
Abstract
Background
Acute lymphoblastic leukemia (ALL) is a heterogeneous form of hematological cancer consisting of various subtypes. We are interested to study the genetic aberration in precursor B-cell ALL with specific t(12;21) translocation in childhood ALL patients. A high resolution 244K array-based Comparative Genomic Hybridization (array-CGH) was used to study eleven ETV6/RUNX1-positive childhood acute lymphoblastic leukemia (ALL) patients.
Result
155 chromosomal aberrations (119 losses, 36 gains) were reported in the array findings, corresponding to 76.8% deletions and 23.2% amplifications. The ETV6 gene deletion occurred in 4 of the patients, corresponding to 45% of the sample. The most common alterations above 1 Mb were deletion 6q (13%), 12p (12%) and 9p (8%), and duplication 4q (6%) and Xq (4%). Other genes important in ALL were also identified in this study including RUNX1, CDKN2A, FHIT, and PAX5. The array-CGH technique was able to detect microdeletion as small as 400 bp.
Conclusion
The results demonstrate the usefulness of high resolution array-CGH as a complementary tool in the investigation of ALL.
Similar content being viewed by others

Background
In childhood B-cell precursor acute lymphoblastic leukemia (ALL), t(12;21)(p13;q22) translocation is the most common chromosomal abnormality and occurs in 20-25% of the cases [1]. This reciprocal translocation, which has a favorable prognosis of more than 80%, leads to the formation of the ETV6/RUNX1 (also known as TEL/AML1) fusion gene [1, 2]. The ETV6/RUNX1 fusion gene was reported in 19% of the Malaysian childhood ALL [3].
Based on the primary ETV6/RUNX1 leukemogenic model, the translocation arises in utero and the rearrangement alone is not sufficient to develop the disease itself [4]. Other secondary genetic alterations (or "hits") are required to trigger the disease progression; however the role of the additional aberrations has not been fully determined [4]. The secondary events such as copy number alterations and point mutations have been suggested to occur postnatally [5]. Major gene targets that are affected in the ETV6/RUNX1 fusion involve genes for stem cell development or lineage specification in hematopoiesis [6]. Such fusion in B-cell ALL leads to the activation of kinase or alteration of transcriptional regulations [6].
Various techniques to screen and study chromosomal aberrations in ALL have been developed in recent years. Conventional cytogenetics is routinely used in the initial assessment for the purpose of classification of specific leukemia [7]. Complementary techniques to conventional cytogenetics such as fluorescence in situ hybridization (FISH), and reverse transcriptase - polymerase chain reaction (RT-PCR) can be used as screening tools for ETV6/RUNX1-positive patients [8]. FISH study is useful to identify specific translocation, but it is limited to the type of probe used to bind the genomic region of interest, and is not genome-wide [9]. Array comparative genomic hybridization (array CGH) has been applied to study copy number alterations and genomic imbalances for evaluation of patients with ALL [10]. Pathogenic chromosomal abnormalities have been reported in patients using the array-based platform, suggesting the usefulness of this technique for diagnostic services.
In the present study, eleven ETV6/RUNX1-positive childhood ALL patients confirmed by RT-PCR were investigated using high resolution array-based comparative genomic hybridization (Agilent 244K Human Genome CGH Microarray).
Results
Array CGH
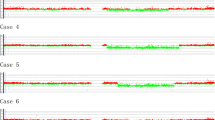
Based on the array-CGH data, a total of 155 genomic aberrations (36 gains, 119 losses, excluding copy number polymorphisms) were identified in all eleven patients, including a patient (no. 4) with duplication of whole chromosome 16. The aberrations ranged from 400 bp to 91.2 Mb. The number of aberrations per patient ranged from 3 to 58, with mean of 14 aberrations per patient. Of the 119 deletions detected, 39 were above 1 Mb and 80 were below 1 Mb. Of the 36 gains detected, 12 were above 1 Mb and 24 were below 1 Mb. In agreement with previous report, we found more deletions (76.8%) than amplifications (23.2%) [11]. Five out of 11 patients (45%) have a deletion of ETV6 gene. The detected aberrations included previously reported loss/gains that are related to ALL, such as 9p13.2 loss involving PAX5 in patient no. 2 [12]; 9p21.3 loss involving CDKN2A in 4 patients (nos. 3, 4, 6 and 7) and MLLT3 in patient no. 3 and 4. Only one patient (no. 11) showed a 0.05 Mb deletion on RUNX1 gene. Three patients (nos. 8, 9 and 10) showed no gross genomic imbalances.
