
Abstract
Background
Microdeletion syndromes are generally identified because they usually give rise to specific phenotypic features; many of these deletions are mediated by duplicons or LCRs. The phenotypes associated with subtelomeric deletions are also becoming recognised. However, reciprocal duplication events at these loci are less easily recognised and identified, as they may give rise to milder phenotypic features, and the individuals carrying them may not therefore be referred for appropriate testing. 403 patients with developmental delay and/or dysmorphism, referred to our Genetics Centre for karyotyping and Fragile X expansion testing, were assessed for chromosome imbalance by Multiplex Ligation-dependent Probe Amplification (MLPA). Two MLPA kits were used, one containing probes for the subtelomere regions, and one containing probes for common microdeletion loci. 321 patients were tested with both kits, 75 with the subtelomere kit alone, and 7 with the microdeletion kit alone.
Results
32 patients had abnormal results; the overall abnormality detection rate was 2.5% for karyotype analysis and 7.2% for MLPA testing; 5.5% of subtelomere tests and 2.1% of microdeletion tests gave abnormal results. Of the abnormal MLPA results, 5 were in cases with cytogenetically visible abnormalities; of the remaining, submicroscopic, changes, 3 results were established as de novo and 8 were inherited; parental samples were not available for the remaining cases. None of the patients was found to have a Fragile X expansion.
Conclusion
Karyotype analysis in combination with MLPA assays for subtelomeres and microdeletion loci may be recommended for this patient group.
Similar content being viewed by others


Background
Microdeletion syndromes are generally caused by recurrent, duplicon- or LCR-mediated, contiguous gene deletions, which are identified because they usually give rise to specific phenotypic features. Similarly, the phenotypes associated with subtelomeric deletions are becoming recognised [1]. However, the reciprocal duplication events at these loci may give rise to milder phenotypic features, and the individuals carrying these duplications may not be referred for appropriate testing.
Multiplex Ligation-dependent Probe Amplification (MLPA) [2] is a molecular method for assessing copy number at multiple loci in the genome, and has advantages over Fluorescence In Situ Hybridization (FISH) in terms of throughput of testing, multiplexing of loci, and unequivocal identification of duplications as well as deletions. Previous reports of the use of MLPA for screening individuals with idiopathic mental retardation and/or dysmorphism include cohorts of 51, 75, 210 and 455 tested for subtelomere imbalance [3–6], 258 patients tested for imbalance at microdeletion loci [7], and 58 patients tested for both subtelomere and microdeletion loci [8]. At our Centre, such patients are routinely referred for karyotype analysis and Fragile X testing.
We have carried out testing for imbalance at microdeletion sites and in the subtelomere regions of chromosomes in a cohort of 403 patients referred with developmental delay and/or dysmorphism, with the aim of assessing the prevalence of imbalance at these loci in patients in this referral group, and to provide diagnoses for those who had normal karyotypes and no Fragile X expansion mutations. This cohort did not include patients whose referrals included requests for specific microdeletion tests.
Results
A total of 403 patients was tested, 321 with subtelomere and microdeletion kits, 75 with the subtelomere kit(s) alone, and 7 with the microdeletion kit alone. 32 patients had abnormal results: 10 patients had cytogenetically visible chromosome abnormalities (detailed in Table 1). Of these, one had an inherited balanced rearrangement (#8), one an apparently balanced inversion (#5), one carried Yq heterochromatin on the chromosome 21 short arm (#6), one had a structurally abnormal Y chromosome (#1), and two had numerical sex chromosome abnormalities (#9 and #10). None of these findings is considered likely to be associated with the referral indications of developmental delay and dysmorphism, although gene disruption at the breakpoints of case #5 cannot be excluded. Clinically significant unbalanced karyotypes, representing 1.0% (4/403) of the patient group, comprised a derivative chromosome 10 (#3) and an abnormal chromosome 20 (#4), both also identified by MLPA, and two interstitial deletions (#2 and #7).
Table 2 shows the abnormalities detected by the microdeletion MLPA kit. Although parents were only available for inheritance studies for patients #4 and #11, it is very likely that the other abnormalities in this group also arose de novo, and were associated with the clinical phenotype of these patients. The finding in patient #4 was consistent with the abnormal karyotype, whilst patient #16, with a single probe duplication in the DiGeorge critical region, also carried a deletion of chromosome 8 short arm material (See Table 3).
