
Abstract
Background
This proof-of-principle study demonstrates the usefulness and robustness of a novel array based method for the elucidation of genetic causes underlying early pregnancy loss. A combined microarray utilizing comparative genomic hybridization and single nucleotide polymorphism detection (CGH + SNP) was used for parallel genome-wide identification of copy number and heterozygosity status of 70 products of conception. Results of samples with previously determined aneuploidies were juxtaposed to those of a second cohort appearing normal after routine genetic diagnostics.
Results
All chromosomal imbalances were confirmed, in one sample of the aneuploid panel additional monosomy X was discovered. Genome-wide uniparental disomy causing a complete hydatidiform mole was identified in another sample. No specimen featured microaberrations of obvious clinical relevance. Among cases with presumable euploidy, one microdeletion and a single region of homozygosity were assigned unclear clinical significance.
Conclusions
The results prove the utility of combined imbalance and homozygosity mapping for routine workup of these challenging specimens. Moreover parallel screening at submicroscopic resolution facilitates the detection of novel genetic alterations underlying spontaneous abortion.
Similar content being viewed by others


Background
About 8 to 20% of clinically confirmed pregnancies abort spontaneously in the first trimester [1, 2]. About half of these early abortions can be explained by chromosome aneuploidy. Non-genetic contributing factors include immunological disorders, thyroid or blood coagulation conditions, acute or chronic diseases of the mother, stress or environmental influence. In order to determine the best medical treatment for all possible causes underlying embryonic demise, genetic analyses of products of conception (POCs) were introduced to diagnostic routines decades ago. Since then, clinical geneticists have been facing two major diagnostic concerns: First of all, mosaic constellations in embryo or placenta remain a profound analytical problem related to POC specimen [3, 4]. Mosaics can only be reliably assessed by comparative analysis of different embryonic and/or chorionic cell types, which is usually not feasible in a diagnostic setting [5]. Moreover, there always is the suspicion of mutations that lie beneath detection limits and resolution of routinely applied protocols. Studies conducted in this respect aimed at the designation of clinically relevant segmental imbalances (deletions or duplications) and uniparental disomy (UPD) [6–10]. Molecular karyotyping by array based comparative genomic hybridization (aCGH) or single nucleotide polymorphism (SNP) detection enabled genome-wide profiling at even higher resolution [11–19]. The results showed in unison that segmental chromosomal aberrations seem non-recurrent and sporadic events in spontaneous abortions.
Most often distal gains or losses arise from the presence of chromosome derivatives from a balanced translocation carrier parent. In this clinical context, the introduction of precise imbalance mapping meant significant progress. In other cases the pathogenic relevance of microimbalances was sometimes hard to evaluate at the state of knowledge on the human genome. The same applied to genomic segments displaying UPD, which in general can be associated with genomic imprinting disorders and autosomal recessive conditions [20]. UPD often shows in mosaic constellations [21]. Potentially, all these features could contribute to early fetal loss. The occurrence of UPD increases with elevating aneuploidy risks due to advancing maternal age [22]. Systematic studies concordantly reported low frequencies of UPD in POCs, but discussed UPD as a possibly underestimated pathogenic factor for spontaneous abortion due to detection limits of available methods [23–27]. UPD comprises the concepts of uniparental heterodisomy (UPHD) and isodisomy (UPID). However, the occurrence of either in POCs is unknown [28].
SNP profiling enables the identification of UPID via homozygosity mapping. The analytical challenge lies in the differentiation of pathogenic regions displaying absence of heterozygosity (AOH) from benign long contiguous stretches of homozygosity occuring frequently in the human genome [29, 30].
The major advantage of array based cytogenomic approaches is that they overcome cell culture failure and- most often- overgrowth by maternal cells [31]. Thus, microarray screening has been repeatedly suggested as first tier test in the analytical workup of missed abortions [32–34].