As summarized in Table 1, chromosome 2, 11, 16, 17, 19, 20, 21, and 22 did not have any aberrations above 1 Mb. The most common alterations above 1 Mb were deletion 6q (13%), 12p (12%) and 9p (8%), and amplification 4q (6%) and Xq (4%). The gene annotations are according to the University of California Santa Cruz Genome Browser on Human March 2006 Assembly (NCBI36/hg18).
FISH
All five patients (nos. 1, 3, 5, 6 and 9) showed positive fusion signals of ETV6/RUNX1. Two patients (nos. 1 and 5) showed one fusion signal, but no green signal, indicating a loss of ETV6 gene (Figure 1). Patient no.3 showed a single fusion signal and three red signals, indicating a duplication of RUNX1 and a loss of ETV6 gene in 55% interphase cells scored (Figure 1). Interestingly, two fusion signals were identified in patient no. 6. Patient no. 9 showed a typical fusion profile for t(12;21) translocation (Figure 1).

FISH in 4 patients. (a) Case no. 1 shows 1 red,1 residual red and 1 fusion signal, indicative of loss of ETV6; (b) Case no. 3 shows 3 red and 1 fusion signal, indicative of extra RUNX1 and loss of ETV6; (c) Case no. 6 shows 2 fusion,1 red,1 residual red and 1 green signal; (d) Case no. 9 shows a typical FISH profile for t(12:21).
The status of the ETV6 and RUNX1 genes in the patients based on array-CGH and FISH findings are as shown in Table 2.
Discussion
Our data have demonstrated that the 244K oligonucleotide array-CGH platform is a powerful tool to detect additional copy number alterations in ETV6/RUNX1-positive patients. A total of 155 aberrations were identified, including microdeletions as small as 400 bp. Many known or potential genes related to leukemia were also identified using this method. These data supported the secondary leukemogenic model that additional aberrations are necessary for leukemogenesis. According to our array data, 5 out of 11 patients (45%) showed deletion involving ETV6 gene from as small as 0.2 Mb to 19.4 Mb. We found more deletions (76.8%) than amplifications (23.2%), which is in agreement with a previous study [11]. Among the deletions, 32.7% were larger than 1 Mb, while 33.3% of the amplifications were larger than 1 Mb.
Patient no. 2 harbored a 0.09 Mb deletion on 9p13.2 that involved the PAX5 gene. PAX5 is important in the normal development of B cells, in which loss of a wild-type PAX5 allele would cause differentiation arrest in ALL [12]. Deletion of the tumor suppressor CDKN2A gene located at 9p21.3 was found in 36% (4/11) of our patients. The CDKN2A deletion is suggested to occur more frequently in T-ALL than in precursor B-ALL [13]. The deletion is thought to vary by cytogenetic subgroup and the prognostic value of the incidence is yet to be determined [14]. One patient (no. 7) was found to have a gross deletion (1.0 Mb) on 3p14.2 region that included the FHIT gene, which is proposed as a putative tumor-suppressor gene. The deletion on this particular gene was found to be correlated with a low clinical remission rate and poor overall survival [15–17].
Several putative target genes within the commonly gained region, including cryptic Xq duplications were also found in patient no. 3 and 6, both females. The sizes of the gains on the two patients were 2.1 Mb and 91 Mb, respectively. This result is discordant with the previous report that males are more common to harbor this aberration [18]. This discrepancy may be explained by the small sample size used in this study. It would be interesting to study the expression level of ETV6/RUNX1 proposed genes, namely the SPANX family genes, on the X chromosome in our female's dataset.
Based on our FISH study on five childhood ALL patients, all samples showed a positive ETV6/RUNX1 fusion signal. Three patients (nos. 1, 3 and 5) showed concordant result with array CGH for ETV6 gene deletion. FISH result for patient no. 3 showed three red signals, suggesting that there was a duplication of the RUNX1 signal, but was not confirmed through the array findings. It has been reported that DNA microarray may fail to detect the chromosomal abnormalities if the abnormal clones are present in fewer than 25% of the cell population [19].
