
Abstract
Background
Melatonin (MLT) has many health implications, therefore it is of valuable importance to develop specific analytical methods for determination of MLT in the presence of its main contaminant, N-{2-[1-({3-[2-(acetylamino)ethyl]-5-methoxy-1H-indol-2-yl}methyl)-5-methoxy-1H-indol-3-yl]ethyl}acetamide (10). For development of these analytical methods, compound 10 had to be prepared in an adequate amount.
Results
Compound 10 was synthesized in six steps starting from 5-methoxyindole-2-carboxylic acid (1). Analytical performance of the proposed spectrofluorimetric methods was statistically validated with respect to linearity, accuracy, precision and specificity. The proposed methods were successfully applied for the assay of MLT in laboratory prepared mixtures containing up to 60 % of compound 10 and in commercial MLT tablets with recoveries not less than 99.00 %. No interference was observed from common pharmaceutical additives and the results were favorably compared with those obtained by a reference method.
Conclusions
This work describes simple, sensitive, and reliable second derivative spectrofluorimetric method in addition to two multivariate calibration methods, principal component regression (PCR) and partial least square (PLS), for the determination of MLT in the presence of compound 10.
Similar content being viewed by others


Background
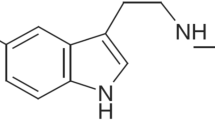
Melatonin (N-acetyl-5-methoxytryptamine, MLT, Figure 1) is primarily produced by the pineal gland in the brain with a marked circadian rhythm normally peaking in the dark to regulate sleep. MLT acts through activation of two G-protein-coupled receptors, designated as MT1 and MT2[1]. In addition, a low-affinity putative MLT binding site called MT3 has been recently characterized as a melatonin-sensitive form of the human enzyme quinine reductase 2 [2]. MLT has found widespread use in the treatment of sleep disorders, other effects described in the literature include its anti- inflammatory, pain modulatory, antitumor, and antioxidant properties [3–8].

Structures of melatonin (MLT) and compound 10.
MLT as well as L-tryptophane (Trp) are naturally occurring indole-based compounds and are sold over-the-counter as dietary supplement in the United States. Contaminants in Trp preparations are the etiological agents for the 1989 outbreak of oesinophilia-myalgia syndrome (EMS), which affected about 1500 people and led to about 30 deaths in the United States [9]. Administration of 15 mg/day MLT for four weeks to cancer patients has led to induction of oesinophilia [10, 11]. Accordingly, Williamson et al.[12] investigated the presence of contaminants structurally related to those found in contaminated Trp preparations. They reported the presence of six structural analogues of Trp contaminants in three different commercial MLT preparations. Two contaminants were identified to be hydroxymelatonin isomers with MH+ = 249 whereas, other four contaminants were identified as melatonin-formaldehyde condensation products with MH+ = 477. Consequently, a tighter control on nutritional supplements sold and used as drugs was recommended.
Compound 10 (Figure 1) is the most abundant regio-isomer from the four melatonin-formaldehyde condensation contaminants that were found in the commercial preparations of MLT [12]. To the best of our knowledge, a preparative methodology for the synthesis of pure 10 has not reported yet. We therefore report herein a preparative method for the synthesis of compound 10, which is required for the development of spectrofluorimetric methods for determination of MLT in the presence of compound 10 in commercial MLT preparations. An evaluation of the literature revealed that only one HPLC/tandem mass spectrometry (LC/MS/MS) method has been published for the determination of MLT in the presence of compound 10[12]. This method offers a high degree of specificity, however its sophisticated instrumentation and cost factor preclude its use in routine analysis. Therefore, it was desirable to develop simple and fast procedures that could be applied in quality control laboratories for the determination of MLT in presence of compound 10. Derivative spectrofluorimetry and multivariate calibration methods such as PCR and PLS are useful means of resolving two overlapping spectra and eliminating matrix interference in the assay of two-component mixtures [13, 14].
The principal advantages of these methods lie in the improved sensitivity and selectivity, in addition to the significant economic advantages over other sophisticated instrumental techniques such as HPLC/tandem mass spectrometry.
Experimental
Melting points were determined using a capillary melting point apparatus (Gallenkamp, Sanyo) and are uncorrected. Column chromatography was carried out on silica gel 60 (0.063–0.200 mm) obtained from Merck. A Bruker AV-400 spectrometer was used to obtain 1 H NMR (400 MHz) and 13 C NMR (100 MHz) spectra. The NMR resonances were assigned by means of HH-COSY, HMQC, and HMBC experiments. EI mass spectra were determined on a Finnigan MAT 8200 spectrometer. IR spectra, recorded as ATR, were obtained by using a Biorad PharmalyzIR FT-IR instrument. Fluorescence measurements were carried out using a Shimadzu (Kyoto, Japan) RF-5301 version 3.0 spectrofluorimeter equipped with a 150 W xenon lamp and 1 cm quartz cells. The slit width of both the excitation and emission monochromators was set at 5 nm. The calibration and linearity of the instrument were frequently checked with standard quinine sulphate (0.01 μg ml-1). Wavelength calibration was performed by measuring λexcitation at 279 nm and λemission at 333 nm; no variation in the wavelength was observed. Elemental analyses were performed by the microanalytical section of the Institute of Inorganic Chemistry, University of Würzburg. All reactions were carried out under an argon atmosphere. MLT was obtained from Merc Inc, New York, USA. Its purity is certified to be 99.5%. Methanol and ethyl acetate were of analytical-reagent grade. Commercially available MLT preparation labeled to contain 3 mg MLT was purchased from the local market.