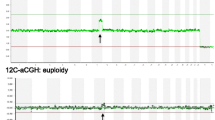
Against this background, we customized a novel CGH + SNP-8x60K microarray for POC analyses. A comparison of this approach to conventional platforms is given in Table 1. Representative hybridization profiles of the novel platform are displayed in Figure 1. The combined detection featured by CGH + SNP-arrays promised a superior robustness with regard to samples of varying quality and thus a possible advantage for POC analyses. With regard to this, the presented feasibility study addressed two key questions: Firstly, if a combined array approach at this resolution was optimal for the reliable detection of known genomic aberrations underlying first trimester fetal demise. Secondly, whether this platform unmasked recurrent small copy number associated or copy number neutral aberrations that could be causative of pregnancy loss in specimens of normal karyotype. To evaluate its utility in a diagnostic setting, the novel test was applied to a representative probe panel from routine diagnostics in our facility.

Genome-wide CGH and SNP hybridization profiles for a sample displaying trisomy 4. Panel A: aCGH data, CN: copy number, Panel B: SNP data, GT: genotype.
Results
High molecular weight genomic DNA was successfully extracted from all 70 POC-samples. In 12 of them protein contamination could not be completely removed by ethanol precipitation resulting in A260/280 < 1.8 (1.740-1.795). Hybridization of respective DNA resulted in notably noisier imbalance profiles (derivate log ratio spread value (DLRS) = 0.16-0.29). However a lower DNA quality was not associated with a general dropout of SNP data (informative SNPs = 32.5-90.6%). Samples with A260/280 > 1.8 and A260/230 > 2 displayed a DLRS <0.2 and more than 80% informative SNPs. Although it was possible to reliably evaluate profiles of lower quality as well, these parameters were determined as optimal for analyses on the novel array platform. The design comprised a single region in 9q22.32, in which coincidental combination of oligonucleotide CGH probes with suboptimal hybridization properties lead to frequent false positive indication of a 47 kb genomic loss.
Microarray data are available in the ArrayExpress database (http://www.ebi.ac.uk/arrayexpress) under accession number E-MTAB-1725. An overview is given in Table 2. Array experiments confirmed all previously known aneuploidies. Additionally, monosomy X was detected in a specimen with 47,XX,+12 karyotype after routine cytogenetics. It had displayed sparse proliferation in culture. Although a low log2-ratio of chromosome X in the respective profile did not suggest it, array and karyotyping findings did not exclude the possible presence of a placental mosaic. For two near-triploid specimen with XXY-constellation, triploidy was indicated by low log2-ratios of gonosomes in aCGH-profiles. Remarkably, in one of them additional loss of chromosome 11 was missed, whereas an additional gain of chromosome 21 in the other sample was called correctly. It was speculated whether different aberration sizes or types of imbalances mislead the algorithm to bivalent copy number determination. Tetraploidy in the two samples with 92,XXXX karyotype was missed.
In the panel of POCs with presumptive normal karyotype one aberration of obvious clinical significance was unmasked: AOH affecting all chromosomes indicated genome-wide UPID and thus the formation of a complete hydatidiform mole in this patient. Interestingly, the algorithm called only 2 heterozygous SNPs as informative (0.03%), which were in fact false calls for a diploid genome build of two identical haploid paternal chromosome sets.
The novel combined microarray did not discover any alterations clearly associated with common microdeletion, −duplication or UPD-related syndromes. Separate from whole chromosome aneuploidy, numbers of detected microimbalances in aneuploid and euploid panels were comparable and varied from 3 to 13 deviations per sample (0.2 kb-4 Mb). Imbalances occurring frequently in both groups overlapped with known polymorphic chromosomal regions. Hybridizations of high quality genomic DNA correlated with low numbers of detected irregularities.
In one sample with normal karyotype a heterozygous loss of 3 Mb was revealed targeting the chromosomal region of 2p24.3. It comprised four genes (TRIB2, FAM84A, NBAS (NAG) and DDX1), whose molecular functions did not imply an obvious cellular cause for miscarriage. However, potential pathogenicity was accredited to the fact that the proximal breakpoint of the deletion mapped close to MYCN. Its gene product, the N-Myc protein, is a key regulator of skeleton morphogenesis and organogenesis during embryogenesis [36, 37]. Breakpoint mapping by FISH with locus specific probes failed due to insufficient cell integrity of the frozen specimen. Thus, it could not be verified, whether the deletion affected regulatory elements of MYCN. Parental samples were unavailable, so the loss was classified as CNC (potentially pathogenic).