Patient no. 6 showed a unique FISH profile where two fusion signals of the ETV6/RUNX1 were detected. Double ETV6/RUNX1 fusion signals were found in 25% of ETV6/RUNX1 positive ALL patients [20]. Previous studies have found that the additional ETV6/RUNX1 fusion signal may have arisen from duplication of the der(21)t(12;21) chromosome [21, 22], duplication of ETV6/RUNX1 fusion gene that was later translocated onto another chromosome [22] or ider(21)(q10)t(12;21)(p12;q22) [23]. In the study by Loncarevic and coworkers (1999), gain of the der(21)t(12;21) chromosome was found exclusively in the relapsed cases [21]. We were not able to ascertain the origin of the extra ETV6/RUNX1 fusion signal in our patient due to non-availability of metaphase cytogenetics. It has however been suggested that secondary changes such as the duplication of fusion signals may contribute to the process of leukemogenesis [22].
Three of the patients, namely patient nos. 4, 10 and 11, had a relapse. Of the three, patient no. 4 had multiple gross deletions as large as 90.8 Mb, whereas patient no. 11 had other gross imbalances larger than 1 Mb. However, the array report for patient no. 10 showed no gross imbalances larger than 1 Mb. We could not determine whether any subsequent aberrations happened after the sample was taken which might trigger the relapse event.
Conclusion
Our study indicates that high resolution oligonucleotide array-CGH is an essential complementary tool in the investigation of the ETV6/RUNX1 positive ALL patients as it helps to complement the findings of FISH and RT-PCR as well as overcoming the limitations of conventional cytogenetics which require cell culture and quality metaphases for analysis. Indeed, array-CGH has revealed additional aberrations which may have pathogenetic implications. A larger cohort is however needed to comprehensively study the genetic diversity of our ETV6/RUNX1- positive ALL cases.
Materials and methods
Patients
Eleven ETV6/RUNX1-positive childhood ALL patients (7 boys and 4 girls) with ages ranging from 2 to 11 years old were selected for this study. All patients were diagnosed as precursor B-ALL with CALLA positivity based on their immuno-phenotyping report. The presence of ETV6/RUNX1 fusion gene in all cases was ascertained by HemaVision® Multiplex RT-PCR System (Bio-Rad Laboratories, Hercules, CA) as part of the routine diagnostic procedure. The study was approved by the Medical Research & Ethics Committee, Ministry of Health Malaysia.
DNAs were extracted from bone marrow aspirates using QIAGEN DNAeasy Blood Kit (Qiagen, Hilden, Germany) according to manufacturer’s instruction. DNA samples subjected to array CGH were of sufficient quality with A260/280 ratio >1.8 as measured by NanoDrop ND-1000 UV–VIS spectrophotometer.
Array-CGH
Array-CGH analysis on the 11 patient samples was carried out using Human Genome CGH 244A Microarray Kit (Agilent Technologies, CA, USA) according to the manufacturer’s protocols. In brief, 1–3 μg of DNA from each patient and reference sample was subjected to restriction digestion using AluI and RsaI restriction enzymes. The reference DNA was commercially obtained from Promega (Promega, Madison, WI) and was gender-matched accordingly. The completion of the digestion for each sample was analyzed using Bioanalyzer before the samples were labeled using Cy3 and Cy5 for patient samples and reference samples, respectively. The yield, the degree of labeling, and the specific activity of the samples were measured using NanoDrop before the two respective samples were combined. The hybridization procedure was carried out at 37 °C for 40 hours and the slides were scanned using Agilent DNA Microarray Scanner. The images from the array-CGH were processed using Agilent Feature Extraction Software (version 9.5.31). The data obtained were analyzed using DNA Analytics v4.0.76 software (Agilent Technologies) with an ADM-2 algorithm with threshold 6.0, and minimum adjacent 3 probes required to be gained or lost for a call to be made. The array-CGH data for all samples have been deposited in Gene Expression Omnibus and are accessible through GEO series accession number GSE32897.
FISH
Five patients with available suspension were selected for FISH studies. The slides for FISH analysis were prepared using cell suspension and were dried at 60°C overnight. The Vysis LSI ETV6/RUNX1 ES dual-color probe was used to identify the translocation pattern for these samples. The probe was added to the slides, hybridized on HyBrite and washed through a series of washes. The slides were viewed under fluorescent microscope, and at least 200 interphase nuclei were analysed for their fusion signal.
