
Abstract
Background
Intracranial aneurysm (IA) is one of the most lethal forms of cerebrovascular diseases characterized by endothelial dysfunction, vascular smooth muscle cell phenotypic modulation, inflammation and consequently loss of vessel cells and extracellular matrix degradation. Besides environmental factors, genetics seem to be a very important factor in the genesis of this disease. Previous mRNA expression studies revealed a large number of differentially expressed genes between IA and control tissue. However, microRNAs (miRNA), small non-coding RNAs which are post-transcriptional regulators of gene expression, have been barely studied. Studying miRNAs could provide a hypothetical mechanism underlying rupture of IA.
Methods
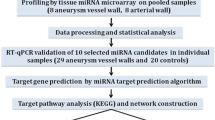
A microarray study was carried out to determine difference in microRNAs and mRNA between patients’ IA tissues and controls. Quantitative RT-PCR assay compared the expression level between two groups (14 IA domes vs. 14 controls) were used for validation. Validated miRNAs were analyzed using Ingenuity Pathway Analysis (IPA) to identify the networks and pathways.
Results
18 miRNAs were confirmed by qPCR to be robustly down-regulated in 14 ruptured IA patients including hsa-mir-133b, hsa-mir-133a, hsa-mir-1, hsa-mir-143-3p, hsa-mir-145-3p, hsa-mir-145-5p, hsa-mir-455-5p, hsa-mir-143-5p, hsa-mir-23b-3p etc., of which 11 miRNAs are clusters: hsa-mir-1/has-mir-133a, hsa-mir-143/hsa-mir-145, hsa-mir-23b/hsa-mir-24-1, and hsa-mir-29b-2/hsa-mir-29c. 12 predicted functions were generated using IPA which showed significant associations with migration of phagocytes, proliferation of mononuclear leukocytes, cell movement of mononuclear leukocytes, cell movement of smooth muscle cells etc.
Conclusion
These data support common disease mechanisms that may be under miRNA control and provide exciting directions for further investigations aimed at elucidating the miRNA mechanisms and targets that may yield new therapies for IA.
Similar content being viewed by others

Background
As one of the most devastating neurological conditions known to date, intracranial aneurysm (IA) has a high mortality rate and undesirable prognosis with spontaneous cerebral hemorrhage, cerebral vasospasm, and oculomotor nerve palsy as the main clinical feature. IA is common result of vascular abnormalities in the brain, with a prevalence of 3.2% in the general population, and an overall risk of rupture around 1.2% in western populations and 2.3% in Japanese series [1]. A significant proportion of aneurysmal patients are around the age of 40–60 [2, 3]. Cigarette smoking, excessive alcohol consumption, hypertension and female gender are significant risk factors for IA formation and growth, and family history of IA has also been suggested to be evidence for genetic causality of cerebral aneurysms. Dysfunction of vessel cells, degeneration of vessel wall and activation of immune system were identified to be the intrinsic factors of IA development [3–6]. Its unpredictable nature and the catastrophic consequences of IA rupture remain a challenge for clinicians. Comprehensive understanding of IA pathobiology is crucial for reasonable management of IA carriers.
Due to the fact that animal models of IA are imperfect and human aneurysmal tissues are difficult to obtain, the molecular mechanisms of IA remain poorly understood. Most studies focus on mRNA expression in aneurysmal and healthy tissue to identify the alteration of gene expression within the vessel wall, which has implied some mechanisms underlying the development of IA. For example, in 2008 Krischek et al. found differentially expressed genes, which indicated that antigen processing was the most significantly associated; another study in 2009 by Shi et al. indicated that misregulated genes were mostly correlated with focal adhesion, ECM-receptor and cell communication etc. Because the large amounts of data created with each study, make a comparison or interpretation of results difficult, Roder et al. (2012) performed a meta-analysis which found seven genes showing altered expression in more than three studies: BCL2, COL1A2, COL3A1, COL5A2, CXCL12, TIMP4, TNC [7–13]. Functional studies on these genes showed that COL1A2, COL3A1, COL5A2, TIMP4, and TNC could modulate processes in the formation of the extracellular matrix (ECM), which have been described in association with IAs [10, 14]. miRNA may be another layer of control in gene expression which modulates pathways and mechanisms of IA, however, expression of miRNA in IA is rarely studied.
A novel direction for IA research is the modulation of miRNA, endogenous approximately 23 nt non-coding RNAs. By binding to the 3’ UTR of complementary protein-coding mRNAs, miRNA primarily acts in the post-transcriptional repression of gene expression in animals and plants. miRNAs are incorporated into the RNA induced silencing complex (RISC) and then inhibit gene expression by either mRNA degradation or inhibiting translation which can thereby regulate up to 75% of the human genome which belong to many biological pathways including immune response and apoptosis [15–19]. Dysregulation of miRNAs have been found to have relevance to tumorigenesis, neurological, cardiovascular and developmental and other diseases [20]. Recent studies have demonstrated that miRNAs play roles in vascular remodelling and atherosclerosis [21, 22]. miRNA may be another layer of control in gene expression which modulates pathways and mechanisms of IA, however, expression of miRNA in IA is rarely studied.
The role of miRNA in the molecular mechanism of IA has been of particular interest. Our study focused on investigating how the differential expression patterns of regulatory microRNAs in IA act as a potential regulator in its pathological mechanism. We generated a microRNA array followed by confirmation of miRNAs individually with qRT-PCR. We identified 18 miRNAs in 14 patients which were significantly down-regulated between IA and control tissue, 11 of these miRNAs in the cluster including hsa-mir-1/has-mir-133a, hsa-mir-143/hsa-mir-145, hsa-mir-23b/hsa-mir-24-1, hsa-mir-29b-2/hsa-mir-29c. Functional analysis indicates these miRNAs are involved with dysfunction and remolding of vascular endothelial cells, vascular smooth muscle cell and involvement of inflammatory/immune processes.
