
Abstract
Background
Acute myeloid leukemia (AML) comprises a spectrum of myeloid malignancies which are often associated with distinct chromosomal abnormalities, and the analysis of such abnormalities provides us with important information for disease classification, treatment selection and prognosis. Some chromosomal abnormalities albeit recurrent are rare such as tetrasomy 8 or isochromosome 5p. In addition, erratic chromosomal rearrangements may occur in AML, sometimes unbalanced and also accompanied by other abnormalities. Knowledge on the contribution of rare abnormalities to AML disease, progression and prognosis is limited.
Here we report a unique case of acute monoblastic leukemia with gain of i(5)(p10), tetrasomy 8, an unbalanced translocation der(19)t(17;19)(q23;p13.3) and mutated NPM1.
Results
Bone marrow cells were examined by conventional karyotyping, fluorescence in situ hybridization (FISH) and mutation analysis at diagnosis and follow-up. At diagnosis we detected trisomy 8, an unbalanced translocation der(19)t(17;19)(q23;p13.3) and mutated NPM1. During the course of the disease we observed clonal evolution with gain of i(5)(p10), tetrasomy 8 and eventually duplication of der(19)t(17;19)(q23;p13.3). By using the der(19)t(17;19) as clonal marker, we found that i(5)(p10) and tetrasomy 8 were secondary genetic events and that tetrasomy 8 had clonally evolved from trisomy 8.
Conclusions
This case of acute monoblastic leukemia presents a combination of rare chromosomal abnormalities including the unbalanced translocation der(19)t(17;19)(q23;p13.3), hitherto un-reported in AML. In addition, our case supports the hypothesis of a step-wise clonal evolution from trisomy 8 to tetrasomy 8 in AML. Reporting and collecting data of rare chromosomal abnormalities will add information to AML disease, progression and prognosis, and may eventually translate to improved patient management.
Similar content being viewed by others


Background
Acute myeloid leukemia (AML) comprises a spectrum of hematologic malignancies with variable outcomes. A hallmark of AML is maturation arrest of myeloid cells, which accumulate in the peripheral blood and bone marrow. The un-differentiated myeloid cells show chromosomal abnormalities in about 55% of cases of adult AML [1]. Some of these abnormalities, e.g. translocations are recurrent and are used for disease classification [1, 2].
Cytogenetics has become mandatory for the diagnosis of AML according to the 2008 revision of the World Health Organization (WHO) classification of tumors of hematopoietic and lymphoid tissues [2]. Cytogenetic analyses complemented with molecular methods and mutation detection are useful for guiding treatment, for monitoring residual disease and for providing prognostic information towards clinical outcomes [3].
Trisomy 8 either as a sole or as an additional abnormality is the most common among numerical abnormalities associated with AML [4]. In contrast, tetrasomy 8 is relatively rare and reported in a few cases in AML, only [5–9]. Tetrasomy 8 seems to be associated with poor prognosis [5, 8]. A clonal relationship between trisomy 8 and tetrasomy 8 has been suggested, however, hardly ever followed up in the cases described [8–10].
Gain of an isochromosome of the short arm of chromosome 5, i.e. i(5)(p10), represents a very rare recurrent abnormality in AML [11, 12]. To date only few cases have been described (Table 1). The presence of i(5)(p10) seems to concomitantly occur with further abnormalities, sometimes in the context of complex karyotypes [13]. In most cases response to chemotherapy was reportedly poor [11–13].
Recurrent translocations in AML are typically balanced and are often associated with a favorable prognosis. Erratic chromosomal rearrangements may occur in AML, sometimes unbalanced and also accompanied by other abnormalities. They are thought to contribute to disease progression and are usually associated with poorer prognosis [3].
We present a unique case of AML with gain of i(5)(p10), tetrasomy 8, unbalanced translocation der(19)t(17;19)(q23;p13.3) and NPM1 mutation. By following the course of the disease we show that gain of i(5)(p10) and tetrasomy 8 represent secondary events in this case and that tetrasomy 8 has clonally evolved from trisomy 8.
Case presentation
In June 2011 a 64 year old male patient was admitted to our hospital with recurrent epistaxis and ecchymosis. The diagnosis of AML, in this case acute monoblastic leukemia, was established. As the patient was not eligible for bone marrow transplantation due to comorbidities, induction therapy (7+3) with daunorubicin and cytarabine followed by three cycles of consolidation treatment with high-dose cytarabine was administered and led to complete hematologic and cytogenetic remission. A first relapse occurred 14 months thereafter, which was treated with induction therapy as before and, again, resulted to complete remission. At 19 months a second relapse occurred with additional infiltration of inguinal lymph nodes and the skin. In January 2013 induction treatment with cisplatin, gemcitabine and dexamethasone was administered, which resulted in hematologic and partial cytogenetic remission at four month thereafter. In July 2013 the patient vastly relapsed and passed away despite of continued consolidation therapy.