Author’s contribution
ZZ designed the experimental study and drafted the manuscript. MFMA and AI carried out the array CGH experiments and performed the data analysis. TSK participated in the FISH analysis and helped draft the manuscript. NMN carried out additional array CGH experiments, performed data analysis and helped draft the manuscript. NRK and EE participated in the design study. EJAR and RO provided the clinical details of the patients, LKY performed FISH analysis. All authors read and approved the final manuscript.
References
Mullighan CG: Genomic profiling of B-progenitor acute lymphoblastic leukemia. Best Pract Res Cl Ha 2011, 24: 489–503. 10.1016/j.beha.2011.09.004
Shurtleff SA, Bujis A, Behm FG, Rubnitz JE, Raimondi SC, Hancock ML, Chan GC, Pui CH, Grosveld G, Downing JR: TEL/AML1 fusion resulting from a cryptic t(12;21) is the most common genetic lesion in pediatric ALL and defines a subgroup of patients with an excellent prognosis. Leukemia 1995, 9(12):1985–1989.
Gill HK, Ten SK, Dhaliwal JS, Moore S, Tan SK, Hassan R, Abdul Karim F, Zakaria Z, Murad S, Mohamed M, Ho CML, Ibrahim H, Abdul Rahman EJ: TEL-AML1 frequency in multi-ethnic Malaysian pediatric acute lymphoblastic leukemia. Cancer Genet Cytogenet 2005, 156: 129–133. 10.1016/j.cancergencyto.2004.04.027
Greaves M: Infection, immune responses and the aetiology of childhood leukaemia. Nat Rev Cancer 2006, 6(3):193–203. 10.1038/nrc1816
van der Weyden L, Giotopoulos G, Rust AG, Matheson LS, van Delft FW, Kong J, Corcoran AE, Greaves MF, Mullighan CG, Huntly BJ, Adams DJ: Modeling the evolution of ETV6-RUNX1–induced B-cell precursor acute lymphoblastic leukemia in mice. Blood 2011, 118(4):1041–1051. 10.1182/blood-2011-02-338848
Zelent A, Greaves M, Enver T: Role of the TEL-AML1 fusion gene in the molecular pathogenesis of childhood acute lymphoblastic leukaemia. Oncogene 2004, 23: 4275–4283. 10.1038/sj.onc.1207672
Wolff DJ, Bagg A, Cooley LD, Dewald GW, Hirsch BA, Jacky PB, Rao KW, Rao PN: Guidance for fluorescence in situ hybridization testing in hematologic disorders. J Mol Diagn 2007, 9(2):134–143. 10.2353/jmoldx.2007.060128
Harbott J, Viehmann S, Borkhardt A, Henze G, Lampert F: Incidence of TEL/AML1 Fusion Gene Analyzed Consecutively in Children with acute lymphoblastic leukemia in relapse. Blood 1997, 90(12):4933–4937.
Dawson A, Yanofsky R, Vallente R, Bal S, Schroedter I, Liang L, Mai S: Array comparative genomic hybridization and cytogenetic analysis in pediatric acute leukemias. Current Onc 2011, 18(5):e210-e217.
Yu S, Bittel DC, Kibiryeva N, Zwick DL, Cooley LD: Validation of the Agilent 244K oligonucleotide array-based comparative genomic hybridization platform for clinical cytogenetic diagnosis. Am J of Clin Path 2009, 132: 349–260. 10.1309/AJCP1BOUTWF6ERYS
Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, Su X, Pui CH, Relling MV, Evans WE, Shurtleff SA, Downing JR: Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature 2007, 446(7137):758–764. 10.1038/nature05690
Heltemes-Harris LM, Willette MJ, Ramsey LB, Qiu YH, Neeley ES, Zhang N, Thomas DA, Koeuth T, Baechler EC, Kornblau SM, Farrar MA: Ebf1 or PAX5 haploinsufficiency synergizes with STAT5 activation to initiate acute lymphoblastic leukemia. J Exp Med 2011, 208(6):1135–1149. 10.1084/jem.20101947
Usvasalo A, Savola S, Räty R, Vettenranta K, Harila-Saari A, Koistinen P, Savolainen ER, Elonen E, Saarinen-Pihkala UM, Knuutila S: CDKN2A deletions in acute lymphoblastic leukemia of adolescents and young adults - an array CGH study. Leuk Res 2008, 32: 1228–1235. 10.1016/j.leukres.2008.01.014
Sulong S, Moorman AV, Irving JA, Strefford JC, Konn ZJ, Case MC, Minto L, Barber KE, Parker H, Wright SL, Stewart AR, Bailey S, Bown NP, Hall AG, Harrison CJ: A comprehensive analysis of the CDKN2A gene in childhood acute lymphoblastic leukemia reveals genomic deletion, copy number neutral loss of heterozygosity, and association with specific cytogenetic subgroups. Blood 2009, 113(1):100–107. 10.1182/blood-2008-07-166801
Wang L, Dong LJ, Tian F, Liu GX, Li CH: Aberrant expression and deletion of FHIT gene in leukemias. J Exp Hematol/Chinese Assoc Pathophysiol 2003, 11(2):153–60.