Methods
Patients and tissue samples
Full-thickness vessel wall samples from 14 ruptured IA domes were prospectively collected from patients (10 female, 4 male, age: 52.7 ± 8.5 ) undergoing microsurgical clipping. 14 middle meningeal artery (MMA) segments with matched sex and age were obtained during standard neurosurgical procedures (traumatic hematoma, tumor resection, IA clipping) as control. Written informed consent for participation in the study was obtained from patients. Tissue samples were snap frozen in liquid nitrogen and directly sent to the laboratory to perform RNA extraction. The collection of the human tissues was approved by Ethical Committee of the Second Xiangya Hospital of Central South, China.
Extract RNA from samples
The total RNA was extracted by Trizol Reagent. RNA concentration and purity were determined using a NanoDrop ND-1000 spectrophotometer (NanoDrop Tech, Rockland, DE), with a 260/280 value >1.8 considered acceptable. RNA samples were further assessed for quality using a Agilent 2100 Bioanalyzer (Agilent Technologies, Foster City, CA) according to the manufacturer’s instructions to ensure an RNA integrity number > 7, and RNA samples for Agilent miRNA Chip: RIN ≥ 6.0 and 28S/18S>0.7 was used.
Determination of specific miRNAs
miRNA microarray profile was performed using Agilent microRNA array 16.0 (3 aneurysmal wall samples and 3 healthy control samples) to identify candidate microRNAs expressed differently between patients’ IA tissues and controls. Agilent Whole Human Genome Oligo Microarray (4 × 44 K) was used for mRNA expression (2 aneurysmal wall samples and 2 healthy control samples). The microarray data can be obtained at the Gene Expression Omnibus (GEO) database (GSE46338 is the reference Series; http://www.ncbi.nlm.nih.gov/geo/).
Confirmation of miRNA expression
miRNA and mRNA profile data were screened, keeping data with a change of more than 2 fold, then we verified the screened miRNA by RTq-PCR (SYBR® PrimeScript™ miRNA RT-PCR Kit (RR716)) according to manufacturer’s recommendation. Quantitative RT-PCR reactions were completed on CFX96™ Real-Time System. The relative expression levels of the miRNAs were calculated using the -ΔΔCT method and relative miRNA levels were normalized to U6 small non-coding RNA. We compared the expression level between two groups (14 IA domes vs. 14 controls). For the data obtained by qRT-PCR, the Mann–Whitney test and Student's t-test were used for the comparison between IA and control, and differences were considered to be significant when p < 0.05. Samples were run in triplicate and the average values were used in subsequent analysis.
Function analysis
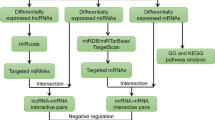
The selected miRNAs were further analyzed to identify the networks and pathways. For this purpose, we used software Ingenuity Pathway analysis (IPA, Ingenuity® Systems; http://www.ingenuity.com). This pathway analysis software identifies the putative targets for the input miRNA(s), integrates with our mRNA microarray profiles data, and then develops the networks and functions among the genes/targets. Before starting the analysis, miRNA targets (confidence was set to "highly predicted" and "experimental observed", species was chosen to "human") were predicted by an integrated database including miRecords, Tarbase and TargetScan Human. Then the high predicted targets were matched and paired with mRNA expression data by the expression pairing function of IPA. We assume that the expression of a given miRNA is anti-correlated with the mRNA expression of its targets. This is a widely accepted and experimentally verified supposition [23]. The results which provide us mainly with bio-functions and canonical pathways associated with our data had been generated automatically using the option of core-analysis in IPA.
Results
Identification of differently expressed miRNAs in IA
Focusing initially miRNA profiling data on IA tissues vs. normal tissues, there were 30 (FDR p < 0.05, ± 2 fold) differentially regulated miRNAs out of 1500 microRNAs. Among the 30 miRNAs identified, 29 were down-regulated in the IA tissue and 1 was upregulated. miRNAs identified in the microarray study were validated using individual real-time qRT-PCR assays. And 18 were found to be significantly different between the IA and control groups of the 14 patients (FC > 2 or FC < -2), and the p-value and FC were calculated (Table 1), miR-142-5p was upregulated in microarray profile, but qRT-PCR result showed no significance between IAs and controls (P = 0.25). The rest of the candidate miRNAs showed an expression tendency consistent with the array result, but without statistical significance.
The expression levels of hsa-mir-1, hsa-mir-7-1-3p, hsa-mir-23b-5p, hsa-mir-23b-3p, hsa-mir-24-1-5p, hsa-mir-28-5p, hsa-mir-28-3p, hsa-mir-29b-2-5p, hsa-mir-29c-5p, hsa-mir-29c-3p, hsa-mir-133a, hsa-mir-133b, hsa-mir-140-3p, hsa-mir-143-5p, hsa-mir-143-3p, hsa-mir-145-5p, hsa-mir-145-3p, hsa-mir-455-5p were down-regulated at least two fold in IA compared with the control group (Figure 1). There are 4 clusters among those miRNAs: hsa-mir-1/has-mir-133a (chr18), hsa-mir-143/hsa-mir-145 (chr5), hsa-mir-23b/hsa-mir-24-1 (chr9), hsa-mir-29b-2/hsa-mir-29c (chr1).

Validation of microarray results by real-time qRT-PCR in a set of samples (n = 14). 18 miRNAs identified as significantly different between IA and controls in the microarray study were evaluated by qRT-PCR in 14 IA and 14 control samples (MMA).
Some miRNAs play a role in the cardiovascular system. For example, miR-1 is induced during smooth muscle cell (SMC) differentiation and increases the expression of SMC-specific contractile proteins. miR-133 is a key regulator of vascular smooth muscle cell phenotypic switch in vitro and in vivo [24]. Also notable, miR-145 is related to the thickness of the vessel wall, and the absence of miR-145 could reduce the vessel thickness and due to hypotrophy of SMCs [25]. miR-145 is down-regulated following vascular injury, during atherosclerosis, and in experimentally induced aneurysms [26, 27].
