
Abstract
Background
Jumping translocations are a rare type of mosaicism in which the same portion of one donor chromosome is translocated to several recipient chromosomes. Constitutional forms of jumping translocations are rare, and the 48 cases reported to date have been associated with both normal and abnormal phenotypes. Concurrence of isochromosome (i) of one arm and translocation of the other is also rare, with seven reported cases. We describe a unique case involving concurrence of i(Yp) and a jumping translocation of Yq to the telomere of chromosomes 12q and 17q, which resulted in five cell lines.
Case presentation
The patient, an otherwise healthy 35-year-old man, was referred for cytogenetic studies because of absolute azoospermia. He had elevated levels of follicle stimulating hormone and luteinizing hormone, consistent with abnormal spermatogenesis, and decreased levels of free testosterone and inhibin B. G-banded chromosome analysis revealed a mosaic male karyotype involving five abnormal cell lines. One of the cell lines showed loss of chromosome Y and presence of i(Yp) as the sole abnormality. Three cell lines exhibited jumping translocation: two involved 17qter, and the other involved 12qter as the recipient and Yq as the common donor chromosome. One of the cell lines with der(17) additionally showed i(Yp). The other der(17) and der(12) cell lines had a missing Y chromosome. All five cell lines were confirmed by FISH. Subtelomric FISH study demonstrated no loss of chromosome material from the recipient chromosomes at the translocation junctions.
Conclusions
We postulate that a postzygotic pericentromeric break of the Y chromosome led to formation of isochromosome Yp, whereas Yq formed a jumping translocation through recombination between its internal telomere repeats and telomeric repeats of recipient chromosomes. This in turn led to either pairing or an exchange at the complimentary sequences. Such translocation junctions appear to be unstable and to result in a jumping translocation. Cryptic deletion or disruption of AZF (azoospermic factor) genes at Yq11 during translocation or defective pairing of X and Y chromosomes during meiosis, with abnormal sex vesicle formation and consequent spermatogenetic arrest, might be the main cause of the azoospermia in our patient.
Similar content being viewed by others

Background
Jumping translocations, a term first introduced by Lejeun and colleagues [1], are a rare type of mosaicism involving the same portion of one donor chromosome translocated to several different recipient chromosomes. They usually occur somatically, in both constitutional and acquired chromosomal abnormalities, and occur in various pathologic conditions. Constitutional forms of jumping translocations are very rare and have varying clinical impact; the 48 cases reported to date have been associated with both normal and abnormal phenotypes [1–11]. Acquired jumping translocations, on the other hand, have been more commonly observed in hematologic malignancies [12].
A jumping translocation probably arises through multiple events of somatic recombination in early development [7, 13]. Regions of repetitive DNA sequences such as centromeres, pericentromeres and telomeres, have been implicated to play a critical role in the events leading to the formation of jumping translocations. Telomere-like repeats, also called interstitial or internal telomeres [13, 14], are generally present in pericentric regions and represent nonfunctional chromosomal elements [15]. These internal telomere repeats have been proposed to be hotspots for recombination [13, 16]. Such hotspots have also been confirmed by evidence pointing toward preferential involvement of the heterochromatin region of donor chromosomes [17] and telomere ends of recipient chromosomes [14]. Additionally, heterochromatic regions may undergo decondensation during early development, leading to increased recombination at the pericentrometric regions of some of the chromosome arms [17]. However, these translocation junctions appear to be unstable and to result in jumping translocations [5]. The fact that not all such rearrangements result in jumping translocations [15, 18, 19] suggests that sequences from both chromosomes at the junction influence the stability of the fusion.