Synthesis
(5-Methoxy-2,3-dihydro-1 H-indol-1-yl)(5-methoxy-1 H-indol-2-yl)methanone (3)
A solution of 5-methoxyindoline (2) (0.94 g, 6.28 mmol) in dry CH2Cl2 (5 ml) was added to a stirred solution of 5-methoxyindole-2-carboxylic acid (1) (1.2 g, 6.28 mmol) and ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDCI·HCl) (1.80 g, 9.42 mmol) in dry CH2Cl2 (15 ml). The reaction mixture was stirred for 18 h at room temperature, extracted with 5 N hydrochloric acid (3 × 5 ml), washed with water (2 × 10 ml), and dried (Na2SO4). The organic layer was evaporated in vacuo, and the residue was recrystallized from isopropanol to yield 1.76 g (87%) of 3 (1.76 g, 87%) as a pale yellow powder mp 234–236°C. FTIR (ATR) ν = 3270, 2935, 1604, 1576, 1406, 797 cm-1. 1 H NMR (DMSO-d 6 ): δ 3.26 (t, 2 H, J = 8.4 Hz, H-3′), 3.79 (s, 3 H, OCH3), 3.81 (s, 3 H, OCH3), 4.52 (t, 2 H, J = 8.4 Hz, H-2´), 6.83 (dd, 1 H, J = 8.8, 2.5 Hz, H-6), 6.93 (d, 1 H, J = 2.5 Hz, H-4′), 6.95-6.96 (m, 1 H, H-6′), 7.04 (s, 1 H, H-3), 7.15 (d, 1 H, J = 2.5 Hz, H-4), 7.43 (d, 1 H, J = 8.8 Hz, H-7), 8.14 (d, 1 H, J = 8.6 Hz, H-7′), 11.58 (br., 1 H, NH). 13 C NMR (DMSO-d 6): δ 28.4 (C-3′), 49.7 (C-2′), 55.3 (OCH3), 55.4 (OCH3), 102.1 (C-4), 104.9 (C-3), 110.7 (C-6′), 111.9 (C-6), 113.1 (C-7), 115.1 (C-4′), 117.6 (C-7′), 127.6, 131.2, 131.4, 133.9, 136.9 (ArC), 153.8, 156.1 (C-5, C-5′), 159.5 (O = C). MS (EI): m/z (%) = 322 (M+, 27), 174 (10), 149 (100), 134 (29). Anal. Calcd for C19H18N2O3: C, 70.79; H, 5.63; N, 8.69. Found: C, 70.41; H, 5.61; N, 8.75.
(5-Methoxy-1 H-indol-1-yl)(5-methoxy-1 H-indol-2-yl)methanone (4)
A mixture of 3 (0.20 g, 0.62 mmol) and 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) (0.19 g, 0.68 mmol) was heated at reflux temperature in ethyl acetate (30 ml) for 18 h. The reaction mixture was evaporated under reduced pressure and the residue was purified by silica gel chromatography (chloroform/methanol/ammonia, 10:1:0.1) to furnish 0.19 g (96%) of 4 as a light red powder mp 178–179°C and. FTIR (ATR) ν = 3290, 2937, 1625, 1517, 1026, 797 cm-1.1 H NMR (CDCl3): δ 3.85 (s, 3 H, OCH3), 3.87 (s, 3 H, OCH3), 6.63 (d, 1 H, J = 3.8 Hz, H-3′), 6.99 (dd, 1 H, J = 9.1, 2.5 Hz, H-6′), 7.03 (dd, 1 H, J = 8.8, 2.2 Hz,, 7.07 (d, 1 H, J = 2.2 Hz, H-4), 7.08-7.09 (m, 2 H, H-3, H-4′), 7.37 (d, 1 H, J = 8.8 Hz, H-7), 7.90 (d, 1 H, J = 3.8 Hz, H-2′), 8.37 (d, 1 H, J = 9.1 Hz, H-7′), 9.70 (br., 1 H, NH). 13 C NMR (CDCl3): δ 55.7 (2 x OCH3), 102.4 (C-4), 103.7 (C-3′), 108.9 (C-3), 109.6 (C-7), 113.0 (C-6′), 113.4 (C-6), 116.9 (C-7′), 117.7 (C-4′), 127.6 (C-2′), 127.8, 129.2, 130.8, 131.6, 132.5 (ArC), 154.9, 156.7 (C-5, C-5′), 160.8 (O = C). MS (EI): m/z (%) = 320 (M+, 70), 173 (53), 147 (100), 119 (29). Anal. Calcd for C19H16N2O3: C, 71.24; H, 5.03; N, 8.75. Found: C, 70.95; H, 5.08; N, 8.68.