Results
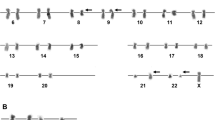
At diagnosis conventional cytogenetics revealed the following karyotype: 47,XY,+8,add(19)(p13)[5]/46,XY[15]. The initial karyotype was defined more precisely with multicolor fluorescence in situ hybridization (24X-FISH) analysis, which yielded to the following karyotype: 47,XY,+8,der(19)t(17;19)(q23;p13.3)[6]/46,XY[19]. At first relapse a marked clonal evolution had occurred with gain of isochromosome 5p and tetrasomy 8. The evolved karyotype was determined as: 49,XY,+i(5)(p10),+8,+8,der(19)t(17;19)(q23;p13.3)[7]/46,XY[14] (Figure 1). At second relapse cytogenetic analysis showed a duplication of the derivative chromosome 19 with loss of the normal chromosome 19 (Figures 1 and 2A). In addition, a second line was detected with trisomy 8, monosomy 13, der(19)t(17;19) and 3 marker chromosomes, however, no i(5)(p10).

Partial karyograms of clonal evolution. GTG-banded partial karyograms of all the involved chromosomes show the step-wise clonal evolution from trisomy 8 to tetrasomy 8, gain of i(5)(p10) and derivative chromosome(s) 19 in the predominant sideline 1 at diagnosis, at first and second relapse (upper panel). The lower panel depicts sideline 2 with trisomy 8, monosomy 13, a derivative chromosome 19 and three marker chromosomes (second relapse). Note, no other aberrant clones have been detected at the respective time points.

GTG-banded karyotype and multicolor-FISH image at second relapse. (A) The predominant aberrant subclone 1 shows gain of i(5)(p10), tetrasomy 8 and two derivative chromosomes 19. (B) Multicolor-FISH analysis confirmed the chromosomal aberrations and unveiled the unbalanced der(19)t(17;19). Arrows point to chromosomal abnormalities.
Triple-color metaphase- and interphase-FISH analysis with a probe mixture for chromosome 8 centromers and for chromosome 5p/5q complemented by 24X-FISH analysis confirmed the respective abnormalities (Figures 2B and 3). In order to narrow the breakpoints of der(19)t(17;19), metaphase-FISH with an E2A specific break-apart probe (D19S883) was used. Fusion signals were observed on both normal and derivative chromosomes 19, which indicated that the breakpoint on chromosome 19 ought to be located distal from the E2A gene locus (Figure 4). The final karyotype was determined as: 49,XY,+i(5)(p10),+8,+8,der(19)t(17;19)(q23;p13.3)x2[16]/49,XY,+8,-13,der(19)t(17;19)(q23;p13.3),+3mar[4]/46,XY[2] (Figure 1).

FISH analysis using locus specific probes for chromosome 5p/5q and a centromer enumeration probe for the identification of chromosome 8. Hybridization signals on an inverted DAPI counterstain metaphase spread show four blue signals indicative of tetrasomy 8, two green signals on the supernumerary isochromosome 5p and two green and two red signals on the normal chromosomes 5. The insert shows a representative interphase-FISH image of sideline 1/subclone 1 with the signal pattern of two red and four green dots (arrow-heads) indicating gain of i(5)(p10) and four blue dots indicating tetrasomy 8 (arrows).

Analysis of the derivative chromosome 19. (A) Partial GTG-banded karyogram showing a normal and a derivative chromosome 19 (left- and right-hand side, respectively). The arrow points to the additive band from 17q23. (B) Metaphase-FISH analysis using the E2A gene specific break-apart probe (green, D19S883; red, RH98588), which shows two normal fusion signals on both chromosomes. The arrow points to the additive chromosomal material on the derivative chromosome 19, which is illuminated by the DAPI counterstain.
The mutation analysis for NPM1 and FLT3 revealed the NPM1-mutation c.959_960insCAGA and no FLT3 internal tandem duplication, respectively. This was also obtained at relapses, and the NPM1 mutation was conserved over the entire course of the disease.