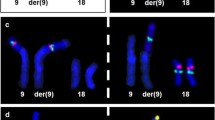
Table 3 shows the subtelomere MLPA abnormalities detected. Five of these confirmed abnormal karyotypes (#3, #4, #6, #9 and #10). There were 3 inherited duplications of 9p (#21, #20 and #25), and three imbalances of 8p (#16, #28, #31), two of which were confirmed by FISH. Patient #24 was found to carry a derivative chromosome 2 from a 2q;22q translocation. Retrospective examination of G-banded chromosomes from this case showed that the translocation was submicroscopic. Patient #18 carried two abnormalities: an inherited duplication of material from the subtelomere region of the chromosome 9 short arm and in addition a structurally abnormal Y chromosome, not originally identified by G-banded chromosome analysis. Nine of the subtelomere anomalies involved only a single MLPA probe, and therefore required confirmation; of these, 4 were confirmed in other family members, one was confirmed by FISH, and 4 remain unconfirmed. Parental or other family samples were only available for 7 of the 17 cases with submicroscopic abnormalities in this group, all of which showed inheritance of the anomalies, although in some cases no information was available on the phenotype of the carrier parent.
None of the cases in the patient cohort carried Fragile X expansion mutations. The overall abnormality detection rate was therefore 0% (0 cases) for Fragile X testing, 2.5% (10 cases) for karyotype analysis and 7.2% (29 cases) for MLPA testing; 5.5% (22 cases) of subtelomere tests and 2.1% (7 cases) of microdeletion tests gave abnormal results.
Discussion
We have previously assessed and validated the commercially available MLPA subtelomere kit, and have described a robust analysis protocol that minimises both false positive and false negative results [6]. Nevertheless, confirmation of single probe abnormalities is prudent. In the data described here, all but 4 of the subtelomere abnormalities were confirmed, either by using a second MLPA subtelomere kit containing probes at nearby loci within the subtelomere, or by FISH, or by confirming the abnormality in another family member. The microdeletion MLPA kit contains several probes within each microdeletion region, and at least two probes within each region showed imbalance for six out of the seven abnormalities reported here. Nevertheless, where FISH probes were available, these were used to confirm the imbalance. Even when subtelomere results were confirmed, their clinical significance was not always clear. Where the same abnormality was found in a parent with apparently normal clinical phenotype, it is likely that the imbalance may represent polymorphic variation; it is interesting that all such abnormalities found in this group were duplications, perhaps reflecting population copy number variation such as that described recently [9–11]. De novo abnormalities, however, are considered very likely to be associated with the patients' developmental delay or dysmorphism.
In one patient (#18), originally assigned a normal karyotype, MLPA revealed imbalance which was visible on retrospective examination of the G-banded chromosomes. This underlines the difficulty of comparing prevalence of such abnormalities in publications from different centres, as prevalence of detection by MLPA (or other molecular cytogenetic methods) will be apparently greater in centres where G-banded chromosome analysis is carried out at lower quality, and will also depend on the selection criteria for the patient cohort. Further studies on these patients are in progress to assess the exact extent of the imbalance.
Of the submicroscopic abnormalities in the subtelomere group, one unbalanced translocation, 5 deletions and 11 duplications were detected, indicating that duplications may be more prevalent. Similarly, there were 3 deletions and 4 duplications in the microdeletion group; here, one might have predicted that those with deletions would be targeted for specific testing because of the characteristic syndromic features associated with such deletions, and therefore would not be included in the tested group. This would, however, depend on the diagnostic skill of the referring physician; the non-specific phenotypes reported here associated with known microdeletions (for instance in patients #12 and #13) underlines the variability in the presentation of these syndromes.
The copy number variation in the human genome reported recently [9–11] is thought to under represent smaller variants [12, 13] and so it seems likely that testing with ~60 base pair probes will uncover further polymorphic variation. For instance, in the data presented here, patients #18, #21 and #25 all carry inherited duplications of 9p; this region may therefore be subject to population polymorphism of no clinical significance, and be an incidental finding in these cases. However, unless carrier parents' phenotypes are carefully assessed, an effect of variable penetrance cannot be completely excluded.
All the patients reported here were referred for Fragile X testing; none carried expansion mutations. At our Centre, Fragile X testing is carried out on average for 800–1000 patients per year, of whom < 1% are found to carry an expansion mutation. This is in contrast to the 7.2% detection rate for submicroscopic imbalance detected by MLPA, although as none of the subtelomere results were established as de novo, only the abnormalities found by microdeletion/duplication testing could be definitively established as of clinical significance.
Previous publications detailing submicroscopic imbalance detected by MLPA in patients with developmental delay/dysmorphism have reported abnormality detection rates of 5.3% [4], 5.9% [3], 6.7% [5] and 5.9% [6] when testing subtelomeric loci, 5.8% [7] when testing microdeletion loci, and 13.8% [8] when testing at both subtelomeric and microdeletion loci. Our overall detection rate by MLPA, for a larger cohort than these previous studies, was 7.2%; however, as discussed above, comparison between different centres is difficult because of the differences in resolution of G-banded chromosome analysis, and in the selection criteria for the patients.