AOH counts per genome varied from 1 to 32. Intervals ranged from 8.4 to 24.2 Mb in size. No POC exhibited an excessive degree of AOH indicating a high grade of parental consanguinity [38]. Segments with AOH occurring in both panels were not assigned any clinical relevance and evaluated as benign.
In two euploid samples AOH mapped to 6q24, overlapping with the UPD critical segment for transient neonatal diabetes mellitus and at the same time with a reported region of biparental apparently benign homozygosity [39]. One of them spanned 16 Mb in 6q23.2q24.3, the other one 14.2 Mb in 6q23.3q25.1. Follow-up by STR was possible in the latter case. It revealed heterozygosity and thus, a technical artifact.
Discussion
The comprehensive analysis of POCs always represents a challenge. It is usually met by a workflow of cytogenetic, molecular cytogenetic and molecular genetic methods depending on their availability in the diagnostic lab. This study demonstrates that the established microarray platform is suitable for the reliable simultaneous detection of imbalances and AOH in abortion material. It facilitates the unification of diagnostic steps in various laboratory workflows, which helps to significantly reduce costs.
Technical utility
A major advantage of molecular karyotyping is its independency from proliferation of starting material in cell culture. It also overcomes in vitro selection against certain aneuploidies, cultural artifacts or maternal cell overgrowth. Maternal contamination of the tissue used for DNA extraction cannot always be precluded even after meticulous preparation of chorionic villi. There are further material-related aspects to consider in diagnostics, especially with regard to mosaicism: While DNA is extracted from all cell types present in the villi sample, karyotyping assesses mainly well-proliferating cells from the extraembryonic mesoderm, and FISH cells of the cytotrophoblast [5].
Good quality of genomic DNA reflected in excellent DLRS-values for aCGH, but did not guarantee a high percentage of informative SNP probes. We suspect postmortem DNA modification and/or degradation resulting in incomplete or faulty restriction digestion not detectable by AGE. Alternative digestion assays with isoschizomers lacking any methylation sensitivity did not alter SNP profiles notably (data not shown).
Our observations suggest that SNP calls were more readily affected by DNA quality than CGH calls. We therefore evaluate the use of a combined CGH + SNP approach superior to hybridizations to sole SNP arrays, because copy numbers are “determined” via more robust CGH, not by SNP based copy number “calling”.
SNP detection on the combined platform is substantially different from that on sole SNP arrays. Instead of direct measurement of both alleles at one locus, only one allele is assessed by aCGH + SNP. Restriction digestion is used to selectively remove one allele. Measurement of the remaining non-digested allele and the copy number state determined by aCGH are used to constitute the presence of heterozygosity. This means the combined chip does not have an equivalent statistic to the B-allele frequency, which is the standard way in which a triploid sample would be detected by a conventional SNP array. Since aCGH is not able to detect polyploidy, the linked SNP analysis proceeds assuming the sample is diploid. Thus, the identification of triploid samples in this study succeeded only for those, in which the presence of a Y chromosome lead to low log2-ratios of gonosomes. For the same reason, combined cytogenomics did not overcome the inability of aCGH to distinguish samples of 46,XX karyotype from tetraploid samples. Like conventional aCGH, the novel method cannot detect ratio changes of sample to reference extending genome-wide.
The combination of aCGH and SNP approaches does not fundamentally emend the detection of mosaicism and chimerism. In contrast to sole SNP arrays, the method is blind to mosaics of both UPHD and UPID [40]. With regard to mosaic constellations of imbalances, low log2-ratios indicating different genomes in the study panel were due to known maternal contamination. Based on our experience with medium resolution aCGH, most often cytogenetically proven placental or fetal mosaics display either as distinct gains or losses or not at all. In routine diagnostics we report them only after verification by karyotyping or FISH.