Integrated analysis of misregulated miRNAs and mRNAs
miRNAs modulate gene expression through both mRNA degradation and translational repression mechanisms, and miRNA-mRNA regulatory networks are highly complex. A dataset of 681 genes created from our mRNA microarray data paired with high predicted and experimentally observed targets to 18 miRNAs, which were used for Ingenuity Pathway Analysis (IPA). IPA results revealed top functions of these 681 common targets. The most impacted biological processes for IA including: migration of phagocytes, proliferation of mononuclear leukocytes, cell movement of mononuclear leukocytes, cell movement of smooth muscle cells, differentiation of macrophages etc. (Table 1). The functions are chosen and arranged by the z-score which indicates the predicted degree of those functions. A positive value means an increase in the function, while a negative value means a decrease the function, and the p-value indicates the significance of each function.
IPA predicted the most impacted biological processes for IA based on the miRNAs and their targets. 54 genes and 11 miRNAs were involved in the top 12 predicted functions, and a network generated by IPA showed the interactions between those miRNAs and mRNA (Figure 2). Some distinctive genes shown in the network targeted by more than 3 miRNAs include Kruppel-like factors 4 (KLF4); inducible T cell co-stimulator (ICOS); CD28, Mitogen-activated protein kinase 1 (MAPK1); and collagen, type IV, alpha 3 (COL4A3). Notably, among the differentially expressed mRNAs predicted to be targeted by the differential miRNAs, were some genes previously experimentally identified to be involved in aneurysm formation or loss of vessel cells, such as TGFBR1, MMPs and IL18 etc. [28, 29].

A network of the interactions of the miRNAs and their target genes (experimental observed or highly predicted). Ingenuity Pathway Analysis® tool was used to generate miRNA–mRNA interactions of miR-1, miR-133, miR-140-3p, miR-143-3p, miR-145-5p and, miR-23a-3p, miR-28-3p, miR-28-5p, miR-29b-3p and their targets in all selected functions. Red and green color represents the molecules to be upregulated and down-regulated respectively.
KLF4 plays roles in cell proliferation, differentiation and survival. Its role, especially in the context of cancer, has been extensively studied, and several studies have explored the role of KLF4 in vascular smooth muscle cell and vascular endothelial cell. ICOS belongs to the CD28 and CTLA-4 cell-surface receptor family, and it plays an important role in cell-cell signaling, immune responses, and regulation of cell proliferation. ICOS also has been found to function in vascular diseases such as atherosclerosis [30]. CD28 is essential for T-cell proliferation and survival, cytokine production, and T-helper type-2 development. A recent study has shown that CD28 influence the atherosclerosis development by co-stimulating T-cell with CD80/86 [31]. MAPK1 is a component of MAP kinase family which is involved in a wide variety of cellular processes such as proliferation, differentiation, and transcription regulation and development. Many researchers have determined the MAPK signaling participate in the biological processes of vascular system such as proliferation of vascular endothelial cell [32] and vascular damage [33]. COL4A3 is a subunit of Type IV collagen and is the major structural component of basement membranes. Investigating these targets in this network may provide new compensatory mechanisms for protecting against IA, and may also be characterized in future studies to contribute to the miRNA-regulated response mechanisms following IA.
The mechanism that underlies the formation and development of IA is complicated and partly understood. In order to demonstrate our result more distinctively, we drew a schematic diagram to list three most impacted aspects of IA [3, 5] in black box (Figure 3). Dysfunction of vascular endothelial cell (VEC), modulation of vascular smooth muscle cell (VSMC) and inflammatory response were identified to be the intrinsic factors of IA development. Several branches are connected to a red box with the name of the relevant function, genes involved in and z-score/p-value. Another green box contain the miRNAs which target the corresponding genes in the function is attached to the red one. Among those functions, migration of phagocytes has the highest z-score about 2.627, which means the function could have the maximum extent of activation based on the genes (CCL2, CCL7, CD40, CSF1, FN1, MMP14, PLAU, PLXND1, SERPINE1, SOCS3) , and those genes are validated or highly predicted targets of mir-145-5p, mir-23a-3p, mir-143-3p, mir-133, mir-28-5p, mir-1, mir-455-5p.

Biological categories of miRNA and predicted functions.
Discussion
IA is the most fatal cerebrovascular system disease. Mechanisms underlying formation, progression, and rupture of IA are complex and involve a multitude of processes that are not completely understood. No safe and effective noninvasive therapies have been applied in clinical practice until recently. Treatments currently available include surgical (clipping) and endovascular (such as coiling), which have similar goals of isolating the aneurysm from blood circulation, but potentially serious complications [4]. Numerous efforts made to uncover the biology of IA have suggested that aneurysm is caused by a combination of hemodynamic stresses and defective vessel wall responses [34]. In recent years, prominent roles for microRNAs (miRNAs) have been revealed in several vascular disorders, several miRNAs have been found to be critical modulators of vascular pathologies, such as atherosclerosis, lipoprotein metabolism, inflammation, arterial remodeling, angiogenesis, smooth muscle cell regeneration, hypertension, apoptosis, neointimal hyperplasia and signal transduction pathways [35]. miRNAs may also serve as novel biomarkers and/or therapeutic targets for vascular disease [36–38]. Determining miRNA regulatory role and investigating the molecular mechanisms will expand our knowledge to better understand IA by analyzing miRNA mediated pathways.