5-Methoxy-1-[(5-methoxy-1 H-indol-2-yl)methyl]-1 H-indole (5)
Compound 4 (0.50 g, 156.03 mmol) was dissolved in dry THF (5 ml) and was added dropwise to a cooled (0°C) suspension of LiAlH4/AlCl3 in dry diethyl ether (prepared by a slow addition of AlCl3 (0.32 g, 2.41 mmol) to a suspension LiAlH4 (0.27 g, 7.13 mmol) in dry diethyl ether (15 ml) at 0°C. The resulting reaction mixture was stirred at 0°C for one hour and at room temperature for another one hour. The reaction was quenched by a slow addition of saturated sodium sulphate solution. The solids were removed by filtration, washed with chloroform (20 ml) and the combined organic phase was dried (Na2SO4) and evaporated under reduced pressure. The residue was purified by silica gel chromatography (chloroform/methanol/ammonia, 10:1:0.1) to produce 0.4 g (83%) of 5 as a light red powder mp 173–174°C. FTIR (ATR) ν = 3384, 2956, 1622, 1485, 795 cm-1.1 H NMR (CDCl3): δ 3.83 (s, 3 H, OCH3), 3.85 (s, 3 H, OCH3), 5.26 (s, 2 H, CH2-N), 6.39 (s, 1 H, H-3) 6.49 (d, 1 H, J = 3.3 Hz, H-3′), 6.81 (dd, 1 H, J = 8.8, 2.3 Hz, H-6′), 6.86 (dd, 1 H, J = 8.8, 2.5 Hz, H-6), 7.01 (d, 1 H, J = 8.8 Hz, H-7′), 7.04-7.05 (m, 2 H, H-2′, H-4), 7.15 (d, 1 H, J = 2.3 Hz, H-4′), 7.20 (d, 1 H, J = 8.8 Hz, H-7), 7.74 (br., 1 H, NH). 13 C NMR (CDCl3): δ 44.0 (CH2-N), 55.8 (OCH3), 55.9 (OCH3), 101.3 (C-3), 101.6 (C-3′), 102.3 (C-4), 102.8 (C-4′), 110.1 (C-6′), 111.6 (C-6), 112.2, 112.3 (C-7, C-7′), 128.4, 128.5 (ArC), 129.2 (C-2′), 131.4, 131.6, 134.7 (ArC), 154.2 (C-5, C-5′). MS (EI): m/z (%) = 306 (M+, 35), 160 (100), 147 (23). Anal. Calcd for C19H18N2O2: C, 74.49; H, 5.92; N, 9.14. Found: C, 74.19; H, 5.95; N, 8.89.
2-{[5-Methoxy-3-(2-nitroethyl)-1 H-indol-1-yl]methyl}-5-methoxy-3-(2-nitroethyl)-1 H-indole (8)
A mixture of 5 (0.250 g, 0.82 mmol), 2-nitroethyl acetate (0.350 g, 2.63 mmol), and tert. butyl catechol (6 mg) in xylene (20 ml) was heated at reflux temperature for 18 h. The reaction mixture was evaporated under vacuum and the residue was purified by silica gel chromatography (chloroform/methanol, 9:0.5) to yield 0.17 g (46%) of 8 as brown viscous oil. FTIR (ATR) ν = 3421, 2965, 1548, 1212, 795 cm-1. 1 H NMR (CDCl3): δ 3.39 (t, 2 H, J = 7.1 Hz, CH 2-CH2-N), 3.44 (t, 2 H, J = 6.8 Hz, CH 2-CH2-N), 3.83 (s, 3 H, OCH3), 3.84 (s, 3 H, OCH3), 4.56 (t, 2 H, J = 6.8 Hz, CH2-CH 2-N), 4.61 (t, 2 H, J = 7.1 Hz, CH2-CH 2-N), 5.30 (s, 2 H, CH2-N), 6.81 (dd, 1 H, J = 8.8, 2.3 Hz, ArH), 6.86 (dd, 1 H, J = 8.8, 2.5 Hz, ArH), 6.88 (s, 1 H, H-2′), 6.80 (d, 1 H, J = 2.4 Hz, ArH), 6.98 (d, 1 H, J = 2.4 Hz, ArH), 7.09 (d, 1 H, J = 8.8 Hz, ArH), 7.14 (d, 1 H, J = 8.8 Hz, ArH), 7.66 (br., 1 H, NH). 13 C NMR (CDCl3): δ 22.6 (CH2-CH2-N), 23.5 (CH2-CH2-N), 42.0 (CH2-N), 55.9 (OCH3), 56.0 (OCH3), 74.9 (CH2-CH2-N), 75.7 (CH2-CH2-N), 100.0, 100.6, 110.4, 112.1, 112.6, 112.8 (ArCH), 126.7 (C-2′), 109.5 (C-3, C-3′), 128.9, 130.6, 131.9, 132.0, 133.4 (ArC), 154.5, 154.6 (C-5, C-5′). MS (EI): m/z (%) = 452 (M+, 17), 233 (58), 186 (100). Anal. Calcd for C23H24N4O6: C, 61.06; H, 5.35; N, 12.38. Found: C, 60.86; H, 5.24; N, 12.49.