We point out that the specimen with the Robertsonian translocation was assigned to the panel of normal samples. Euploidy was confirmed by array analysis. Nevertheless we emphasize in diagnostic reports that mosaicism of low level trisomy cannot fully be excluded.
Diagnostic utility
We established a robust assay for the rapid detection of aneuploidy and UPID in a one step analysis. Genome-wide UPID is associated with complete hydatidiform moles. Hydatidiform moles are at high risk of developing into choriocarcinomas and demand specific medical treatment.
Results of this pilot study show that the chosen probe resolution is reasonable for oligonucleotide detection of segmental aberrations in POCs, because it does not lead to excessive calling of microalterations, which then have to be evaluated with regard to their clinical significance. At medium resolution, partial chromosome imbalances due to derivative chromosomes from balanced reciprocal translocation carrier parents will reliably be picked up since those are usually detectable at the microscopic level. Apart from this, microduplications and microdeletions unambiguously associated with early pregnancy loss seem sporadic events. The clinical evaluation of CNCs will become easier with the ongoing understanding of functional elements in the human genome.
For the evaluation of partial AOH we consider it reasonable to apply the UPD thresholds determined in postnatal diagnostics as a guideline: due to steric reasons in recombination, UPD usually affects stretches of 20 Mb or wider in interstitial and appr. 10 Mb in telomeric regions [39]. Segments of homozygosity smaller than this are more likely to represent benign AOH. As knowledge of epigenetic effects such as imprinting increases, homozygosity mapping will become an important tool in the elucidation of pathogenetic mechanisms in abortions. There is an urgent need for valid databases on UPD effects on fetal demise. Now that it becomes technically possible to generate these data, our platform being a part thereof, they will hopefully arise. Strict differentiation between UPHD and UPID in scientific and medical reports will greatly promote this. Homozygosity profiling as it is presented here, will only detect UPID. For UPHD, both parents have to be included in the array screening as well. In diagnostics this is very expensive, but feasible (though not with current algorithm options of this platform). However, at the current state of medical science it is clear that segmental or complex UPD arises only from co-occurrence of meiotic or mitotic recombination, abnormal segregation, and subsequent correction [41]. Therefore the risk of recurrence is negligible versus the risk of invasive prenatal diagnosis in a future pregnancy.
On the long road towards genetic analysis at base pair resolution, we present a profound diagnostic method that at the same time allows further insight into pathogenic molecular mechanisms resulting in spontaneous abortions. The clinical benefits of POC-analyses remain unquestionable: they permit accurate genetic counseling and the estimation of recurrence risks. Moreover they help to determine the best medical treatment to fulfill the couples’ desire for a healthy child.
Conclusion
We suggest genome-wide CGH + SNP profiling as a first tier test for simultaneous detection of copy number associated and copy number neutral aberrations underlying spontaneous abortion. Although polyploidy and structural rearrangements have to be assessed in further diagnostic steps, the peculiar advantages of this novel cytogenomic method are independency from cell culture proliferation, reliable detection of chromosomal aneuploidy and pathogenic microimbalances as well as genome-wide UPID underlying hydatidiform moles.
Methods
This study was done on 70 samples of fetal tissue or chorionic villi from early pregnancy losses remaining after genetic diagnostics (native n =63, cultivated n =7). Written informed consent was obtained from all patients for the use of material from products of conception after completion of genetic diagnostics and further use in this study. Ethical approval was granted for this study by an interdisciplinary institutional reviewer board of medical and scientific experts of the participating centers. From conventional cytogenetics, karyotypes were known for 60 POCs. Due to cell culture failure, 10 samples were assessed by molecular cytogenetics: 2 of them by fluorescent in situ hybridization (FISH) with probes targeting chromosomes most commonly affected by aneuploidies (13, 16, 18, 21, 22, X, Y) and 8 samples by genome-wide aCGH. If nondistinctive, fetal origin of material had been verified by short tandem repeat marker analysis (STR). Thus, the panel comprised 35 samples previously determined as aneuploid (31 by karyotyping and 4 by aCGH) and 35 samples that appeared euploid after diagnostic routine testing (29 by karyotyping, 2 by FISH and 4 by aCGH, protocols available upon request).