Seven instances of concurrent isochromosome of the short arm, i(Yp), and translocation of the long arm of a single chromosome onto a telomere of a nonhomologous chromosome have been described [20–26]. Concolino and colleagues suggested that alpha-satellite fission of the chromosome can result in an isochromosome of the short arm and deletion of the long arm [27]. Moreover, Petit noted that chromosomal fission within the alpha satellite is likely to form a stable deletion, of one chromosomal arm, whereas fragments with breaks at the junction of alpha and beta sequences may translocate to the telomere of another chromosome [9]. However, one function of telomeres is to prevent chromosomes from fusing with other chromosomal fragments [28]. In several organisms in which telomere shortening is seen, telomeres lose the ability to protect against chromosome fusion well before all telomere sequence is lost. Perhaps a minimum length is needed to form a t-loop; once they are sufficiently short, they can no longer protect the end [29]. Overall, this somatic recombination provides a protective mechanism through which the fragment can be maintained within a cell [30].
Azoospermia, when no sperm can be detected on two separate semen samples, is found in up to 20% of infertile men [30]. Genital tract obstruction and defective spermatogenesis are the principal causes of azoospermia [31]. Chromosomal abnormalities are present in 7% of infertile men and in 10% to 15% of azoospermic men [32, 33]. Among the karyotypic abnormalities found in azoospermic men, sex chromosome abnormalities predominate [34, 35]. Translocations involving the Y chromosome have been reported in association with male infertility and azoospermia [36]. In the most common form, the heterochromatic region of Yq is translocated. A chromosomal breakpoint at band Yq11 is rarely described. Moreover, isochromosome Yp, which results in duplication of the short arm and loss of the long arm, has been reported in association with azoospermia. Two mechanisms have been proposed for male infertility. First, the AZF (azoospermic factor) gene, which is located at Yq11 and is critical for spermatogenesis, may be deleted secondary to a microdeletion, rearrangement, or complete loss as a result of the translocation mechanism [36]. The second explanation is that a defective X–Y pairing during meiosis, with abnormal sex vesicle formation, could lead to spermatogenetic arrest [37–40].
We describe the first reported case involving a de novo isochromosome Yp and jumping translocation of Yq, which resulted in five cell lines and was associated with azoospermia in an infertile man.
Case presentation
Clinical report
The patient was an apparently healthy 35-year-old man with infertility and absolute azoospermia. His physical examination was normal except for slight testicular hypotrophy. No sperm were found in any of the three routine semen analyses, and the elevated serum concentrations of FSH (17 IU/L; normal range 3–7 IU/L) and LH (12 IU/L; normal range 3–8 IU/L) suggested abnormal spermatogenesis. The free testosterone (286 ng/dL; normal range 450–950 ng/dL) and inhibin B (26 pg/mL, normal range 80–270 pg/mL) levels were low. The patient was referred to our laboratory for chromosome analysis.
Material and methods
Cytogenetic and Fluorescence In Situ Hybridization (FISH) studies
The chromosomes of the patient and his parents were studied in peripheral blood mitoses, after culturing and G banding by standard techniques.
The results were confirmed by performing fluorescence in situ hybridization (FISH) with probes for the Yqh region (DYZ1), the sex determining region Y (SRY) gene, and the chromosome X centromere (DXZ1) probe (Vysis/Abbott, Inc., Downers Grove, IL). The translocation junctions of recipient chromosomes were illustrated by chromosome specific subtelomeric probes for chromosomes 12 and 17 (TelVysion; Vysis, Downers Grove, IL, USA). All FISH studies were performed according to the manufacturer’s protocol. Fluorescence images were captured with a Nikon epifluorescence microscope and analyzed by ISIS software (MetaSystems, Altlussheim, Germany).
Results
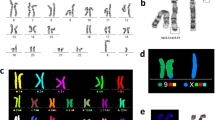
The analysis of 50 metaphases and subsequent FISH studies revealed the presence of five cell lines in the proband, all with an abnormal chromosome Y. One cell line showed loss of chromosome Y (Figure 1; 6% of metaphases) and the other showed i(Yp) with loss of Yq (Figure 2; 6% of metaphases) as the sole abnormality. Two other cell lines represented derivative chromosome 12 (Figure 3; 8% of metaphases) or 17 (Figure 4; 28% of metaphases) due to fusion of Yq to the telomere of their long arms. The fifth cell line showed der(17) and i(Yp) as the second abnormality (Figure 5; 52% of metaphases). Additional count of 30 cells to the routinely 20 cells were in order to reduce the possibility of a 47,XXY cell line in peripheral blood. Both parents had normal G-banded karyotypes.