We found that 18 miRNAs were significantly down-regulated in IA domes of 14 ruptured IA patients. There are 4 clusters among 18 miRNAs according to mirBase (http://www.mirbase.org): hsa-mir-1/has-mir-133a, hsa-mir-143/hsa-mir-145, hsa-mir-23b/hsa-mir-24-1, hsa-mir-29b-2/hsa-mira-29c. We searched the literature for information on the 18 miRNAs. miR-1, miR-133, miR-143, miR-145 are highly expressed miRs in SMCs and have been found to regulate the SMC phenotype [39–44]. miR-1 is induced during SMC differentiation and increases the expression of SMC-specific contractile proteins by targeting KLF4 [45]. Notably, the interaction which is critical for modulation of vascular smooth muscle cell phenotype, between KLF4 and miR-143/145 has also been identified [46, 47]; miR-133 impairs the proliferation of SMCs and inhibits the PDGF-induced switch towards a synthetic SMC phenotype by repressing the transcription factor Sp-1 [48]. Several recent studies describe the involvement of miR-29 in aneurysm formation by post-transcriptionally repressing the expression of extracellular matrix proteins such as collagens, elastin, and fibrillins [49–54]. Several genome-wide linkage studies have determined some disease-related loci such as chr1p34.3–p36.13, chr7q11, chr19q13.3, and chrXp22 underlying the development of IA [55]. One study which is worth noting has identified several loci in familial IAs, miR-133a-1/miR-1-2 locates at chromosomes 18q11.2 which is strongly associated with the development of intracranial aneurysms [56].
miRNAs may function as provital regulators of biological processes during IA development by regulating downstream genes. A meta-analysis of five microarray gene expression studies of 60 samples revealed seven genes: BCL2, COL1A2, COL3A1, COL5A2, CXCL12, TIMP4, TNC that are very likely to be involved in the genesis of IAs [57]. These genes are also upregulated more than two fold in our samples. BCL2 is targeted by miR-143 in cervical cancer, which is involved in apoptosis and tumor formation; miR-1 regulates cardiomyocyte apoptosis by targeting BCL2 [58, 59]. miR-1 is able to inhibit thyroid carcinoma cell proliferation and migration by targeting CCND2, CXCR4 and CXCL12 [60]. COL1A2, COL3A1, COL5A2 are a group of collagen genes in which mutations are associated with several connective diseases such as the involvement of COL3A1 mutations in intracranial aneurysms and Ehlers-Danlos syndrome type IV with aortic and arterial aneurysms [61, 62]. miR-29 targeted several extracellular matrix genes including COL1A2, COL3A1 and COL5A2, and has been validated in nasopharyngeal carcinomas and HTM (human trabecular meshwork) cells [63, 64]. Another study has observed that COL3A1 is targeted by miR-29 as a participant in the mechanism of atrial fibrillation [65].
A break in the delicate balance between local hemodynamic stress and arterial wall integrity may be the reason why IA occurs. Genesis of IA may be triggered by aberrant flow conditions, and a group of activated cells could lead to an unstable situation between "repair and maintain" and "degrade and destroy", following which dysfunction of endothelial cell, and loss of mural cell and inflammatory response may eventually lead IA rupture [3]. Lots of cells and genes are abnormally modulated during the development of IA, investigating those miRNA or mRNA found in our study and their regulating networks may provide new insight of IA pathogenesis.
Analysis generated by IPA on those validated miRNAs and their putative targets revealed that these miRNAs may be involved in the three main pathological processes: loss of vessel cells, phenotypic change of vessel cells, and inflammation of the vessel. Several targets of miRNAs have been reported to function in the loss of vessel cells which is the main characteristic of the late stages of the human aneurysmal disease. For example, Thrombospondin 1 (THBS1), also known as TSP-1, targeted by miR-1 [66], and form a subunit of a disulfide-linked homotrimeric protein. Study of human aortic smooth muscle cells (HASMC) has revealed that TSP-1 is involved in the migration and proliferation of HASMC, moreover, the upregulation of TSP-1 by leptin is depended on JAK2 and MAPK pathways [67]. Activated movement of smooth muscle cell and migration/movement of endothelial cell may imply phenotypic modulation of those cells. Neuropilin 1 (NRP1), validated to be targeted by miR-1 [66], participates in several different types of signaling pathways that control cell migration, for example, NRP1 binding with VEGF is essential for stimulation of endothelial cell migration [68]. One of the crucial players in the pathophysiology of IA is inflammation. Some studies at the transcriptome level are in accordance with the histopathological series that associated endothelial dysfunction, loss of mural cells, inflammatory cell infiltration and degradation of the matrix with sIA wall rupture [69].Though inflammatory macrophages and lymphocytes infiltrate the aneurysm wall, a link between their presence and aneurysm growth with subsequent rupture is not completely understood [70]. 5 out of 12 functions are related to immune response, among those functions, migration of phagocytes, affected by 7 miRNAs and 11 genes, has the highest potential to be activated. Genes validated to interact with miRNA include PNP, MYC, CD276, PIM1, THBS1, F11R and PLAU etc. which are involved in immune response. For example, purine nucleoside phosphorylase (PNP) targeted by miR-1 [71], is associated with T-cell (cell-mediated) immunity, B-cell immunity and antibody responses [72].
Functional analysis revealed some molecules targeted miRNAs with high prediction, validation of the relationship between the miRNA and these predicted targets are necessary for extending the molecular network of IA. Some upregulated genes expressed in intracranial arteries including NLR family, tumor necrosis factor (ligand) superfamily, interleukin, fibronectin and chemokine are predicted targets of down-regulated miRNAs in our study. Previous studies have implied their importance in IA [11, 57]. NLRP1, predicted target of miR-143-3p, is a member of the Ced-4 family of apoptosis proteins that could induce caspase-1 activation through the assembly of inflammasomes, multiprotein complexes, which are critical for generating mature proinflammatory cytokines including IL-1β and IL18. IL18 is also a predicted target of miR-143-3p, which increase early stage apoptosis of cultured HUVEC (umbilical vein endothelial cells) cells [73], and increase the death of VEC [74]. Another apoptosis related gene is TNFSF15, predicted target of miR-145-5p, which belongs to the tumor necrosis factor (TNF) ligand family acts as an autocrine factor to induce apoptosis in endothelial cells by activating NF-kappaB and MAP kinases [75]. FN1, targeted by miR-1, is involved in cell adhesion and migration processes. Two cytokine, CCL2 and CCL7, were targeted by miR-1, miR-23a-3p and miR-143-3p respectively. CCL2 displays chemotactic activity for monocytes and basophils which has been implicated in the pathogenesis of atherosclerosis which is characterized by monocytic infiltrates [76]. CCL7 is a secreted chemokine which attracts macrophages during inflammation and metastasis [77]. Although their interactions with miRNAs are predicted, validation the interactions in IA tissue could unearth the pivotal role of miRNAs in the pathogenesis of IA.