Specimens of dissected fetal tissue or cultured cells were thawed and washed three times in phosphate buffered saline (Invitrogen, Carlsbad, USA). Genomic DNA was extracted utilizing the Maxwell®16 LEV Blood DNA kit (Promega Corp., Madison, USA), MagNA Pure kit (Roche, Grenzach-Wyhlen, Germany) or QIAamp DNA Blood Mini kit (QIAGEN, Hilden, Germany) according to respective manufacturers’ protocols. Concentration and quality of genomic DNA was assessed spectrophotometrically (NanoDrop® ND 1000, peqlab Biotechnologie GmbH, Erlangen, Germany). If necessary, ethanol/sodium acetate precipitation was performed. High molecular weight of genomic DNA as well as complete restriction digestion were verified by agarose gel electrophoresis (AGE) according to standard laboratory protocols.
Sample-DNA was hybridized to custom CGH + SNP-8×60K microarrays (BlueGnome Ltd., Cambridge, UK). The design was genome-wide, but targeted known disease genes and regions (design identification: MUNCH005). It was made up of 59 090 60mer oligonucleotides mapped to the human genome according to the Genome Reference Consortium Human Build 37 of February 2009 (GRCh37/hg19). The practical resolution for aCGH was appr. 200 kb corresponding to three consecutive deviating probes with average spacing of 66 kb. The design comprised 18% SNP-probes enabling the detection of stretches of homozygosity of 13 Mb or wider. Respective male or female genomic DNA of known haplotype derived from single individuals displaying no clinical phenotype was used as reference (SNPRef DNA, commercially not available, BlueGnome Ltd.). Wherever applicable, samples were cohybridized with sex-matched references according to the CytoChip Oligo reference manual v 1.8 (BlueGnome Ltd.). Slides were scanned in double pass mode at 3 μm resolution with a G2565CA microarray scanner (Agilent Technologies, Santa Clara, USA). Data were analyzed with software BlueFuseMulti versions 2.5 to 3.0 (BlueGnome Ltd.). In addition, aCGH and SNP-profiles were inspected manually and simultaneously in a 3 Mb sliding window. Sex-mismatch derived copy number deviations and benign AOH of chromosome Y in male samples served as internal controls. Detected imbalances were assigned either benign copy number variation (CNV), pathogenic copy number alteration (CNA) or copy number change of uncertain clinical significance (CNC) [42]. Homozygous segments were rated as benign, pathogenic, or AOH of uncertain clinical significance. The following online databases were used in the evaluation process: Database of Genomic Variants Beta (DGVbeta, http://dgvbeta.tcag.ca/dgv/app/home), Decipher (DECIPHER, http://decipher.sanger.ac.uk), International Standards for Cytogenomic Arrays (ISCA, http://www.iscaconsortium.org) and the UPD-database of the University Medical Centre of Jena, Germany (http://ssmc-tl.com/Start.html).
For verification of homozygosity in the 6q24 chromosomal region, STR analyses were carried out utilizing the PowerPlex® ES system (Promega Corp).
Abbreviations
- aCGH:
-
Array based comparative genomic hybridization
- AGE:
-
Agarose gel electrophoresis
- AOH:
-
Absence of heterozygosity
- CGH:
-
Comparative genomic hybridization
- CNA:
-
Copy number aberration
- CNC:
-
Copy number change
- CNV:
-
Copy number variant
- DLRS:
-
Derivative log ratio spread
- DNA:
-
Deoxyribonucleic acid
- FISH:
-
Fluorescence in situ hybridization
- POC:
-
Product of conception
- SNP:
-
Single nucleotide polymorphism
- STR:
-
Short tandem repeat
- UPD:
-
Uniparental disomy
- UPHD:
-
Uniparental heterodisomy
- UPID:
-
Uniparental isodisomy.