Iwai T, Yokota S, Nakao M, Nakazawa N, Taniwaki M, Kimura T, Sonoda Y, Kaneko H, Okuda T, Azuma H, Oka T, Takeda T, Watanabe A, Kikuta A, Asami K, Sekine I, Matsushita T, Tsuchiya T, Mimaya J, Koizumi S, Ohta S, Miyake M, Takaue Y, Iwai A, Fujimoto T: Frequent Aberration of FHIT gene expression in acute leukemias. Cancer Res 1998, 58: 5182.
Sugimoto K, Yamada K, Miyagawa K, Hirai H, Oshimi K: Decreased or altered expression of the FHIT gene in human leukemias. Stem Cells 1997, 15(3):223–228. 10.1002/stem.150223
Lilljebjorn H, Heidenblad M, Nilsson B, Lassen C, Horvat A, Heldrup J, Behrendtz M, Johansson B, Andersson A, Fioretos T: Combined high-resolution array-based comparative genomic hybridization and expression profiling of ETV6/RUNX1-positive acute lymphoblastic leukemias reveal a high incidence of cryptic Xq duplications and identify several putative target genes within the commonly gained region. Leukemia 2007, 21(10):2137–44. 10.1038/sj.leu.2404879
Rabin KR, Man TK, Yu A, Folsom MR, Zhao YJ, Rao PH, Plon SE, Naeem RC: Clinical utility of array comparative genomic hybridization for detection of chromosomal abnormalities in pediatric acute lymphoblastic leukemia. Pediatr Blood Cancer 2008, 51: 171–720. 10.1002/pbc.21488
Al-Sweedan SA, Neglia JP, Steiner ME, Bostrom BC, Casey T, Hirsch BA: Characteristics of patients with TEL-AML1-positive acute lymphoblastic leukemia with single or multiple fusions. Pediatr Blood Cancer 2007, 48: 510–514. 10.1002/pbc.20911
Loncarevic IF, Roitzheim B, Ritterbach J, Viehmann S, Borkhardt A, Lampert F, Harbott J: Trisomy 21 is a recurrent secondary aberration in childhood acute lymphoblastic leukemia with TEL/AML1 gene fusion. Genes Chromosomes Cancer 1999, 24: 272–277. 10.1002/(SICI)1098-2264(199903)24:3<272::AID-GCC13>3.0.CO;2-U
Jarosova M, Holzerova M, Mihal V, Blatny J, Lakoma I, Trka J, Pikalova Z, Hrusak O, Indrak K: Additional evidence of genetic changes in children with ALL and TEL/AML1 fusion gene. Leukemia 2002, 16: 1873–1875. 10.1038/sj.leu.2402589
Andreasson P, Johansson B, Strombeck B, Donner M, Mitelman F, Hoglund M: Childhood acute lymphoblastic leukemia with ider (21)(q10)t(12;21)(p12;q22): a new recurrent abnormality showing ETV6/CBFA2 fusion. Br J Haematol 1997, 98: 216–218. 10.1046/j.1365-2141.1997.1652982.x
Acknowledgements
The authors thank the Director General of Health, Malaysia, for permission to publish this scientific paper. We would also like to thank the Deputy Director General of Health (Research and Technical Support) and the Director of the Institute for Medical Research for their support. This research was funded by the Ministry of Health Malaysia (JPP-IMR 07–042).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
All authors declare no competing interest.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zakaria, Z., Ahid, M.F.M., Ismail, A. et al. Chromosomal Aberrations in ETV6/RUNX1-positive Childhood Acute Lymphoblastic Leukemia using 244K Oligonucleotide Array Comparative Genomic Hybridization. Mol Cytogenet 5, 41 (2012). https://doi.org/10.1186/1755-8166-5-41
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8166-5-41