N-{2-[1-({3-[2-(Acetylamino)ethyl]-5-methoxy-1 H-indol-2-yl}methyl)-5-methoxy-1 H-indol-3-yl]ethyl}acetamide (10)
A mixture of 8 (0.17 g, 0.38 mmol) and 10% Pd/C (70 mg) in absolute ethanol (10 ml) was hydrogenated under 4 mbar pressure in Parr shaker device at ambient temperature for 18 h. The reaction mixture was filtered off and the filtrate was evaporated under reduced pressure to furnish 0.15 g of 9 as pale yellow viscous oil. Crude 9 (0.15 g, 0.38 mmol) was acetylated using acetic anhydride (0.36 ml, 3.82 mmol) and triethylamine (0.38 ml, 2.66 mmol) in dry DCM (10 ml) at room temperature for 18 h. The solvent was evaporated under vacuum and the residue was purified by silica gel chromatography (chloroform/methanol/ammonia, 10:1:0.1) to yield 0.11 g (63%) of 10 as a beige powder mp 88–90°C and was FTIR (ATR) ν = 3286, 2924, 1635, 1216, 794 cm-1.1 H NMR (CDCl3): δ 1.76 (s, 3 H, CH3), 1.80 (s, 3 H, CH3), 2.84 (t, 2 H, J = 6.6 Hz, CH 2-CH2-N), 2.96 (t, 2 H, J = 6.8 Hz, CH 2-CH2-N), 3.39-3.44 (m, 2 H, CH2-CH 2-N), 3.48-3.53 (m, 2 H, CH2-CH 2-N), 3.81 (s, 3 H, OCH3), 3.82 (s, 3 H, OCH3), 5.25 (s, 2 H, CH2-N), 5.74 (t, 1 H, J = 5.7 Hz, NH), 5.83 (t, 1 H, J = 5.6 Hz, NH), 6.78 (dd, 1 H, J = 8.8, 2.3 Hz, ArH), 6.81 (dd, 1 H, J = 8.8, 2.5 Hz, ArH), 6.87 (s, 1 H, H-2′), 6.99 (d, 2 H, J = 2.3 Hz, ArH), 7.11 (d, 1 H, J = 8.8 Hz, ArH), 7.14 (d, 1 H, J = 8.8 Hz, ArH), 8.24 (br., 1 H, NH). 13 C NMR (CDCl3): δ 23.1 (CH3), 23.2 (CH3), 24.3 (CH2-CH2-N), 25.3 (CH2-CH2-N), 39.8 (CH2-CH2-N), 40.2 (CH2-CH2-N), 41.9 (CH2-N), 55.9 (2 x OCH3), 100.5, 100.9, (ArCH), 110.1, 110.3 (C-3, C-3′), 111.9, 112.3, 112.4, 112.5 (ArCH), 126.2 (C-2′), 128.5, 128.8, 130.9, 131.4, 131.9 (ArC), 154.1, 154.2 (C-5, C-5′), 170.3 (O = C), 170.5 (O = C). MS (EI): m/z (%) = 476 (M+, 31), 417 (16), 245 (100), 203 (41), 186 (64). Anal. Calcd for C27H32N4O4: C, 68.05; H, 6.77; N, 11.76. Found: C, 68.37; H, 6.59; N, 11.66.
Analysis
Preparation of MLT and compound 10 standard solutions
Stock solutions of MLT (100 μg ml-1) and compound 10 (300 μg ml-1) were prepared by dissolving 10 mg and 30 mg of MLT and compound 10, respectively, in 100 ml methanol. Appropriate volumes of these stock solutions were diluted to give working solutions of 4 and 3 μg ml-1for MLT and compound 10, respectively. Stock and working solutions were stable for at least two weeks when stored refrigerated at 4°C.
Preparation of MLT tablets sample solutions
Ten tablets were weighed and finely powdered. An accurately weighed portion of the powder equivalent to 3 mg of MLT was extracted with ethyl acetate and the extract was filtered. The extract was evaporated and reconstituted in methanol to obtain final concentration of 4 μg ml-1 MLT. Aliquots of tablet extract were diluted with methanol to obtain final concentration of 120 ng ml-1 and the samples were subjected to the analysis according to the Calibration procedures.