References
Warburton D, Fraser FC: Spontaneous abortion risks in man: data from reproductive histories collected in a medical genetics unit. Am J Hum Genet 1964, 16: 1–25.
Wang X, Chen C, Wang L, Chen D, Guang W, French J: Conception, early pregnancy loss, and time to clinical pregnancy: a population-based prospective study. Fertil Steril 2003,79(3):577–584.
Jobanputra V, Esteves C, Sobrino A, Brown S, Kline J, Warburton D: Using FISH to increase the yield and accuracy of karyotypes from spontaneous abortion specimens. Prenat Diagn 2011,31(8):755–759.
Vorsanova SG, Kolotii AD, Iourov IY, Monakhov VV, Kirillova EA, Soloviev IV, Yurov YB: Evidence for high frequency of chromosomal mosaicism in spontaneous abortions revealed by interphase FISH analysis. J Histochem Cytochem 2005,53(3):375–380.
Lebedev I: Mosaic aneuploidy in early fetal losses. Cytogenet Genome Res 2011,133(2–4):169–183.
Daniely M, Aviram-Goldring A, Barkai G, Goldman B: Detection of chromosomal aberration in fetuses arising from recurrent spontaneous abortion by comparative genomic hybridization. Hum Reprod 1998,13(4):805–809.
Kondo Y, Tsukishiro S, Tanemura M, Sugiura-Ogasawara M, Suzumori K, Sonta S: Maternal uniparental disomy of chromosome 16 in a case of spontaneous abortion. J Hum Genet 2004,49(4):177–181.
Hu Y, Chen X, Chen LL, Xu ZF, Wang X, Cui H: Comparative genomic hybridization analysis of spontaneous abortion. Int J Gynaecol Obstet 2006,92(1):52–57.
Azmanov DN, Milachich TV, Zaharieva BM, Michailova GI, Dimitrova VG, Karagiozova ZH, Maznejkova VT, Chernev TA, Toncheva DI: Profile of chromosomal aberrations in different gestational age spontaneous abortions detected by comparative genomic hybridization. Eur J Obstet Gynecol Reprod Biol 2007,131(2):127–131.
Sazhenova EA, Lebedev IN: Epimutations of the KCNQ1OT1 imprinting center of chromosome 11 in early human embryo lethality. Genetika 2008,44(12):1609–1616.
Schaeffer AJ, Chung J, Heretis K, Wong A, Ledbetter DH, Less Martin C: Comparative genomic hybridization-array analysis enhances the detection of aneuploidies and submicroscopic imbalances in spontaneous miscarriages. Am J Hum Genet 2004,74(6):1168–1174.
Shimokawa O, Harada N, Miyake N, Satoh K, Mizuguchi T, Niikawa N, Matsumoto N: Array comparative genomic hybridization analysis in first-trimester spontaneous abortions with ’normal’ karyotypes. Am J Med Genet A 2006,140(18):1931–1935.
Borovik CL, Perez ABA, da Silva LRJ, Krepischi-Santos ACV, Costa SS, Rosenberg C: Array-CGH testing in spontaneous abortions with normal karyotypes. Genet Mol Biol 2008, 31: 416–422.
Zhang YX, Zhang YP, Gu Y, Guan FJ, Li SL, Xie JS, Shen Y, Wu BL, Ju W, Jenkins EC, Brown WT, Zhong N: Genetic analysis of first-trimester miscarriages with a combination of cytogenetic karyotyping, microsatellite genotyping and arrayCGH. Clin Genet 2009,75(2):133–140.
Deshpande M, Harper J, Holloway M, Palmer R, Wang R: Evaluation of array comparative genomic hybridization for genetic analysis of chorionic villus sampling from pregnancy loss in comparison to karyotyping and multiplex ligation-dependent probe amplification. Genet Test Mol Biomarkers 2010,14(3):421–424.
Rajcan-Separovic E, Diego-Alvarez D, Robinson WP, Tyson C, Qiao Y, Harvard C, Fawcett C, Kalousek D, Philipp T, Somerville MJ, Stephenson MD: Identification of copy number variants in miscarriages from couples with idiopathic recurrent pregnancy loss. Hum Reprod 2010,25(11):2913–2922.