Limitation
This study has several limitations. One limitation is that only end-stage of disease tissues are available, as only the human IA samples are large enough or ruptured and need surgical intervention can be obtained. IA dome contains different cell type and tissues, the contribution of misregulated genes in each cell type should be further determined. Our functional analyses were based on miRNA targets which include highly predicted and experimentally validated, so these highly predicted interactions should be validated in future research.
Conclusion
Our data clearly showed the differential expression of 18 miRNAs in IA tissue from a control group of human MMA tissue. Bio-informatic analysis by IPA indicates that miRNAs target genes which may play a role in functional changes in VEC and VSMC, and activation of inflammatory response, and loss of cells in vessel wall. Our study was in line with many previous studies, several candidates need to be studied thoroughly to uncover the role of miRNA in IA. Our study provides novel evidence identifying miRNAs involved with response of the rupture of IA and gives us a deeper understanding on pathology of IA, miRNA found in this study may be a notably potential entry point to reveal pathology of IA from another perspective.
References
Juvela S: Prevalence of and risk factors for intracranial aneurysms. Lancet Neurol. 2011, 10 (7): 595-597. 10.1016/S1474-4422(11)70125-9.
Tulamo R, Frösen J, Paetau A, Seitsonen S, Hernesniemi J, Niemelä M, et al: Lack of complement inhibitors in the outer intracranial artery aneurysm wall associates with complement terminal pathway activation. Am J Pathol. 2010, 177: 3224-3232. 10.2353/ajpath.2010.091172.
Frösen J, Tulamo R, Paetau A, Laaksamo E, Korja M, Laakso A, Niemelä M, Hernesniemi J: Saccular intracranial aneurysm: pathology and mechanisms. Acta Neuropathologica. 2012, 123: 773-786. 10.1007/s00401-011-0939-3.
Krings T, Mandell DM, Kiehl TR, Geibprasert S, Tymianski M, Alvarez H, ter Brugge KG, Hans FJ: Intracranial aneurysms: from vessel wall pathology to therapeutic approach. Nat Rev Neurol. 2011, 7 (10): 547-559. 10.1038/nrneurol.2011.136.
Chalouhi N, Ali MS, Jabbour PM, Tjoumakaris SI, Gonzalez LF, Rosenwasser RH, Koch WJ, Dumont AS: Biology of intracranial aneurysms: role of inflammation. J Cereb Blood Flow Metab. 2012, 32 (9): 1659-1676. 10.1038/jcbfm.2012.84.
Steiner T, Juvela S, Unterberg A, Jung C, Forsting M, Rinkel G: European stroke organization guidelines for the management of intracranial aneurysms and subarachnoid haemorrhage. Cerebrovasc Dis. 2013, 35 (2): 93-112. 10.1159/000346087.
Aoki T, Nishimura M: The development and the use of experimental animal models to study the underlying mechanisms of CA formation. J Biomed Biotechnol. 2011, 2011: 535921.
Krischek B, Kasuya H, Tajima A, Akagawa H, Sasaki T, Yoneyama T, Ujiie H, Kubo O, Bonin M, Takakura K, Hori T, Inoue I: Network-based gene expression analysis of intracranial aneurysm tissue reveals role of antigen presenting cells. Neuroscience. 2008, 154: 1398-1407. 10.1016/j.neuroscience.2008.04.049.
Krischek B, Tatagiba M: The influence of genetics on intracranial aneurysm formation and rupture: current knowledge and its possible impact on future treatment. Adv Tech Stand Neurosurg. 2008, 33: 131-147. 10.1007/978-3-211-72283-1_3.
Li L, Yang X, Jiang F, Dusting GJ, Wu Z: Transcriptome-wide characterization of gene expression associated with unruptured intracranial aneurysms. European Neurology. 2009, 62: 330-337. 10.1159/000236911.
Shi C, Awad IA, Jafari N, Lin S, Du P, Hage ZA, et al: Genomics of human intracranial aneurysm wall. Stroke. 2009, 40: 1252-1261. 10.1161/STROKEAHA.108.532036.
Marchese E, Vignati A, Albanese A, Nucci CG, Sabatino G, Tirpakova B, et al: Comparative evaluation of genome-wide gene expression profiles in ruptured and unruptured human intracranial aneurysms. J Biol Regul & Homeost Agents. 2010, 24: 185-195.
Pera J, Korostynski M, Krzyszkowski T, Czopek J, Slowik A, Dziedzic T, et al: Gene expression profiles in human ruptured and unruptured intracranial aneurysms: what is the role of inflammation?. Stroke. 2010, 41: 224-231. 10.1161/STROKEAHA.109.562009.
Hsia HC, Schwarzbauer JE: Meet the tenascins: multifunctional and mysterious. J Biol Chem. 2005, 280: 26641-26644. 10.1074/jbc.R500005200.
Lewis BP, Burge CB, Bartel DP: Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell. 2005, 120: 15-20. 10.1016/j.cell.2004.12.035.
Frösen J, Piippo A, Paetau A, Kangasniemi M, Niemelä M, Hernesniemi J, et al: Growth factor receptor expression and remodeling of saccular cerebral artery aneurysm walls: implications for biological therapy preventing rupture. Neurosurgery. 2006, 58: 534-541.
Bartel DP: MicroRNAs: target recognition and regulatory functions. Cell. 2009, 136 (2): 215-233. 10.1016/j.cell.2009.01.002.