Calibration procedures
Second derivative method
Aliquots equivalent to 20–220 ng ml-1 MLT were accurately transferred from its standard working solution into separate series of 5-ml volumetric flasks then completed to volume with methanol. The emission spectra of the prepared standard solutions were scanned from 300 to 450 nm using λexcitation at 279 nm and stored in the computer. The second derivative of stored emission spectra of MLT were computed with Δλ = 10 nm. The amplitude of the second derivative peak of MLT was measured at 324.0 nm. The calibration graph was constructed by relating the peak amplitudes at 324.0 nm to the corresponding concentrations of MLT and the regression equation for the data was computed.
PCR and PLS chemometric models
Four level, two factor calibration design [15] was used for construction of 16 samples of the calibration set by transferring different volumes of MLT and compound 10 from their standard working solutions into 5-ml volumetric flasks then completed to volume with methanol (Table 1).
The emission spectra of the calibration set were scanned from 300 to 380 nm using λexcitation at 279 nm and stored in the computer. Mean centering of the data proved to be the best preprocessing method for getting the optimum results.
Constructing the PCR and PLS models
The calibration set fluorescence intensities and their corresponding concentrations were used to build the PCR and PLS models using PLS-Toolbox 2.0 software for the calculations.
Selection of the optimum number of factors to build the PCR and PLS models
The cross validation method, leaving out one sample at a time, was used to select the optimum number of factors [16]. Given a set of 16 calibration samples, the PCR and PLS calibrations were performed on 15 samples. By using this calibration, the concentration of the sample left out was predicted. This process was repeated a total of 16 times until each sample had been left out once. The predicted concentrations were then compared with the known concentrations. The root mean square error of cross validation (RMSECV) was calculated in the same manner each time a new factor was added to the model. The maximum number of factors used to calculate the optimum RMSECV was selected to be nine. The method described by Haland and Thomas [17] was used for selecting the optimum number of factors.
Assay of laboratory prepared mixtures
Aliquots of MLT and compound 10 were transferred from their standard working solutions into a series of 5-ml measuring flasks, completed to volume with methanol and mixed well. For determination of MLT, by the proposed methods, the Calibration procedures were applied.
Results and discussion
MLT dietary supplement tablets are mainly used in treatment of sleep disorders and are sold over-the-counter. It was reported that commercial MLT preparations contain six contaminants; four of them (MLT-formaldehyde condensation contaminants) are thought to be responsible for induction of oesinophilia when MLT was administered in a high dose for long time [10–12]. Compound 10 is the most abundant regio-isomer from these four formaldehyde condensation contaminants [12]. Daily value of MLT intake is not established yet and therefore, it is of sizable importance to develop simple, accurate, and sensitive methods for the routine analysis of MLT in the presence of its main contaminant, compound 10.
To develop such analytical methods, a considerable amount of pure compound 10 was required to be prepared. Synthesis of the target compound 10 was carried out according to the synthetic pathway depicted in Scheme 1. Thus, the commercially available 5-methoxyindole-2-carboxylic acid (1) was allowed to react with the nucleophile 5-methoxyindoline (2) [18] in the presence of coupling reagent ethyl-3-(3-dimethylaminopropyl)-carbodiimide hydrochloride (EDCI.HCl) in DCM at room temperature to furnish the amide 3 in good yield. Subsequent oxidation of indoline ring of 3 was accomplished using 2,3-dichloro-5,6-dicyanobenzoquinone (DDQ) in ethyl acetate [19] at reflux temperature to yield the di-indole derivative 4. Trials to reduce amide bond in 4 using LiAlH4 in THF or in diethyl ether led to cleavage of the amide bond. Reduction of the amide bond in compound 4 was successfully achieved using LiAlH4/AlCl3 (3/1) mixture in THF/diethyl ether solvent system at 0°C for one hour and then at room temperature for another one hour. Introduction of the aminoethyl side chains into positions 3 and 3′ of compound 5 via adopting our previously reported procedure [20] was unsuccessful. Briefly, compound 5 was subjected to Mannich reaction using dimethylamine and formaldehyde in glacial acetic acid produced the Mannich base 6. Subsequent quaternization of 6 with methyl iodide followed by substitution with potassium cyanide in the presence of dicyclohexyl[18]-crown[6] did not yield the anticipated compound 7 which might be reduced to its respective diamine derivative that could produce the target compound 10 upon acetylation. Accordingly, another strategy was adopted to synthesize 10. Thus, 2-nitroethyl acetate [21] was reacted with 5 in xylene at reflux temperature to yield the di-nitro derivative 8 which was catalytically hydrogenated in Parr shaker device at 4 mbar pressure to furnish compound 9. Acetylation of 9 using acetic anhydride and triethylamine in DCM produced the target compound 10. Assigned structures of the synthesized compounds were characterized by 1 H NMR, 13 C NMR, and MS spectral data whereas, purity was determined via microanalyses.