A metaphase showing loss of chromosome Y. The left side shows the G-banded karyotype and the right side is the inverted DAPI image of metaphase using Yqh (green), SRY gene (red), and X centromere (green) probes.

A metaphase showing duplication of Yp resulting in isochromosome Yp and loss of Yq. The left side shows the G-banded karyotype and the side image is the inverted DAPI image of metaphase using Yqh (green), SRY gene (red), and X centromere (green) probes.

A metaphase showing translocation of Yq to 12qter resulting in derivative chromosome 12 and loss of Y chromosome. The left side shows the G-banded karyotype and the right side is the inverted DAPI image of metaphase using Yqh (green), SRY gene (red), and X centromere (green) probes.

A metaphase showing translocation of Yq to 17qter resulting in derivative chromosome 17 and loss of Y chromosome. The left side shows the G-banded karyotype and the right side is the inverted DAPI image of metaphase using Yqh (green), SRY gene (red), and X centromere (green) probes.

A metaphase showing translocation of Yq to 17qter resulting in derivative chromosome 17 and duplication of Yp resulting in isochromosome Yp. The left side shows the G-banded karyotype and the right side is the inverted DAPI image of metaphase using Yqh (green), SRY gene (red), and X centromere (green) probes.
The hybridization of two SRY probes at i(Yp) in a mirror image configuration confirmed that this chromosome consisted of two short arms (Figures 2 and 5). The translocation of the long arm of Y into two different telomeric regions (17q and 12q) was confirmed using the Yqh probe (Figures 3, 4, and 5). Detection of the 12q and 17q subtelomeric probe signals confirmed that there was no loss of chromosomal materials at the translocation junctions (Figure 6).

Inverted Dapi image of metaphases using subtelomeric probes for the long arm (green) and short arm (red) of chromosome 12 (right image) and chromosome 17 (left image). Subtelomeric probe signals (green) at the fusion junctions of both recipient chromosomes shows that there is no loss of chromosomal materials at these regions.
Based on the results obtained from the chromosome and FISH analysis, the karyotype of our patient was designated as:
Mos46,X,idic(Y)(q11.1),der(17)t(Y;17)(q11.1;q25.3)[26]/45,X,der(17)t(Y;17)(q11.1;q25.3)[14]/45,X,der(12)t(Y;12)(q11.1;q24.33)[4]/46,X,i(Y)(q11.1)[3]/45,X[3].
Conclusions
Constitutional jumping translocations are very rare, and only 7 cases to date have involved concurrent isochromosomes of one arm and translocations of the other [20–26]. The present case is unique, in that it exhibited concurrence of i(Yp) and jumping translocation of Yq as a result of fusion to the telomeres of chromosomes 12q and 17q.
Characteristics common to all eight reported cases (including the present case) of jumping chromosome with concurrent isochromosome and translocation of a single chromosome include a de novo origin, an initial centromeric or pericentromeric break, a monocentric appearance of both the isochromosome and the derivative, and absence of demonstrated chromatin loss. FISH study using subtelomeric probes demonstrated that both recipient chromosomes were intact, with no loss of chromosome material. Moreover, both parents had normal G-banded karyotypes, indicating that the chromosomal abnormalities in their son were de novo. Our case differed from the prior seven cases in having jumping translocation rather than a stable translocation.
These aberrations must have been initiated by a pericentromeric break of Y chromosome at Yq11.1, yielding two chromosomal fragments. The short arm carrying the centromere evolved into an isochromosome, and the acentric long arm fused to the telomeres of the recipient chromosomes. This is possibly the result of a natural mechanism that maintains both fragments throughout cell divisions. We postulate that the pericentromeric region of chromosome Y had a complementary sequence to the telomeric repeat of chromosomes 12 and 17, which led to either pairing or an exchange at the complimentary sequences. In either case, Reddy and colleagues [5] proposed that a single-stranded DNA or a double/triple-stranded DNA at the junction might cause the instability observed in jumping translocations.