At diagnosis the bone marrow aspirate showed 81% monoblasts which stained positive for HLA-DR, CD33, CD56, CD36, CD64, and negative for MPO, Tdt and CD117, and the CBC was as follows: WBC 26.7 G/L (47% blasts), Hgb 14.1 g/dl, and platelets 56 G/L. The immunophenotyping and the cytological analysis were consistent with acute monoblastic leukemia.
Discussion
We described a case of acute monoblastic leukemia with a unique combination of rare aberrations, i.e. gain of i(5)(p10), tetrasomy 8, der(19)t(17;19)(q23;p13.3) and NPM1 mutation. To the best of our knowledge such a case has not been reported previously.
In myeloid malignancies the occurrence of i(5)(p10) is very rare and to date only 23 cases have been reported in the literature (Table 1). The occurrence of i(5)(p10) was described in 16 cases of AML, in 6 cases of MDS and in one case of CML. Interestingly, gain of i(5)(p10), which results to a supernumerary chromosome 5, has exclusively been reported in only 12 cases of AML, however, not in MDS (Table 1). In 8 of the 12 cases with AML, i(5)(p10) was accompanied by trisomy 8. In 7 of the 12 cases the cells were of monoblastic/monocytic lineage (FAB M5). The gain of i(5)(p10) resulted to tetrasomy 5p and disomy 5q, which was also observed in our case. In contrast, in the cases with MDS (6 cases) and in other cases of AML (4 cases) an i(5)(p10) replaced a normal chromosome 5 and, thus, led to trisomy 5p and monosomy 5q, perhaps similar to a 5q- syndrome [13, 25]. It is not clear, whether these differences translate into differing phenotypes of the diseases, as e.g. the cytological pattern is not completely consistent in the cases described (Table 1). With two exceptions, i(5)(p10) was always observed in the context of at least one other chromosomal abnormality, often in the context of a complex karyotype (Table 1). Thus, i(5)(p10) may represent a secondary cytogenetic event in the majority of cases [11, 13]. Our case supports this notion, since gain of i(5)(p10) was not detected at diagnosis but was observed in the wake of clonal evolution.
In the past, the incidence of i(5)(p10) could have been underestimated by conventional G-banding as i(5)(p10) might have been misinterpreted as 5q-, which is frequent in patients with MDS or AML. By using locus specific probes towards 5p and 5q, FISH analysis allowed clear discrimination between 5q- and i(5)(p10) both on metaphases and on interphase nuclei (Figure 3). This approach is useful not only in this case but also in the routine diagnostic work-up of other cases.
Tetrasomy 8 has been reported in about 120 cases to date [5]. It was observed mostly in AML of monocytic/monoblastic lineage, less frequently in MDS and very rarely in MPD [5]. In the cases described the clone with tetrasomy 8 was often seen in the presence of trisomy 8, which favoured the hypothesis of a stepwise clonal evolution by consecutive mitotic non-disjunctions from disomy 8 to trisomy 8 and to tetrasomy 8 or polysomy 8 [5, 6, 8]. Alternatively, tetrasomy 8 may evolve from simultaneous non-disjunction of both homologous chromosomes during a single cell division [8]. In our case clonal evolution was monitored at three consecutive timepoints by using the derivative chromosome 19 as clonal marker. Thus, we demonstrated that tetrasomy 8 has evolved from trisomy 8. Clonal evolution of tetrasomy 8 in AML was previously reported in two cases, only. The one case by Kameoka et al. was a MDS with trisomy 8 as the sole abnormality, and when progression into AML occurred, tetrasomy 8 was found [9]. A chromosomal marker was not available as clonal control. In the other case by Takahashi et al. AML showed the translocation t(9;11)(p22;q23) and trisomy 8 at diagnosis [10]. During clonal evolution tetrasomy 8 was detected and the t(9;11)(p22;q23) served as clonal control. Together with our case these findings support the hypothesis that tetrasomy 8 usually evolves stepwise by consecutive non-disjunctions of chromosome 8 in AML [8].