Synthetic pathway for preparation of compound 10. Reagents and conditions: i) EDCI.HCl, DCM, rt, 18h; ii) DDQ, ethyl acetate, reflux, 18h; iii) LiAlH4/AlCl3, THF/Et2O, 0°C-rt, 2h; iv) dimethyl amine, HCHO, CH3COOH; v) 1. MeI, CH2CL2, 2. KCN, dicyclohexyl[18]-crown[6], MeCN; vi) 2-nitroethyl acetate, tert. butyl catechol, xylene, reflux, 18h; vii) H2, Pd/C, 4 mbar, rt, 18h; viii) acetic anhydride, Et3N, DCM, rt, 18h.
Spectrofluorometric technique affords a higher sensitivity when compared with chromatographic ones. Both MLT and compound 10 exhibited native fluorescence in methanol with λ emission of 333 after excitation at 279 nm showing great similarity in their emission spectra. This fact hindered the direct determination of MLT in the presence of compound 10 (Figure 2).

Zero order excitation and emission spectra of 40 ng ml -1 of MLT and compound 10 in methanol.
In order to ascertain whether determination of MLT in the presence of compound 10 was feasible, the influence of the variables potentially affecting the fluorescence intensity or the position of the emission maxima was studied. The stability of the drug solutions was checked as a function of the preparation time and pH. The fluorescence intensity of MLT and compound 10 solutions maintained at 4°C was found not to vary within two weeks after preparation. Fluorescence intensity and fluorescence range of MLT and compound 10 were stable in pH ranges 2.8-11.2 as they are indole derivatives [22]. Also the effect of diluting solvent was checked and it was found that methanol and ethanol gave higher sensitivity than water. Fluorescence intensity for both MLT and compound 10 was stable for at least 2 h.
Second derivative method
By examining first and second-order derivative curves of both MLT and compound 10 emission spectra (Δλ = 10 nm) (Figures 3 and 4), it was clear that MLT can be determined solely by measuring peak amplitude of the second-order derivative at 324.0 nm where compound 10 gave zero response (zero- crossing point). The proposed method was validated according to ICH-guidelines [23] regarding linearity, sensitivity, accuracy, specificity, repeatability and reproducibility.

First order derivative emission spectra of 40 ng ml -1 of MLT (–) and compound 10 (…) in methanol using λ excitation at 279 nm.

Second order derivative emission spectra of 40 ng ml -1 of MLT (–) and compound 10 (…) in methanol using λ excitation at 279 nm.
Linearity and sensitivity
A linear correlation was obtained between peak amplitude of second derivative spectra at 324 nm and concentrations of MLT in a range of 20–220 ng ml-1 (Figure 5).

Second order derivative emission calibration spectra of MLT in methanol (20-220 ng ml -1 ) using λ excitation at 279 nm.
The regression equation was:

Where P is the peak amplitude of the second derivative spectrum of MLT at 324.0 nm, C is the concentration of MLT in ng ml-1 and r is the correlation coefficient. LOD and LOQ were calculated according to the following equations [23]:

Where, σ is the standard deviation of the intercept of regression line and S is the slope of regression line of the calibration curve. All results are shown in Table 2.
Accuracy
The accuracy of the proposed method was tested by analyzing triplicate samples of MLT solutions. The recovery % (mean ± SD) was 100.70 ± 1.772. These results revealed excellent accuracy (Table 2). The results obtained by applying the proposed method for determination of MLT in tablets were statistically compared with those results obtained by the reference spectrophotometric method [24]. It was concluded that with 95% confidence, there is no significant difference between them, since the calculated t and F values are less than the theoretical values [25] (Table 3).
Repeatability and reproducibility
Intra-assay precision was assessed by analyzing varying concentrations of MLT (40, 60 and 80 ng ml-1) in triplicate in one assay batch. The inter-assay precision was assessed by analyzing the same concentrations in triplicate on 3 successive days (Table 2). The average Recovery % around 100% and low SD indicates high accuracy and high precision of the proposed method, respectively.
Specificity
MLT was determined in laboratory prepared mixtures containing different percentages of compound 10. The recovery % (mean ± SD) of 101.09 ± 1.701 proved the high specificity of the proposed method for quantifying MLT in presence up to 60% of compound 10 (Table 4). Specificity was also investigated by observing any possible interferences from excepients in commercial MLT tablets, such as talc, magnesium stearate, dicalcium phosphate, and microcrystalline cellulose. These excipients did not interfere with the proposed method as indicated from the obtained good recovery values for the analysis of commercial MLT tablets (Table 3).