Five cell lines evolved from the time of pericentromeric break until regaining complete stability by forming an isochromosome and recombining with the telomeres of two other chromosomes over the course of multiple cell divisions. One of the cell lines showed complete loss of chromosome Y; two cell lines maintained the translocation of Yq to 12q or 17q in the presence or absence of i(Yp); and one cell line showed i(Yp) and deletion of Yq. The events that led to stability probably occur only during early development [3], which resulted in a limited number of cell lines. The zygotic origin of isochromosomes and chromatin instability during early zygote division [30] lends support to this assumption. So, it can also be postulated that this is a de novo process and a post-fertilization event.
The azoospermia and resulting infertility in our patient might be as a result of 1) cryptic deletion or disruption of AZF genes at the Yq11.1 breakpoint (translocation’s junction); 2) defective pairing of X and Y chromosomes during meiosis, which could have led to abnormal sex vesicle development and consequent spermatogenetic arrest.
Ethical approval and consent
These studies were performed on anonymized samples received in the clinical laboratory and thus were exempted from the requirement for consent by an opinion for the Western institutional review board.
References
Lejeun J, Maunoury C, Prieur M, Van Den Akker J: Translocation sauteuse (5p;15q), (8q;15q). Ann Genet 1979, 22: 210–213.
Reddy KS: The conundrum of a jumping translocation (JT) in CVS from twins and review of jumping translocations. Am J Med Genet 2010, 11: 2924–2936.
Gross SJ, Tharapel AT, Phillips OP, Shulman LP, Pivnick EK, Park VM: A jumping Robertsonian translocation: A molecular and cytogenetic study. Hum Genet 1996, 98: 291–296. 10.1007/s004390050209
Von Ballestrem CL, Boavida MG, Zuther C, Carreiro MH, David D, Gal A, Schwinger E: Jumping translocation in a phenotypically normal female. Clin Genet 1996, 49: 156–159.
Reddy KS, Murphy T: Fusion of 9 beta-satellite and telomere (TTAGGG)n sequences results in a jumping translocation. Hum Genet 2000, 107: 268–275. 10.1007/s004390000360
Devriendt K, Petit P, Matthijs G, Vermeesch JR, Holvoet M, Muelenaere AD, Marynen P, Cassiman JJ, Fryns JP: Trisomy 15 rescue with jumping translocation of distal 15q in Prader–Willi syndrome. J Med Genet 1997, 34: 395–399. 10.1136/jmg.34.5.395
Vermeesch JR, Petit P, Speleman F, Devriendt K, Fryns JP, Marynen P: Interstitial telomeric sequences at the junction site of a jumping translocation. Hum Genet 1997, 99: 735–737. 10.1007/s004390050440
Jewett T, Marnane D, Stewart W, Hayworth-Hodge R, Finklea L, Klinepeter K, Rao PN, Pettenati MJ: Jumping translocation with partial duplications and triplications of chromosomes 7 and 15. Clin Genet 1998, 53: 415–420.
Petit P, Devriendt K, Vermeesch JR, de Cock P, Fryns JP: Unusual de novo t(13;15)(q12.1;p13) translocation leading to complex mosaicism including jumping translocation. Ann Genet 1998, 41: 22–26.
