld JA, Traylor R, Theisen A, Bader PI, Ladda RL, Sell SL, Steinraths M, Surti U, McGuire M, Williams S, Farrell SA, Filiano J, Schnur RE, Coffey LB, Tervo RC, Stroud T, Marble M, Netzloff M, Hanson K, Aylsworth AS, Bamforth JS, Babu D, Niyazov DM, Ravnan JB, Schultz RA, Lamb AN, Torchia BS, Bejjani BA, Shaffer LG: High-resolution array CGH defines critical regions and candidate genes for microcephaly, abnormalities of the corpus callosum, and seizure phenotypes in patients with microdeletions of 1q43q44. Hum Genet. 2012, 131: 145-156. 10.1007/s00439-011-1073-y.
Mercati O, Danckaert A, André-Leroux G, Bellinzoni M, Gouder L, Watanabe K, Shimoda Y, Grailhe R, de Chaumont F, Bourgeron T, Cloëz-Tayarani I: Contactin 4, -5 and -6 differentially regulate neuritogenesis while they display identical PTPRG binding sites. Biol Open. 2013, 2: 324-334. 10.1242/bio.20133343.
Kost MV, Alimov AA, Sarafanov AG, Tikchomirova TP, Gumeniuk RR, Timofeeva MJA, Zelenin AV: 5S rRNA gene hybridizes to human chromosome 1 at two sites (1q42.11-->q42.13 and 1q43). Cytogenet Cell Genet. 1995, 68: 82-84. 10.1159/000133896.
Akalin I, Bozdag S, Spielmann M, Basaran SY, Nanda I, Klopocki E: Partial trisomy 1q41-qter and partial trisomy 9pter-9q21.32 in a newborn infant: an array CGH analysis and review. Am J Med Genet Part A. 2014, 164A: 490-494.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1755-8794/7/55/prepub
Acknowledgments
We thank Linda Beatriz Muñoz Martínez for technical support in the cytogenetic analysis.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The authors declare that they have no competing interest.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Cervantes, A., García-Delgado, C., Fernández-Ramírez, F. et al. Trisomy 1q41-qter and monosomy 3p26.3-pter in a family with a translocation (1;3): further delineation of the syndromes. BMC Med Genomics 7, 55 (2014). https://doi.org/10.1186/1755-8794-7-55
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8794-7-55