Apart from trisomy 8 our case presented with an unbalanced translocation der(19)t(17;19)(q23;p13.3) at diagnosis. Although both breakpoint regions (17q23 and 19p13, respectively) are intermittently involved in various translocations and other aberrations in hematologic malignancies, a literature search (PubMed and Mitelman database) on t(17;19)(q23;p13) in AML did not return a single hit, which suggests that the observed derivative chromosome 19 ought be erratic in AML [31, 32]. There is only one case of ALL described, which shows a balanced translocation t(17;19)(q23;p13) as sole abnormality [33]. Translocations, however, with a breakpoint at 19p13, often with involvement of E2A, have been reported in approximately 400 cases in AML and in the context of various fusion partners such as MLL on 11q23 and others [32]. In an approach to define the breakpoints on the molecular level in our case we used an E2A (TCF3) specific break-apart probe for chromosome-band 19p13 and obtained fusion signals only. This indicated absence of a chromosomal break within this gene locus (Figure 4). Therefore, we assume that the breakpoint is located distal from this locus (D19S883), if not within the sub-telomeric region. This also precludes other genes within this region with potential in leukemogenesis, such as MLLT1 or LYL1, as those are located proximal to E2A. Distal to E2A are the genes FSTL3 (FLRG), PTBP1 and STK11, and there is only one report of involvement of FSTL3 with CCND1 in a case with B-CLL [34].
The band 17q23 has been identified as a “fragile site” in cancer with higher incidence of chromosomal breakage [35]. Nevertheless translocations involving 17q23 are rare in hematologic diseases and have been reported in AML in only 59 cases, of which 7 cases showed trisomy 8, too. Apart from protein coding genes in 17q23, there are four miRNA genes (miR-21, miR-301, miR-142s, miR-142as) located and involvement in neoplastic diseases has been suggested [36, 37]. The translocation der(19)t(17;19)(q23;p13.3) in our case is new and further studies are warranted to unveil the potential involvement of genes in this unbalanced translocation. The underlying molecular event could have provided a proliferative advantage to the cells or assisted in genomic instability, since der(19)t(17;19)(q23;p13.3) was eventually found duplicated in the predominant clone.
AMLs with mutated NPM1 harbour chromosomal abnormalities in only about 15% of cases and the most frequent abnormalities are +8, –4 or –Y [38, 39]. As mutated NPM1 is retained during clonal evolution, it is thought that NPM1 mutations represent a founder mutation, and additional mutations or chromosomal abnormalities are thought to be secondary. In our case the NPM1 mutation did not change during clonal evolution. However, as we observed trisomy 8 and der(19)t(17;19)(q23;p13.3) at diagnosis, it is not clear which of the abnormalities conferred the founder.
By using the International Prognostic Scoring System in AML our case initially classified as “Intermediate-II”, as trisomy 8 is not a favourable prognostic factor, but does not score as an adverse prognostic factor either [1, 3]. However, the prognostic relevance of der(19)t(17;19)(q23;p13.3) is not known and, hence, this scoring was only provisional. At first relapse our case reclassified as “Adverse”, as the criteria of a complex karyotype were met with the addition of a fourth chromosome 8 and gain of i(5)(p10). Tetrasomy 8 has been described as a poor prognostic factor, however, much less information is available for i(5)(p10) [5, 11, 13]. On their own, tetrasomy 8 and i(5)(p10) are frequently part of a complex karyotype and may reflect genomic instability. Genes located on chromosome 8 and 5p may be over-expressed by gene dosage effects and, thus, could contribute to a possible proliferative advantage of the cells. In vitro, a proliferative advantage of tetrasomy 8 over trisomy 8 has been observed in AML [40]. Cases with either tetrasomy 8 or i(5)(p10) reportedly show a more aggressive course of the disease, shorter survival times and poor response to chemotherapy [11, 13, 25]. Those abnormalities are held as factors of poor prognosis and our case falls in line with those case reports.
Conclusion
We described the first case of acute monoblastic leukemia with gain of i(5)(p10), tetrasomy 8, an unbalanced translocation der(19)t(17;19)(q23;p13.3) and NPM1 mutation. During the follow-up of the disease we observed that gain of i(5)(p10) and tetrasomy 8 represented secondary genetic events in this case. By using the der(19)t(17;19) as clonal marker we formally demonstrated that tetrasomy 8 has evolved from trisomy 8. This case is only the third, illustrating a step-wise clonal evolution from trisomy 8 to tetrasomy 8 in AML.
Reporting and collecting data on rare chromosomal abnormalities of AML will add information to pathogenesis, disease progression and prognosis, and may eventually translate to improved patient management.
Materials and methods
Conventional karyotyping
Bone marrow aspirates were cultured for 24 h and 48 h in bone marrow culture medium (MarrowMax™, GIBCO, Paisley, UK). Metaphase chromosomes were prepared and GTG-banded by standard techniques. A minimum of 20 metaphases were karyotyped according to the International System for Human Cytogenetic Nomenclature (ISCN) 2013 [41].