Farrell SA, Winsor EJ, Markovic VD: Moving satellites and unstable chromosome translocations: Clinical and cytogenetic implications. Am J Med Genet 1993, 46: 715–720. 10.1002/ajmg.1320460624
Duvall E, Van Den Ended A, Vanhaesebrouck P, Speleman F: Jumping translocation in a newborn boy with dup(4q) and severe hydrops fetalis. Am J Med Genet 1994, 52: 214–217. 10.1002/ajmg.1320520217
Berger R, Bernard OA: Jumping translocations. Genes Chromosomes Cancer 2007, 46: 717–723. 10.1002/gcc.20456
Meyne J, Baker RJ, Hobart HH, Hsu TC, Ryder OA, Ward OG, Wiley JE, Wurster-Hill DH, Yates TL, Moyzis RK: Distribution of non-telomeric sites of the (TTAGGG)n telomeric sequence in vertebrate chromosomes. Chromosoma 1990, 99: 3–10. 10.1007/BF01737283
Gray BA, Bent-Williams A, Wadsworth J, Maiese RL, Bhatia A, Zori RT: Fluorescence in situ hybridization assessment of the telomeric regions of jumping translocations in a case of aggressive B-cell non- Hodgkin lymphoma. Cancer Genet Cytogenet 1997, 98: 20–27. 10.1016/S0165-4608(96)00409-8
Park VM, Gustashaw KM, Wathen TM: The presence of interstitial telomeric sequences in constitutional chromosome abnormalities. Am J Hum Genet 1992, 50: 914–923.
Hastie ND, Allshire RC: Human telomeres: fusion and interstitial sites. Trends Genet 1989, 5: 326–331.
Sawyer JR, Tricot G, Mattox S, Jagannath S, Barlogie B: Jumping translocations of chromosome 1q in multiple myeloma: Evidence for a mechanism involving decondensation of pericentromeric heterochromatin. Blood 1998, 91: 1732–1741.
Rossi E, Floridia G, Casali M, Danesino C, Chiumello G, Bernardi F, Magnani I, Papi L, Mura M, Zuffardi O: Types, stability, and phenotypic consequences of chromosome rearrangements leading to interstitial telomeric sequences. J Med Genet 1993, 30: 926–931. 10.1136/jmg.30.11.926
Rivera H, Vásquez AI, Perea FJ: Centromere-telomere (12;8p) fusion, telomeric 12q translocation, and i(12p) trisomy. Clin Genet 1999, 55: 122–126. 10.1034/j.1399-0004.1999.550209.x
Smith G, McCaa A, Kelly TE: Trisomy 9p with an isochromosome of 9p. Hum Genet 1978, 42: 93–97. 10.1007/BF00291631
Herva R, Koivisto M: Trisomy 9p with i(9p) and t(pq18p). Hum Genet 1979, 50: 237–240. 10.1007/BF00399387
Sandig KR, Mucke J, Veit H: Trisomy 9p resulting from de novo 9/15 translocation and 9p isochromosome. Hum Genet 1979, 52: 175–178.
Leschot NJ, Lim KS: “Complete” trisomy 5p; de novo translocation t(2;5)(q36;p11) with isochomosome 5p. Case report and review of the literature. Hum Genet 1979, 46: 271–278. 10.1007/BF00273310
Orye E, Benoit Y, Van Mele B: Complete trisomy 5p owing to de novo translocation t(5;22)(q11;p11) with isochromosome 5p associated with a familial pericentric of chromosome 2, inv 2(p21q11). J Med Genet 1983, 20: 394–396. 10.1136/jmg.20.5.394
Andre MJ, Aurias A, de Berranger P, Gillot F, Lefrance G, Lejeun J: Trisomie 4p de novo par isochromosome 4p. Ann Genet 1976, 19: 127–131.
Rivera H, Garcia-Esquivel L, Jimenez M, Vaca G, Ibarra B, Cantu JM: Centric fission, centromere-telomere fusion and ischromosome formation: a possible origin of a de novo 12p trisomy. Clin Genet 1987, 31: 393–398.