Korja M, Silventoinen K, McCarron P, Zdravkovic S, Skytthe A, Haapanen A, et al: Genetic epidemiology of spontaneous subarachnoid hemorrhage: Nordic twin study. Stroke. 2010, 41: 2458-2462. 10.1161/STROKEAHA.110.586420.
Lynam-Lennon N, Maher SG, Reynolds JV: The roles of microRNA in cancer and apoptosis. Biol Rev Camb Philos Soc. 2009, 84: 55-71. 10.1111/j.1469-185X.2008.00061.x.
Esteller M: Non-coding RNAs in human disease. Nat Rev Genet. 2011, 12 (12): 861-874. 10.1038/nrg3074.
Small EM, Frost RJ, Olson EN: MicroRNAs add a new dimension to cardiovascular disease. Circulation. 2010, 121 (8): 1022-1032. 10.1161/CIRCULATIONAHA.109.889048.
van Rooij E: The art of microRNA research. Circ Res. 2011, 108: 219-234. 10.1161/CIRCRESAHA.110.227496.
Guo H, Ingolia NT, Weissman JS, Bartel DP: Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010, 466: 835-840. 10.1038/nature09267.
Torella D, Iaconetti C, Catalucci D, Ellison GM, Leone A, Waring CD, Bochicchio A, Vicinanza C, Aquila I, Curcio A, Condorelli G, Indolfi C: MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ Res. 2011, 109 (8): 880-893. 10.1161/CIRCRESAHA.111.240150.
Xin M, Small EM, Sutherland LB, et al: MicroRNAs miR-143 and miR-145 modulate cytoskeletal dynamics and responsiveness of smooth muscle cells to injury. Genes Dev. 2009, 23: 2166-2178. 10.1101/gad.1842409.
Cheng Y, Liu X, Yang J, et al: MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ Res. 2009, 105: 158-166. 10.1161/CIRCRESAHA.109.197517.
Elia L, Quintavalle M, Zhang J, Contu R, Cossu L, Latronico MV, Peterson KL, Indolfi C, Catalucci D, Chen J, Courtneidge SA, Condorelli G: The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: correlates with human disease. Cell Death Differ. 2009, 16 (12): 1590-1598. 10.1038/cdd.2009.153.
van Rooij E, Sutherland LB, Thatcher JE, et al: Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A. 2008, 105: 13027-13032. 10.1073/pnas.0805038105.
Siefert SA, Sarkar R: Matrix metalloproteinases in vascular physiology and disease. Vascular. 2012, 20 (4): 210-216. 10.1258/vasc.2011.201202.
Afek A, Harats D, Roth A, Keren G, George J: A functional role for inducible costimulator (ICOS) in atherosclerosis. Atherosclerosis. 2005, 183 (1): 57-63. 10.1016/j.atherosclerosis.2005.03.040.
Ewing MM, Karper JC, Abdul S, de Jong RC, Peters HA, de Vries MR, Redeker A, Kuiper J, Toes RE, Arens R, Jukema JW, Quax PH: T-cell co-stimulation by CD28-CD80/86 and its negative regulator CTLA-4 strongly influence accelerated atherosclerosis development. Int J Cardiol. 2013, in press
Fournier NM, Lee B, Banasr M, Elsayed M, Duman RS: Vascular endothelial growth factor regulates adult hippocampal cell proliferation through MEK/ERK- and PI3K/Akt-dependent signaling. Neuropharmacology. 2012, 63 (4): 642-652. 10.1016/j.neuropharm.2012.04.033.
de Nigris F, Rienzo M, Sessa M, Infante T, Cesario E, Ignarro LJ, Al-Omran M, Giordano A, Palinski W, Napoli C: Glycoxydation promotes vascular damage via MAPK-ERK/JNK pathways. J Cell Physiol. 2012, 227 (11): 3639-3647. 10.1002/jcp.24070.
Krings T, Geibprasert S, ter Brugge KG: Pathomechanisms and treatment of pediatric aneurysms. Childs Nerv Syst. 2010, 26: 1309-1318. 10.1007/s00381-009-1054-9.
Small EM, Olson EN: Pervasive roles of microRNAs in cardiovascular biology. Nature. 2011, 469: 336-342. 10.1038/nature09783.
van Rooij E, Olson EN: MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat Rev Drug Discov. 2012, 11 (11): 860-872. 10.1038/nrd3864.
Jamaluddin MS, Weakley SM, Zhang L, Kougias P, Lin PH, Yao Q, Chen C: miRNAs: roles and clinical applications in vascular disease. Expert Rev Mol Diagn. 2011, 11 (1): 79-89. 10.1586/erm.10.103.
Thum T: MicroRNA therapeutics in cardiovascular medicine. EMBO Mole Med. 2012, 4: 3-14. 10.1002/emmm.201100191.
Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Høydal M, Autore C, Russo MA, Dorn GW, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G: MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007, 13 (5): 613-618. 10.1038/nm1582.
Chen J, Yin H, Jiang Y, Radhakrishnan SK, Huang ZP, Li J, Shi Z, Kilsdonk EP, Gui Y, Wang DZ, Zheng XL: Induction of microRNA-1 by myocardin in smooth muscle cells inhibits cell proliferation. Arterioscler Thromb Vasc Biol. 2011, 31 (2): 368-375. 10.1161/ATVBAHA.110.218149.
Bostjancic E, Zidar N, Stajer D, Glavac D: MicroRNAs miR-1, miR-133a, miR-133b and miR-208 are dysregulated in human myocardial infarction. Cardiology. 2010, 115: 163-169. 10.1159/000268088.
Long X, Miano JM: Transforming growth factor-beta1 (TGF-beta1) utilizes distinct pathways for the transcriptional activation of microRNA 143/145 in human coronary artery smooth muscle cells. J Biol Chem. 2011, 286 (34): 30119-30129. 10.1074/jbc.M111.258814.