Fluorescence in situ hybridization (FISH)
Polysomy 8 and i(5)(p10) were simultaneously analyzed by using a probe mixture of the Vysis CEP 8 probe (D8Z2, SpectrumAqua) and the Vysis LSI 5p/5q Dual Color probe (EGR1/D5S23,D5S721, SpectrumOrange/SpectrumGreen, Abbott Molecular, Illinois, USA). The E2A (TCF3) gene locus on chromosome 19 was analyzed with the XL E2A break-apart probe (D19S883/RH98588, Metasystems, Altlußheim, Germany). Metaphase chromosomes were also analyzed with the 24XCyte multicolor FISH probe set (Metasystems). All hybridizations were performed on a ThermoBrite instrument (Abbott Molecular) with denaturation at 78°C for 2 minutes followed by over-night incubation at 37°C and subsequent standard washing procedures, which were completed by counterstaining with DAPI (4,6 diamidino-2-phenylindole). For interphase FISH cells from 24 h cultures were used, and more than 200 nuclei were analyzed. All of the FISH analyses were performed with a Zeiss Axio-Imager 1 M fluorescence microscope in combination with the Metafer 4 software (Metasystems).
Mutation analysis
DNA and RNA were prepared from bone marrow aspirates by standard procedures and subjected to NPM1 and FLT3 gene mutation analysis as described [42, 43].
Morphology and Immunophenotyping
Peripheral blood cells were examined by an automated hematologic analyzer (Sysmex, XE-5000, Vienna, Austria). Bone marrow smears were stained with Wright-Giemsa and analyzed according to routine clinical laboratory procedures. Immunophenotyping of bone marrow cells was performed on an 8-color Navios™ Flow Cytometer (Beckman Coulter, Germany) according to the manufacturer’s instructions.
Consent
The patient gave written informed consent for the publication of this case including accompanying images prior to his passing away. A copy of this statement is available for review by the Editor-in-Chief of this journal.
References
Döhner H, Estey EH, Amadori S, Appelbaum FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson RA, Lo-Coco F, Naoe T, Niederwieser D, Ossenkoppele GJ, Sanz MA, Sierra J, Tallman MS, Löwenberg B, Bloomfield CD, European LeukemiaNet: Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood 2010, 115: 453–474. 10.1182/blood-2009-07-235358
Swerdlow SH, Campo E, Harris NL, Jaffe ES, Pileri SA, Stein H, Thiele J, Vardiman JW: In WHO classification of tumours of haematopoietic and lymphod tissues. Lyon: IARC; 2008.
Mrózek K, Marcucci G, Nicolet D, Maharry KS, Becker H, Whitman SP, Metzeler KH, Schwind S, Wu YZ, Kohlschmidt J, Pettenati MJ, Heerema NA, Block AW, Patil SR, Baer MR, Kolitz JE, Moore JO, Carroll AJ, Stone RM, Larson RA, Bloomfield CD: Prognostic significance of the European LeukemiaNet standardized system for reporting cytogenetic and molecular alterations in adults with acute myeloid leukemia. J Clin Oncol 2012, 30(36):4515–4523. 10.1200/JCO.2012.43.4738
Paulsson K, Johansson B: Trisomy 8 as the sole chromosomal aberration in acute myeloid leukemia and myelodysplastic syndromes. Pathol Biol 2007, 55(1):37–48. 10.1016/j.patbio.2006.04.007
Beyer V, Mühlematter D, Parlier V, Cabrol C, Bougeon-Mamin S, Solenthaler M, Tobler A, Pugin P, Gregor M, Hitz F, Hess U, Chapuis B, Laurencet F, Schanz U, Schmidt PM, van Melle G, Jotterand M: Polysomy 8 defines a clinico-cytogenetic entity representing a subset of myeloid hematologic malignancies associated with a poor prognosis: report on a cohort of 12 patients and review of 105 published cases. Cancer Genet Cytogenet 2005, 160(2):97–119. 10.1016/j.cancergencyto.2004.12.003