PCR and PLS chemometric methods
Two chemometric methods – PCR and PLS – were applied for the determination of MLT in the presence of compound 10. PCR and PLS methods involve the decomposition of the experimental data, such as spectrofluorimetric data in this case, into systematic variations (principal components or factors) that explain the observed variance in data. The purpose of both methods is to build a calibration model between the concentration of the analyte under study (MLT in our case) and the factors of the data matrix. The main difference between PLS and PCR methods is in the process of the decomposition of the experimental data. PCR performs the decomposition of data matrix into principal component without using the information about the analyte concentration. On the other hand, PLS performs the decomposition using both spectrum data matrix and analyte concentration [16]. The first step in the determination of MLT in presence of compound 10 by PCR and PLS methods, involves constructing the calibration matrix for the binary mixture. In this study calibration set was optimized with the aid of the multilevel multifacor design method [15]. Table 1 shows the composition of the 16 calibration samples. The emission spectra of these mixtures were collected and examined, the near zero fluorescence intensity after 380 nm accounted for the rejection of this part from the spectra. The selection of the optimum number of factors for the PCR and PLS methods was a very important pre-construction step: if the number of factors retained was more than required, more noise would be added to the data; if the number retained was too small, meaningful data that could be necessary for the calibration might be discarded. In this study, the leave-one out cross-validation method was used and the RMSECV values of different developed models were compared. Two factors were found suitable for both PCR and PLS methods. To validate the predictive ability of the suggested models, PCR and PLS methods were employed to predict the concentration of MLT in five laboratory-prepared mixtures (validation samples) containing different percentages of compound 10, where satisfactory results were obtained (Table 4). The predicted concentrations of the validation samples were plotted against the known concentrations to determine whether the model accounted for the concentration variation in the validation set. Plots were expected to fall on a straight line with a slope of 1 and zero intercept. MLT, in all samples, lay on a straight line and the equations of these lines were y = 0.994 x − 0.205 (r = 0.9995) for PCR and y = 0.997 x − 0.205 (r = 0.9995) for PLS. Both plots had a slope of almost 1 and an intercept close to zero. The proposed PCR and PLS methods were successfully used for the determination of MLT in commercial preparations (Table 3). The results obtained by applying PCR and PLS methods for determination of MLT in tablets were statistically compared with those results obtained by the reference spectrophotometric method [24]. It was concluded that with 95% confidence, there is no significant difference between them, since the calculated t and F values are less than the theoretical values [25] (Table 3).
Conclusion
In this study, simple and sensitive spectrofluorimetric methods were developed for the analysis of MLT in the presence of its main contaminant, compound 10. Reviewing the literature exposed that there are no reports for such analysis except a sophisticated HPLC/Ms/Ms method. Accordingly, compound 10 was synthesized in adequate quantities starting from 5-methoxyindole-2-carboxylic acid (1) to be used for the development of appropriate spectrofluorimetric methods for routine analysis of MLT in commercial preparations. The proposed spectrofluorimetric methods combine the rapidness and simplicity advantages of traditional spectrometric methods together with other important analytical merits, such as sensitivity and specificity. Moreover, simplicity was illustrated by the minimum requirement of chemicals and solvents since methanol was the only organic solvent used in the procedure. The suggested methods were validated and can be applied for routine quality control analysis of MLT commercial tablets without prior separation or interference from impurities/excipients.
References
Csernus V, Mess B: Biorhythms and pineal gland. Neuroendocrinol Lett 2003, 24:404–411.
Nosjean O, Ferro M, Coge F, Beauverger P, Henlin J-M, Lefoulon F, Fauchere J-L, Delagrange P, Canet E, Boutin JA: Identification of the Melatonin-binding SiteMT3 as the Quinone Reductase 2. J Biol Chem 2000, 275:31311–31317.
Genovese T, Mazzon E, Muia C, Bramanti P, De Sarro A, Cuzzocrea S: Attenuation in the evolution of experimental spinal cord trauma by treatment with melatonin. J Pineal Res 2005, 38:198–208.
Peres MFP: Melatonin, the pineal gland and their implications for headache disorders. Cephalalgia 2005, 25:403–411.
Witt-Enderby PA, Radio NM, Doctor JS, Davis VL: Therapeutic treatments potentially mediated by melatonin receptors: potential clinical uses in the prevention of osteoporosis, cancer and as an adjuvant therapy. J Pineal Res 2006, 41:297–305.
Blask DE, Sauer LA, Dauchy RT: Melatonin as a Chronobiotic/Anticancer Agent: Cellular, Biochemical, and Molecular Mechanisms of Action and their Implications for Circadian-Based Cancer Therapy. Curr Top Med Chem 2002, 2:113–132.
Mills E, Wu P, Seely D, Guyatt G: Melatonin in the treatment of cancer: a systematic review of randomized controlled trials and meta-analysis. J Pineal Res 2005, 39:360–366.