Boucher JM, Peterson SM, Urs S, Zhang C, Liaw L: The miR-143/145 cluster is a novel transcriptional target of Jagged-1/Notch signaling in vascular smooth muscle cells. J Biol Chem. 2011, 286 (32): 28312-28321. 10.1074/jbc.M111.221945.
Rangrez AY, Massy ZA, Metzinger-Le Meuth V, Metzinger L: miR-143 and miR-145: molecular keys to switch the phenotype of vascular smooth muscle cells. Circulation: Cardiovasc Genet. 2011, 4: 197-205. 10.1161/CIRCGENETICS.110.958702.
Xie C, Huang H, Sun X, Guo Y, Hamblin M, Ritchie RP, Garcia-Barrio MT, Zhang J, Chen YE: MicroRNA-1 regulates smooth muscle cell differentiation by repressing Kruppel-like factor 4. Stem Cells Dev. 2011, 20 (2): 205-210. 10.1089/scd.2010.0283.
Cordes KR, Sheehy NT, White MP, et al: miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009, 460: 705-710.
Davis-Dusenbery BN, Chan MC, Reno KE, Weisman AS, Layne MD, Lagna G, Hata A: Down-regulation of Kruppel-like factor-4 (KLF4) by microRNA-143/145 is critical for modulation of vascular smooth muscle cell phenotype by transforming growth factor-beta and bone morphogenetic protein 4. J Biol Chem. 2011, 286 (32): 28097-28110. 10.1074/jbc.M111.236950.
Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, Olson EN: microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev. 2008, 22 (23): 3242-3254. 10.1101/gad.1738708.
Maegdefessel L, Azuma J, Toh R, Merk DR, Deng A, Chin JT, Raaz U, Schoelmerich AM, Raiesdana A, Leeper NJ, McConnell MV, Dalman RL, Spin JM, Tsao PS: Inhibition of microRNA-29b reduces murine abdominal aortic aneurysm development. J Clin Invest. 2012, 122 (2): 497-506. 10.1172/JCI61598.
Maegdefessel L, Azuma J, Tsao PS: MicroRNA-29b regulation of abdominal aortic aneurysm development. Trends Cardiovasc Med. 2013, in press
Boon RA, Seeger T, Heydt S, Fischer A, Hergenreider E, Horrevoets AJ, Vinciguerra M, Rosenthal N, Sciacca S, Pilato M, van Heijningen P, Essers J, Brandes RP, Zeiher AM, Dimmeler S: MicroRNA-29 in aortic dilation: implications for aneurysm formation. Circ Res. 2011, 109 (10): 1115-1119. 10.1161/CIRCRESAHA.111.255737.
Fort A, Borel C, Migliavacca E, Antonarakis SE, Fish RJ, Neerman-Arbez M: Regulation of fibrinogen production by microRNAs. Blood. 2010, 116 (14): 2608-2615. 10.1182/blood-2010-02-268011.
Sinha S, Dutta S, Datta K, Ghosh AK, Mukhopadhyay D: Von Hippel-Lindau gene product modulates TIS11B expression in renal cell carcinoma: impact on vascular endothelial growth factor expression in hypoxia. J Biol Chem. 2009, 284 (47): 32610-32618. 10.1074/jbc.M109.058065.
Boon RA, Dimmeler S: MicroRNAs and aneurysm formation. Trends Cardiovasc Med. 2011, 21 (6): 172-177. 10.1016/j.tcm.2012.05.005.
Ruigrok YM, Rinkel GJ: Genetics of intracranial aneurysms. Stroke. 2008, 39 (3): 1049-1055. 10.1161/STROKEAHA.107.497305.
Ruigrok YM, Rinkel GJ: From GWAS to the clinic: risk factors for intracranial aneurysms. Genome Med. 2010, 2: 61-10.1186/gm182.
Roder C, Kasuya H, Harati A, Tatagiba M, Inoue I, Krischek B: Meta-analysis of microarray gene expression studies on intracranial aneurysms. Neuroscience. 2012, 201: 105-113.
Liu L, Yu X, Guo X, Tian Z, Su M, Long Y, Huang C, Zhou F, Liu M, Wu X, Wang X: miR-143 is down-regulated in cervical cancer and promotes apoptosis and inhibits tumor formation by targeting Bcl-2. Mol Med Rep. 2012, 5 (3): 753-760.
Tang Y, Zheng J, Sun Y, Wu Z, Liu Z, Huang G: MicroRNA-1 regulates cardiomyocyte apoptosis by targeting Bcl-2. Int Heart J. 2009, 50 (3): 377-387. 10.1536/ihj.50.377.
Leone V, D'Angelo D, Rubio I, de Freitas PM, Federico A, Colamaio M, Pallante P, Medeiros-Neto G, Fusco A: miR-1 is a tumor suppressor in thyroid carcinogenesis targeting CCND2, CXCR4, and SDF-1alpha. J Clin Endocrinol Metab. 2011, 96 (9): E1388-E1398. 10.1210/jc.2011-0345.
Kuivaniemi H, Prockop DJ, Wu Y, Madhatheri SL, Kleinert C, Earley JJ, Jokinen A, Stolle C, Majamaa K, Myllylä VV, et al: Exclusion of mutations in the gene for type III collagen (COL3A1) as a common cause of intracranial aneurysms or cervical artery dissections: results from sequence analysis of the coding sequences of type III collagen from 55 unrelated patients. Neurology. 1993, 43 (12): 2652-2658. 10.1212/WNL.43.12.2652.
Kontusaari S, Tromp G, Kuivaniemi H, Ladda RL, Prockop DJ: Inheritance of an RNA splicing mutation (G + 1 IVS20) in the type III procollagen gene (COL3A1) in a family having aortic aneurysms and easy bruisability: phenotypic overlap between familial arterial aneurysms and Ehlers-Danlos syndrome type IV. Am J Hum Genet. 1990, 47 (1): 112-120.