Marosi C, Muhm M, Argyriou-Tirita A, Pehamberger H, Pirc-Danoewinata H, Geissler K, Locker G, Grois N, Haas OA: Tetrasomy 8 in acute monoblastic leukemia (AML-M5a) with myelosarcomatosis of the skin. Cancer Genet Cytogenet 1993, 71(1):50–54. 10.1016/0165-4608(93)90201-V
Kim J, Park TS, Song J, Lee KA, Lee SG, Cheong JW, Choi JR: Tetrasomy 8 in a patient with acute monoblastic leukemia. Korean J Lab Med 2008, 28(4):262–266. 10.3343/kjlm.2008.28.4.262
Tsirigotis P, Papageorgiou S, Abatzis D, Athanatou S, Girkas C, Pappa V, Pangalos C, Papageorgiou E, Dervenoulas J, Raptis S: Acute myelogenous leukemia with tetrasomy 8 is a disease with a poor prognosis. Cancer Genet Cytogenet 2005, 161(1):78–81. 10.1016/j.cancergencyto.2004.12.016
Kameoka J, Funato T, Obara Y, Kadowaki I, Yokoyama H, Kimura T, Tomiya Y, Yamada M, Ishikawa I, Takagawa M, Sasaki O, Kimura J, Harigae H, Miura I, Meguro K, Kaku M, Sasaki T: Clonal evolution from trisomy into tetrasomy of chromosome 8 associated with the development of acute myeloid leukemia from myelodysplastic syndrome. Cancer Genet Cytogenet 2001, 124(2):159–164. 10.1016/S0165-4608(00)00347-2
Takahashi T, Tsukuda H, Kimura H, Yoshimoto M, Tsujisaki M: Extramedullary relapse of AML with t(9;11)(p22;q23) associated with clonal evolution from trisomy 8 into tetrasomy 8. Intern Med 2009, 49(5):447–451.
Schoch C, Bursch S, Kern W, Schnittger S, Hiddemann W, Haferlach T: Gain of an isochromosome 5p: a new recurrent chromosome abnormality in acute monoblastic leukemia. Cancer Genet Cytogenet 2001, 127: 85–88. 10.1016/S0165-4608(00)00424-6
Panani AD: Gain of an isochromosome 5p: a rare recurrent abnormality in acute myeloid leukemia. Vivo 2006, 20: 359–360.
Herry A, Douet-Guilbert N, Morel F, Le Bris MJ, Guéganic N, Berthou C, De Braekeleer M: Isochromosome 5p and related anomalies: a novel recurrent chromosome abnormality in myeloid disorders. Cancer Genet Cytogenet 2010, 200(2):134–139. 10.1016/j.cancergencyto.2010.04.006
Choi WT, Folsom MR, Azim MF, Meyer C, Kowarz E, Marschalek R, Timchenko NA, Naeem RC, Lee DA: C/EBPbeta suppression by interruption of CUGBP1 resulting from a complex rearrangement of MLL. Cancer Genet Cytogenet 2007, 177(2):108–114. 10.1016/j.cancergencyto.2007.07.002
Calabrese G, Fantasia D, Spadano A, Morizio E, Di Bartolomeo P, Palka G: Karyotype refinement in five patients with acute myeloid leukemia using spectral karyotyping. Haematologica 2000, 85(11):1219–1221.
Herry A, Douet-Guilbert N, Morel F, Le Bris MJ, De Braekeleer M: Redefining monosomy 5 by molecular cytogenetics in 23 patients with MDS/AML. Eur J Haematol 2007, 78(6):457–467. 10.1111/j.1600-0609.2007.00847.x
Tamura S, Kanamaru A: De-novo acute myeloid leukemia with trilineage myelodysplasia (AML/TMDS) and myelodysplastic remission marrow (AML/MRM). Leuk Lymphoma 1995, 16: 263–270. 10.3109/10428199509049765
Goldschmidt N, Yehuda-Gafni O, Abeliovich D, Slyusarevsky E, Rund D: Interstitial insertion of RARα gene into PML gene in a patient with acute promyelocytic leukemia (APL) lacking the classic t(15;17). Hematology 2010, 15(5):332–337. 10.1179/102453310X12647083621083
El-Rifai W, Elonen E, Larramendy M, Ruutu T, Knuutila S: Chromosomal breakpoints and changes in DNA copy number in refractory acute myeloid leukemia. Leukemia 1997, 11: 958–963. 10.1038/sj.leu.2400706
Schmidt HH, Strehl S, Thaler D, Strunk D, Sill H, Linkesch W, Jäger U, Sperr W, Greinix HT, König M, Emberger W, Haas OA: RT-PCR and FISH analysis of acute myeloid leukemia with t(8;16)(p11;p13.3) and chimeric MOZ and CBP transcripts: breakpoint cluster region and clinical implications. Leukemia 2004, 18: 1115–1121. 10.1038/sj.leu.2403353
Yunis JJ: Recurrent chromosomal defects are found in most patients with acute nonlymphocytic leukemia. Cancer Genet Cytogenet 1984, 11(2):125–137. 10.1016/0165-4608(84)90106-7