Rajcan-Separovic E, Qiao Y, Tyson C, Harvard C, Fawcett C, Kalousek D, Stephenson M, Philipp T: Genomic changes detected by array CGH in human embryos with developmental defects. Mol Hum Reprod 2010,16(2):125–134.
Gao J, Liu C, Yao F, Hao N, Zhou J, Zhou Q, Zhang L, Liu X, Bian X, Liu J: Array-based comparative genomic hybridization is more informative than conventional karyotyping and fluorescence in situ hybridization in the analysis of first-trimester spontaneous abortion. Mol Cytogenet 2012,5(1):33.
Lathi RB, Massie JAM, Loring M, Demko ZP, Johnson D, Sigurjonsson S, Gemelos G, Rabinowitz M: Informatics enhanced SNP microarray analysis of 30 miscarriage samples compared to routine cytogenetics. PLoS One 2012,7(3):31282.
Engel E: A new genetic concept: uniparental disomy and its potential effect, isodisomy. Am J Med Genet 1980,6(2):137–143.
Bruno DL, White SM, Ganesamoorthy D, Burgess T, Butler K, Corrie S, Francis D, Hills L, Prabhakara K, Ngo C, Norris F, Oertel R, Pertile MD, Stark Z, Amor DJ, Slater HR: Pathogenic aberrations revealed exclusively by single nucleotide polymorphism (SNP) genotyping data in 5000 samples tested by molecular karyotyping. J Med Genet 2011,48(12):831–839.
Kotzot D: Advanced parental age in maternal uniparental disomy (UPD): implications for the mechanism of formation. Eur J Hum Genet 2004,12(5):343–346.
Shaffer LG, McCaskill C, Adkins K, Hassold TJ: Systematic search for uniparental disomy in early fetal losses: the results and a review of the literature. Am J Med Genet 1998,79(5):366–372.
Smith MJ, Creasy MR, Clarke A, Upadhyaya M: Sex ratio and absence of uniparental disomy in spontaneous abortions with a normal karyotype. Clin Genet 1998,53(4):258–261.
Fritz B, Aslan M, Kalscheuer V, Ramsing M, Saar K, Fuchs B, Rehder H: Low incidence of UPD in spontaneous abortions beyond the 5th gestational week. Eur J Hum Genet 2001,9(12):910–916.
Tsukishiro S, Li QY, Tanemura M, Sugiura-Ogasawara M, Suzumori K, Sonta S: Paternal uniparental disomy of chromosome 14 and unique exchange of chromosome 7 in cases of spontaneous abortion. J Hum Genet 2005,50(3):112–117.
Toutain J, Labeau-Gaüzere C, Barnetche T, Horovitz J, Saura R: Confined placental mosaicism and pregnancy outcome: a distinction needs to be made between types 2 and 3. Prenat Diagn 2010,30(12–13):1155–1164.
Kotzot D, Utermann G: Uniparental disomy (UPD) other than 15: phenotypes and bibliography updated. Am J Med Genet A 2005,136(3):287–305.
Kearney HM, Kearney JB, Conlin LK: Diagnostic implications of excessive homozygosity detected by SNP-based microarrays: consanguinity, uniparental disomy, and recessive single-gene mutations. Clin Lab Med 2011,31(4):595–613.
Li LH, Ho SF, Chen CH, Wei CY, Wong WC, Li LY, Hung SI, Chung WH, Pan WH, Lee MT, Tsai FJ, Chang CF, Wu JY, Chen YT: Long contiguous stretches of homozygosity in the human genome. Hum Mutat 2006,27(11):1115–1121.
Benkhalifa M, Kasakyan S, Clement P, Baldi M, Tachdjian G, Demirol A, Gurgan T, Fiorentino F, Mohammed M, Qumsiyeh MB: Array comparative genomic hybridization profiling of first-trimester spontaneous abortions that fail to grow in vitro. Prenat Diagn 2005,25(10):894–900.