Conclusion
In conclusion, this paper reports the largest cohort to date of patients with developmental delay and/or dysmorphism, referred for the investigation of chromosome abnormality and tested for submicroscopic imbalance using MLPA. The importance of robust analysis protocols, confirmation of single probe findings, and appropriate family studies is emphasised. A comprehensive review [14] of available methodologies for assessing imbalance at subtelomeres concluded that MLPA is currently the method of choice, although the use of real-time PCR and microarrays may become more widespread as affordable commercial platforms become available [15]. Interestingly, a recent comparison of approaches to the investigation of patients with developmental delay [8] suggests that a strategy of replacing karyotyping with MLPA as a first test, followed by microarray analysis, may prove effective in terms of detection rate and cost-effectiveness. Of course, in the long term, it has been suggested that microarrays as a first test may prove to be the most efficient as the cost of this technology falls further [16]. In the UK, karyotype analysis as the "gold standard" and first test for children with developmental delay and/or dysmorphism is currently considered to be Best Professional Practice. The findings presented here suggest that the abnormality detection rate using a strategy of karyotype analysis followed by MLPA for subtelomeres and microdeletion loci will give a diagnostic yield considerably in excess of that gained by a strategy of karyotype analysis and Fragile X testing, and may be the best approach until microarray testing is generally validated as routine diagnostic tool, and becomes economical enough to replace karyotyping as the first test for these patients.
Methods
Karyotype analysis
Karyotype analysis was carried out on G-banded chromosomes prepared from peripheral lymphocytes by standard laboratory procedures. Preparations were generally analysed at quality 6 G-banding. Fragile X testing was performed with a biplex PCR to amplify the repeat regions at the FRAXA and FRAXE loci, using standard fluorescent PCR amplification. Products were then analysed using an ABI 3730 analyser (Applied Biosystems, USA) [17].
MLPA analysis
MLPA analysis was carried out using kits (P036, P036B, P064, P064B and P069; MRC-Holland, The Netherlands), according to the manufacturer's protocols. Initial subtelomere testing used kit P069, which was followed by testing with kit P036 or P036B if an abnormality was detected. Analysis was according to our previous publication [6]. The MLPA microdeletion P064B kit contains probes for the following regions; 1p36 telomeric region, 7q11.23 Williams syndrome region, 17p11.2 Smith-Magenis region, 17p13.3 Miller-Dieker region, 22q11.21 DiGeorge region, 15q11.2 Prader-Willi/Angelman's region, 20p12.2 Alagille region, 7p21 Saethre-Chotzen syndrome, 5q35.3 Sotos syndrome region.
Fluorescence in situ hybridization
Fluorescence in situ hybridization (FISH) confirmation used subtelomere and microdeletion probes from Abbott-Vysis (ToTelVysion; UK), Cytocell (Aquarius; UK) or MP Biomedicals (QBiogene; CA, USA), according to manufacturers' protocols. Subtelomere probes for confirmation were chosen according to the available information on genomic position relative to that of the MLPA probes.
References
de Vries BB, White SM, Knight SJ, Regan R, Homfray T, Young ID, Super M, McKeown C, Splitt M, Quarrell OW, Trainer AH, Niermeijer MF, Malcolm S, Flint J, Hurst JA, Winter RM: Clinical studies on submicroscopic subtelomeric rearrangements: a checklist. J Med Genet 2001, 38(3):145–150. 10.1136/jmg.38.3.145
Schouten JP, McElgunn CJ, Waaijer R, Zwijnenburg D, Diepvens F, Pals G: Relative quantification of 40 nucleic acid sequences by multiplex ligation-dependent probe amplification. Nucleic Acids Res 2002, 30(12):e57. 10.1093/nar/gnf056
Northrop EL, Ren H, Bruno DL, McGhie JD, Coffa J, Schouten J, Choo KH, Slater HR: Detection of cryptic subtelomeric chromosome abnormalities and identification of anonymous chromatin using a quantitative multiplex ligation-dependent probe amplification (MLPA) assay. Hum Mutat 2005, 26(5):477–486. 10.1002/humu.20243
Rooms L, Reyniers E, van Luijk R, Scheers S, Wauters J, Ceulemans B, Van Den Ende J, Van Bever Y, Kooy RF: Subtelomeric deletions detected in patients with idiopathic mental retardation using multiplex ligation-dependent probe amplification (MLPA). Hum Mutat 2004, 23(1):17–21. 10.1002/humu.10300