Sengupta S, den Boon JA, Chen IH, Newton MA, Stanhope SA, Cheng YJ, Chen CJ, Hildesheim A, Sugden B, Ahlquist P: MicroRNA 29c is down-regulated in nasopharyngeal carcinomas, up-regulating mRNAs encoding extracellular matrix proteins. Proc Natl Acad Sci USA. 2008, 105 (15): 5874-5878. 10.1073/pnas.0801130105.
Luna C, Li G, Qiu J, Epstein DL, Gonzalez P: Role of miR-29b on the regulation of the extracellular matrix in human trabecular meshwork cells under chronic oxidative stress. Mol Vis. 2009, 15: 2488-2497.
Dawson K, Wakili R, Ordög B, Clauss S, Chen Y, Iwasaki Y, Voigt N, Qi XY, Sinner MF, Dobrev D, Kääb S, Nattel S: MicroRNA29: a mechanistic contributor and potential biomarker in atrial fibrillation. Circulation. 2013
Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N: Widespread changes in protein synthesis induced by microRNAs. Nature. 2008, 455 (7209): 58-63. 10.1038/nature07228.
Chavez RJ, Haney RM, Cuadra RH, Ganguly R, Adapala RK, Thodeti CK, Raman P: Upregulation of thrombospondin-1 expression by leptin in vascular smooth muscle cells via JAK2- and MAPK-dependent pathways. Am J Physiol Cell Physiol. 2012, 303 (2): C179-C191. 10.1152/ajpcell.00008.2012.
Herzog B, Pellet-Many C, Britton G, Hartzoulakis B, Zachary IC: VEGF binding to NRP1 is essential for VEGF stimulation of endothelial cell migration, complex formation between NRP1 and VEGFR2, and signaling via FAK Tyr407 phosphorylation. Mol Biol Cell. 2011, 22 (15): 2766-2776. 10.1091/mbc.E09-12-1061.
Guo F, Li Z, Song L, Han T, Feng Q, Guo Y, et al: Increased apoptosis and cysteinyl aspartate specific protease-3 gene expression in human intracranial aneurysm. J Clin Neurosc. 2007, 14: 550-555. 10.1016/j.jocn.2005.11.018.
Ferns SP, Sprengers ME, van Rooij WJ, Rinkel GJ, van Rijn JC, Bipat S, Sluzewski M, Majoie CB: Coiling of intracranial aneurysms: a systematic review on initial occlusion and reopening and retreatment rates. Stroke. 2009, 40: e523-e529. 10.1161/STROKEAHA.109.553099.
Nohata N, Hanazawa T, Kikkawa N, Sakurai D, Sasaki K, Chiyomaru T, Kawakami K, Yoshino H, Enokida H, Nakagawa M, Okamoto Y, Seki N: Identification of novel molecular targets regulated by tumor suppressive miR-1/miR-133a in maxillary sinus squamous cell carcinoma. Int J Oncol. 2011, 39 (5): 1099-1107.
Somech R, Lev A, Simon AJ, Hanna S, Etzioni A: T- and B-cell defects in a novel purine nucleoside phosphorylase mutation. J Allergy Clin Immunol. 2012, 130 (2): 539-542. 10.1016/j.jaci.2012.03.038.
Zhou G, Zhou Z, Ge S, Liu D, Zhang R, Xu G, Zhu W, Yin Q, Chen AF, Liu X: IL-18 accelerates the cell apoptosis by up-regulating cysteinyl leukotriene 2 receptor expression in human umbilical vein endothelial cells at the early stage of administration. Vascul Pharmacol. 2009, 32: 1659-1676.
Chandrasekar B, Boylston WH, Venkatachalam K, Webster NJ, Prabhu SD, Valente AJ: Adiponectin blocks interleukin-18-mediated endothelial cell death via APPL1-dependent AMP-activated protein kinase (AMPK) activation and IKK/NF-kappaB/PTEN suppression. J Biol Chem. 2008, 283: 24889-24898. 10.1074/jbc.M804236200.
Duan L, Yang G, Zhang R, Feng L, Xu C: Advancement in the research on vascular endothelial growth inhibitor (VEGI). Target Oncol. 2012, 7 (1): 87-90. 10.1007/s11523-012-0206-0.
Qian BZ, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW: CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011, 475 (7355): 222-225. 10.1038/nature10138.
Ben-Baruch A, Xu L, Young PR, Bengali K, Oppenheim JJ, Wang JM: Monocyte chemotactic protein-3 (MCP3) interacts with multiple leukocyte receptors. C-C CKR1, a receptor for macrophage inflammatory protein-1 alpha/Rantes, is also a functional receptor for MCP3. J Biol Chem. 1995, 270 (38): 22123-22128. 10.1074/jbc.270.38.22123.
Pre-publication history
The pre-publication history for this paper can be accessed here:http://www.biomedcentral.com/1755-8794/6/36/prepub
Acknowledgements
This work was supported in part by National Natural Science Foundation of China (grant number: 81071028, 81172513, 81130021), Program for New Century Excellent Talents (grant number: 7603230006), the Major State Basic Research Development Program of China (973 Program) (grant number: 2011CB510000, 2012CB944600).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
YJ and MZ recruited patients, obtained tissue samples from cases and controls, verified clinical information, designed experiment, analyzed data, and critically reviewed the manuscript. HH, JC and HZ prepared RNA samples for microarray and RTq-PCR, and ran the RTq-PCR assay. JL contributed to the microarray data handling, functional analysis, as well as drafting and editing the manuscript. RD contributed to the experimental design, data analysis, drafting and editing of the manuscript, and obtained funding for the study. All authors read and approved the final manuscript.
Yugang Jiang, Mingming Zhang contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Jiang, Y., Zhang, M., He, H. et al. MicroRNA/mRNA profiling and regulatory network of intracranial aneurysm. BMC Med Genomics 6, 36 (2013). https://doi.org/10.1186/1755-8794-6-36
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8794-6-36