Robberecht C, Schuddinck V, Fryns JP, Vermeesch JR: Diagnosis of miscarriages by molecular karyotyping: benefits and pitfalls. Genet Med 2009,11(9):646–654.
Menten B, Swerts K, Delle Chiaie B, Janssens S, Buysse K, Philippé J, Speleman F: Array comparative genomic hybridization and flow cytometry analysis of spontaneous abortions and mors in utero samples. BMC Med Genet 2009, 10: 89.
Shaffer LG, Dabell MP, Fisher AJ, Coppinger J, Bandholz AM, Ellison JW, Ravnan JB, Torchia BS, Ballif BC, Rosenfeld JA: Experience with microarray-based comparative genomic hybridization for prenatal diagnosis in over 5000 pregnancies. Prenat Diagn 2012,32(10):976–985.
Shaffer LG, McGowan-Jordan J, Schmid M: ISCN 2013: An International System for human Cytogenetic Nomenclature. Basel: Karger; 2013.
Zhou ZQ, Shung CY, Ota S, Akiyama H, Keene DR, Hurlin PJ: Sequential and coordinated actions of c-Myc and N-Myc control appendicular skeletal development. PLoS One 2011,6(4):18795.
van Bokhoven H, Celli J, van Reeuwijk J, Rinne T, Glaudemans B, van Beusekom E, Rieu P, Newbury-Ecob RA, Chiang C, Brunner HG: MYCN haploinsufficiency is associated with reduced brain size and intestinal atresias in Feingold syndrome. Nat Genet 2005,37(5):465–467.
McMullan DJ, Bonin M, Hehir-Kwa JY, de Vries BBA, Dufke A, Rattenberry E, Steehouwer M, Moruz L, Pfundt R, de Leeuw N, Riess A, Altug-Teber O, Enders H, Singer S, Grasshoff U, Walter M, Walker JM, Lamb CV, Davison EV, Brueton L, Riess O, Veltman JA: Molecular karyotyping of patients with unexplained mental retardation by SNP arrays: a multicenter study. Hum Mutat 2009,30(7):1082–1092.
Papenhausen P, Schwartz S, Risheg H, Keitges E, Gadi I, Burnside RD, Jaswaney V, Pappas J, Pasion R, Friedman K, Tepperberg J: UPD detection using homozygosity profiling with a SNP genotyping microarray. Am J Med Genet A 2011,155A(4):757–768.
Conlin LK, Thiel BD, Bonnemann CG, Medne L, Ernst LM, Zackai EH, Deardorff MA, Krantz ID, Hakonarson H, Spinner NB: Mechanisms of mosaicism, chimerism and uniparental disomy identified by single nucleotide polymorphism array analysis. Hum Mol Genet 2010,19(7):1263–1275.
Kotzot D: Complex and segmental uniparental disomy updated. J Med Genet 2008,45(9):545–556.
Gardner McKinlay RJ, Sutherland GR: Chromosome abnormalities and genetic counselling. Oxford Monographs on medical genetics 4th edition. 2012, 61.
Acknowledgements
The authors gratefully acknowledge all women who kindly gave their consent to the re-analysis of specimen. We express our deep gratitude to Dr. Birgit Becker and Dr. Sonja Bingemann, who supported this study with their excellent expertise. No funding was obtained.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
SB, BS, FS, JP, MS and CNSH declare no conflict of interests. AC and MB are employees of BlueGnome Ltd., an Illumina company.
Authors’ contributions
SB outlined the study, participated in array design and analysis and drafted the manuscript. BS, FS, JP and MS carried out hybridizations, acquisition of data, interpretation and documentation. AC and MB designed the array platform and developed the analysis algorithm. CNSH supervised the study and finally approved the manuscript to be published. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Bug, S., Solfrank, B., Schmitz, F. et al. Diagnostic utility of novel combined arrays for genome-wide simultaneous detection of aneuploidy and uniparental isodisomy in losses of pregnancy. Mol Cytogenet 7, 43 (2014). https://doi.org/10.1186/1755-8166-7-43
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8166-7-43