Koolen DA, Nillesen WM, Versteeg MH, Merkx GF, Knoers NV, Kets M, Vermeer S, van Ravenswaaij CM, de Kovel CG, Brunner HG, Smeets D, de Vries BB, Sistermans EA: Screening for subtelomeric rearrangements in 210 patients with unexplained mental retardation using multiplex ligation dependent probe amplification (MLPA). J Med Genet 2004, 41(12):892–899. 10.1136/jmg.2004.023671
Ahn JW, Ogilvie CM, Welch A, Thomas H, Madula R, Hills A, Donaghue C, Mann K: Detection of subtelomere imbalance using MLPA: validation, development of an analysis protocol, and application in a diagnostic centre. BMC Med Genet 2007, 8: 9. 10.1186/1471-2350-8-9
Kirchhoff M, Bisgaard AM, Bryndorf T, Gerdes T: MLPA analysis for a panel of syndromes with mental retardation reveals imbalances in 5.8% of patients with mental retardation and dysmorphic features, including duplications of the Sotos syndrome and Williams-Beuren syndrome regions. Eur J Med Genet 2007, 50(1):33–42. 10.1016/j.ejmg.2006.10.002
Kriek M, Knijnenburg J, White SJ, Rosenberg C, den Dunnen JT, van Ommen GJ, Tanke HJ, Breuning MH, Szuhai K: Diagnosis of genetic abnormalities in developmentally delayed patients: a new strategy combining MLPA and array-CGH. Am J Med Genet A 2007, 143(6):610–614.
Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, Gonzalez JR, Gratacos M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Armengol L, Conrad DF, Estivill X, Tyler-Smith C, Carter NP, Aburatani H, Lee C, Jones KW, Scherer SW, Hurles ME: Global variation in copy number in the human genome. Nature 2006, 444(7118):444–454. 10.1038/nature05329
Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, Scherer SW, Lee C: Detection of large-scale variation in the human genome. Nature genetics 2004, 36(9):949–951. 10.1038/ng1416
Tuzun E, Sharp AJ, Bailey JA, Kaul R, Morrison VA, Pertz LM, Haugen E, Hayden H, Albertson D, Pinkel D, Olson MV, Eichler EE: Fine-scale structural variation of the human genome. Nature genetics 2005, 37(7):727–732. 10.1038/ng1562
Estivill X, Armengol L: Copy number variants and common disorders: filling the gaps and exploring complexity in genome-wide association studies. PLoS genetics 2007, 3(10):1787–1799. 10.1371/journal.pgen.0030190
Conrad DF, Hurles ME: The population genetics of structural variation. Nature genetics 2007, 39(7 Suppl):S30–6. 10.1038/ng2042
Rooms L, Reyniers E, Kooy RF: Subtelomeric rearrangements in the mentally retarded: a comparison of detection methods. Hum Mutat 2005, 25(6):513–524. 10.1002/humu.20185
Rauch A, Hoyer J, Guth S, Zweier C, Kraus C, Becker C, Zenker M, Huffmeier U, Thiel C, Ruschendorf F, Nurnberg P, Reis A, Trautmann U: Diagnostic yield of various genetic approaches in patients with unexplained developmental delay or mental retardation. Am J Med Genet A 2006, 140(19):2063–2074.
Koolen DA, Sistermans EA, Nilessen W, Knight SJ, Regan R, Liu YT, Kooy RF, Rooms L, Romano C, Fichera M, Schinzel A, Baumer A, Anderlid BM, Schoumans J, van Kessel AG, Nordenskjold M, de Vries BB: Identification of non-recurrent submicroscopic genome imbalances: the advantage of genome-wide microarrays over targeted approaches. Eur J Hum Genet 2007.
Whibley A, Fratter CS, Abbs SJ, Yau SC: A fluorescent PCR method for allele detection and accurate sizing in Fragile X syndrome. J Med Genet 2005, 42(Supplement 1):S99.
Acknowledgements
We acknowledge the Guy's and St Thomas' Charity for funding JWA and the Cytogenetics Department for their contribution.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
JWA developed the analysis protocol, carried out the MLPA testing and data analysis, and assisted in drafting the paper; KM and CO conceived the project and obtained the funding; KM analysed the data and assisted in drafting the paper; ZD provided critical input in the karyotype interpretation; CO analysed the data and wrote the paper.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ahn, J.W., Mann, K., Docherty, Z. et al. Submicroscopic chromosome imbalance in patients with developmental delay and/or dysmorphism referred specifically for Fragile X testing and karyotype analysis. Mol Cytogenet 1, 2 (2008). https://doi.org/10.1186/1755-8166-1-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1755-8166-1-2