Gervais C, Murati A, Helias C, Struski S, Eischen A, Lippert E, Tigaud I, Penther D, Bastard C, Mugneret F, Poppe B, Speleman F, Talmant P, VanDen AJ, Baranger L, Barin C, Luquet I, Nadal N, Nguyen-Khac F, Maarek O, Herens C, Sainty D, Flandrin G, Birnbaum D, Mozziconacci MJ, Lessard M, Groupe Francophone de Cytogénétique Hématologique: Acute myeloid leukaemia with 8p11 (MYST3) rearrangement: an integrated cytologic, cytogenetic and molecular study by the groupe francophone de cytogénétique hématologique. Leukemia 2008, 22(8):1567–1575. 10.1038/leu.2008.128
Slovak ML, Nemana L, Traweek ST, Stroh JA: Acute monoblastic leukemia (FAB-M5b) with t(8;14)(p11;q11.1). Cancer Genet Cytogenet 1991, 56(2):237–242. 10.1016/0165-4608(91)90176-U
Flach J, Dicker F, Schnittger S, Schindela S, Kohlmann A, Haferlach T, Kern W, Haferlach C: An accumulation of cytogenetic and molecular genetic events characterizes the progression from MDS to secondary AML: an analysis of 38 paired samples analyzed by cytogenetics, molecular mutation analysis and SNP microarray profiling. Leukemia 2011, 25(4):713–718. 10.1038/leu.2010.304
Douet-Guilbert N, Basinko A, De Braekeleer E, Guéganic N, Bovo C, Le Bris MJ, Morel F, Eveillard JR, Berthou C, Herry A, De Braekeleer M: Isolated 5p isochromosome in myelodysplastic syndrome: report of the first case. Leuk Res 2011, 35(11):193–197. 10.1016/j.leukres.2011.07.008
Jimenez-Sousa MA, Ferro MT, Talavera M, Villalon C, Cabello P, Laraña J, Herrera P, Garcia Sagredo JM: Myelodysplastic syndrome with isochromosome 5p and trisomy 8 after treatment of a multiple myeloma. Cancer Genet Cytogenet 2010, 203: 345–347. 10.1016/j.cancergencyto.2010.09.010
Lessard M, Hélias C, Struski S, Perrusson N, Uettwiller F, Mozziconacci MJ, Lafage-Pochitaloff M, Dastugue N, Terré C, Brizard F, Cornillet-Lefebvre P, Mugneret F, Barin C, Herry A, Luquet I, Desangles F, Michaux L, Verellen-Dumoulin C, Perrot C, Van den Akker J, Lespinasse J, Eclache V, Berger R, Groupe Francophone de Cytogénétique Hématologique: Fluorescence in situ hybridization analysis of 110 hematopoietic disorders with chromosome 5 abnormalities: do de novo and therapy-related myelodysplastic syndrome-acute myeloid leukemia actually differ? Cancer Genet Cytogenet 2007, 176(1):1–21. 10.1016/j.cancergencyto.2007.01.013
Christodoulou J, Schoch C, Schnittger S, Haferlach T: Myelodysplastic syndrome (RARS) with +i(12p) abnormality in a patient 10 months after diagnosis and successful treatment of a mediastinal germ cell tumor (MGCT). Ann Hematol 2004, 83: 386–389. 10.1007/s00277-003-0787-x
Reddi DM, Lu CM, Fedoriw G, Liu YC, Wang FF, Ely S, Boswell EL, Louissaint A Jr, Arcasoy MO, Goodman BK, Wang E: Myeloid neoplasms secondary to plasma cell myeloma: an intrinsic predisposition or therapy-related phenomenon? A clinicopathologic study of 41 cases and correlation of cytogenetic features with treatment regimens. Am J Clin Pathol 2012, 138(6):855–866. 10.1309/AJCPOP7APGDT9JIU
Markovic VD, Bouman D, Bayani J, Al-Maghrabi J, Kamel-Reid S, Squire JA: Lack of BCR/ABL reciprocal fusion in variant Philadelphia chromosome translocations: a use of double fusion signal FISH and spectral karyotyping. Leukemia 2000, 14(6):1157–1160. 10.1038/sj.leu.2401718
Pubmed Database. Available at: [accessed 26.06.2013] http://www.ncbi.nlm.nih.gov/pubmed/
Mitelman F, Johansson B, Mertens F (Eds): In Mitelman Database of Chromosome Aberrations in Cancer. 2013. Available at: Chromosomes/Mitelman [accessed 26.06.2013] http://cgap.nci.nih.gov/