Concolino D, Cinti R, Morrica M, Andria G, Striscioglio P: Centric fission of chromosome 9in a boy with trisomy 9p. Am J Med Genet 1998, 79: 35–37. 10.1002/(SICI)1096-8628(19980827)79:1<35::AID-AJMG9>3.0.CO;2-H
Huang B, Martin CL, Sandlin CJ, Wang S, Ledbetter DH: Mitotic and meiotic instability of a telomere association involving the Y chromosome. Am J Med Genet 2004, 129A: 120–123. 10.1002/ajmg.a.30146
Greider CW: Telomers do D-loop-T-loop. Cell 1999, 97: 419–422. 10.1016/S0092-8674(00)80750-3
Robinson WP, Bernasconi F, Basaran S, Yuksel-Apak M, Neri G, Serville F, Balicek P, Haluza R, Farah LMS, Luleci G, Schinzel AA: A somatic origin of homologous Robertsonian translocations and isochromosomes. Am J Hum Genet 1994, 54: 290–302.
Jarow JP, Espeland MA, Lipshultz LI: Evaluation of the azoospermic patient. J Urol 1989, 142: 62–65.
De Braekeleer M, Dao TN: Cytogenetic studies in male infertility: a review. Hum Reprod 1991, 6: 245–250.
Van Assche E, Bonduelle M, Tournaye H, Joris H, Verheyen G, Devroey P, Van Steirteghem A, Liebaers I: Cytogenetics of infertile men. Hum Reprod 1996, 11(Suppl 1):1–24.
Karaer K, Ergun MA, Weise A, Ewers E, Liehr T, Kosyakova N, Mkrtchyan H: The case of an infertile male with an uncommon reciprocal X-autosomal translocation: how does this affect male fertility? Genet Couns 2010, 21: 397–404.
Brisset S, Izard V, Misrahi M, Aboura A, Madoux S, Ferlicot S, Schoevaert D, Soufir JC, Frydman R, Tachdjian G: Cytogenetic, molecular and testicular tissue studies in an infertile 45, X male carrying an unbalanced (Y;22) translocation: case report. Hum Reprod 2005, 20: 2168–2172. 10.1093/humrep/dei034
Hsu LY: Phenotype/karyotype correlations of Y chromosome aneuploidy with emphasis on structural aberrations in postnatally diagnosed cases. Am J Med Genet 1994, 53: 108–140. 10.1002/ajmg.1320530204
Vogt PH, Edelmann A, Kirsch S, Henegariu O, Hirschmann P, Kiesewetter F, Kohn FM, Schill WB, Farah S, Ramos C, et al.: Human Y chromosome azoospermia factors (AZF) mapped to different subregions in Yq11. Hum Mol Genet 1996, 5: 933–943. 10.1093/hmg/5.7.933
Laurent C, Chandley AC, Dutrillaux B, Speed RM: The use of surface spreading in the pachytene analysis of a human t (Y;17) reciprocal translocation. Cytogenet Cell Genet 1982, 33: 312–318. 10.1159/000131777
Delobel B, Djlelati R, Gabriel-Robez O, Croquette MF, Rousseaux-Prevost R, Rousseaux J, Rigot JM, Rumpler Y: Y-Autosome translocation and infertility: usefulness of molecular, cytogenetic and meiotic studies. Hum Genet 1998, 102: 98–102. 10.1007/s004390050660
Buonadonna AL, Cariola F, Caroppo E, Di Carlo A, Fiorente P, Valenzano MC, D'Amato G, Gentile M: Molecular and cytogenetic characterization of an azoospermic male with a de-novo Y;14 translocation and alternate centromere inactivation. Hum Reprod 2002, 17: 564–569. 10.1093/humrep/17.3.564
Acknowledgement
The authors would like to express their thanks to Jeff Radcliff (Quest Diagnostics) for assistance in editorial review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
The authors performed analysis, interpretation of the results, drafting and finalizing the manuscript. All authors read and approved the final manuscript.
Morteza Hemmat, Omid Hemmat contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hemmat, M., Hemmat, O. & Boyar, F.Z. Isochromosome Yp and jumping translocation of Yq resulting in five cell lines in an infertile male: a case report and review of the literature. Mol Cytogenet 6, 36 (2013). https://doi.org/10.1186/1755-8166-6-36
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8166-6-36