Lawler SD, Khokhar MT, Davies H, Williams GJ, Powles R: Cytogenetic studies of leukemic recurrence in recipients of bone marrow allografts. Cancer Genet Cytogenet 1990, 47(2):249–263. 10.1016/0165-4608(90)90034-8
Hayette S, Gadoux M, Martel S, Bertrand S, Tigaud I, Magaud JP, Rimokh R: FLRG (follistatin-related gene), a new target of chromosomal rearrangement in malignant blood disorders. Oncogene 1998, 16(22):2949–2954. 10.1038/sj.onc.1201807
Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, Croce CM: Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci 2004, 101(9):2999–3004. 10.1073/pnas.0307323101
Garzon R, Volinia S, Liu CG, Fernandez-Cymering C, Palumbo T, Pichiorri F, Fabbri M, Coombes K, Alder H, Nakamura T, Flomenberg N, Marcucci G, Calin GA, Kornblau SM, Kantarjian H, Bloomfield CD, Andreeff M, Croce CM: MicroRNA signatures associated with cytogenetics and prognosis in acute myeloid leukemia. Blood 2008, 111(6):3183–3189. 10.1182/blood-2007-07-098749
Si ML, Zhu S, Wu H, Lu Z, Wu F, Mo YY: miR-21-mediated tumour growth. Oncogene 2007, 25: 1–5.
Haferlach C, Mecucci C, Schnittger S, Kohlmann A, Mancini M, Cuneo A, Testoni N, Rege-Cambrin G, Santucci A, Vignetti M, Fazi P, Martelli MP, Haferlach T, Falini B: AML with mutated NPM1 carrying a normal or aberrant karyotype show overlapping biologic, pathologic, immunophenotypic, and prognostic features. Blood 2009, 114(14):3024–3032. 10.1182/blood-2009-01-197871
Thiede C, Koch S, Creutzig E, Steudel C, Illmer T, Schaich M, Ehninger G: Prevalence and prognostic impact of NPM1 mutations in 1485 adult patients with acute myeloid leukemia (AML). Blood 2006, 107(10):4011–4020. 10.1182/blood-2005-08-3167
Yan J, Marceau D, Drouin R: Tetrasomy 8 is associated with a major cellular proliferative advantage and a poor prognosis. Two cases of myeloid hematologic disorders and review of the literature. Cancer Genet Cytogenet 2001, 125(1):14–20. 10.1016/S0165-4608(00)00352-6
Schaffer LG, McGowan-Jordan J, Schmid M (Eds): ISCN 2013: an international system for human cytogenetic nomenclature. Basel: S. Karger; 2013.
Döhner K, Schlenk RF, Habdank M, Scholl C, Rücker FG, Corbacioglu A, Bullinger L, Fröhling S, Döhner H: Mutant nucleophosmin (NPM1) predicts favorable prognosis in younger adults with acute myeloid leukemia and normal cytogenetics: interaction with other gene mutations. Blood 2005, 106(12):3740–3746. 10.1182/blood-2005-05-2164
Murphy KM, Levis M, Hafez MJ, Geiger T, Cooper LC, Smith BD, Small D, Berg KD: Detection of FLT3 internal tandem duplication and D835 mutations by a multiplex polymerase chain reaction and capillary electrophoresis assay. J Mol Diagn 2003, 5(2):96–102. 10.1016/S1525-1578(10)60458-8
Acknowledgements
We thank Renate Marschon, Krankenhaus der Barmherzigen Schwestern Linz, for the analysis of NPM1 and FLT3 gene mutations and Lana Harder, Institut für Tumorgenetik Nord, Kiel, Germany, for helpful discussions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declared that they have no competing interests.
Authors’ contributions
CP and GH performed the cytogenetic studies, DV and MF collected and compiled clinical data, JB and CP designed the study, HS participated in the design of the study and its coordination, JB and CP drafted the manuscript and all authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Paar, C., Herber, G., Voskova, D. et al. A case of acute myeloid leukemia (AML) with an unreported combination of chromosomal abnormalities: gain of isochromosome 5p, tetrasomy 8 and unbalanced translocation der(19)t(17;19)(q23;p13). Mol Cytogenet 6, 40 (2013). https://doi.org/10.1186/1755-8166-6-40
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8166-6-40