Sofic E, Rimpapa Z, Kundurovic Z, Sapcanin A, Tahirovic I, Rustembegovic A, Cao G: Antioxidant capacity of the neurohormone melatonin. J Neural Transm 2005, 112:349–358.
Swygert LA, Back EE, Auerbach SB, Sewell LE, Falk H: Oesinophilia-myalgia syndrome case-associated contaminants in commercially available 5-hydroxytryptophane. J Rheumatol 1993, 20:1711–1717.
Lissoni P, Ardizzoia A, Tisi E, Rossini F, Barni S, Tancici G, Conti A, Maestroni GJ: Amplification of eosinophilia by melatonin during the immunotherapy of cancer with interleukin-2. J Biol Regul Homeost Agents 1993, 7:34–36.
Lissoni P, Barni S, Tancini G, Rovelli F, Ardizzoia A, Conti A, Maestroni GJ: A study of the mechanisms involved in the immunostimulatory action of the pineal hormone in cancer patients. Oncology 1993, 50:399–402.
Williamson BL, Tomlinson AJ, Mishra PK, Gleich GJ, Naylor S: Structural Characterization of Contaminants Found in Commercial Preparations of Melatonin: Similarities to Case-Related Compounds from l-Tryptophan Associated with Eosinophilia-Myalgia Syndrome. Chem Res Toxicol 1998, 11:234–240.
Murillo JA, García LF: Application of First Derivative Fluorescence Spectrometry to the Simultaneous Determination of Paracetamol and Salicylamide in Pharmaceuticals. Anal Lett 1996, 29:423–438.
Ni Y, Gong X: Simultaneous spectrophotometric determination of mixtures of food colorants. Anal Chem Acta 1997, 354:163–171.
Brereton RG: Multilevel multifactor designs for multivariate calibration. Analyst 1997, 122:1521–1529.
Kramer R: Chemometric techniques for quantitative analysis. Marcel Dekker Inc, New York; 1998.
Haland DM, Thomas EV: Partial least-squares methods for spectral analyses. 2. Application to simulated and glass spectral data. Anal Chem 1988, 60:1202–1208.
Gangjee A, Vasudevan A, Queener SF: Synthesis and Biological Evaluation of Nonclassical 2,4-Diamino-5-methylpyrido[2,3-d]pyrimidines with Novel Side Chain Substituents as Potential Inhibitors of Dihydrofolate Reductases. J Med Chem 1997, 40:479–485.
Lakatosh SA, Luzikov YN, Preobrazhenskaya MN: Synthesis of 6H-pyrrolo[3′,4′:2,3] [1,4]diazepino[6,7,1-hi]indole-8,10(7H,9H)-diones using 3-bromo-4-(indol-1-yl)maleimide scaffold. Org Biomol Chem 2003, 1:826–833.
Attia MI, Witt-Enderby PA, Julius J: Synthesis and pharmacological evaluation of pentacyclic 6a,7-dihydrodiindole and 2,3-dihydrodiindole derivatives as novel melatoninergic ligands. Bioorg Med Chem 2008, 16:7654–7661.
Bartoli G, Bosco M, Dalpozzo R, Marcantoni E, Massaccesi M, Sambri L: Zn(ClO4)2·6H2O as a Powerful Catalyst for a Practical Acylation of Alcohols with Acid Anhydrides. Eur J Org Chem 2003, 2003:4611–4617.
Sorouraddin MH, Rashidi MR, Kalhor EG, Zeynali KA: Simultaneous spectrofluorimetric and spectrophotometric determination of melatonin and pyridoxine in pharmaceutical preparations by multivariate calibration methods. Il Farmaco 2005, 60:451–458.
ICH: Validation of Analytical procedures: Methodology (Q2AR1). International Conference on Harmonization, Food and Drug Administration, USA; . November 1996 and November 2005
The Merck Index, An Encyclopedia of Chemicals, Drugs and Biologicals In . 12th edition. ,; 1996:. 5860
Hinchen JD: Practical statistics for chemical Research. 1st edition. , London; 1969.
Acknowledgement
The authors thank the Deanship of Scientific Research and the Research Center of the College of Pharmacy, King Saud University for supporting this study. Authors appreciate the effort of PD Dr. D. P. Zlotos, Pharmaceuitical Institute, University of Würzburg for offering the opportunity to conduct synthesis of compound 10.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
HD designed the proposed analytical method, carried out the analytical experimental work, analyzed the data statistically, participated in the results and discussion, and wrote the analytical part of the manuscript. MA proposed, planned the work, synthesized compound 10, participated in the results and discussion, and wrote the synthesis part of the manuscript. All authors read and approved the final manuscript.
An Erratum for this chapter can be found at http://dx.doi.org/10.1186/s13065-016-0203-y
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 2.0 International License (https://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Darwish, H.W., Attia, M.I. & Zlotos, D.P. New spectrofluorimetric methods for determination of melatonin in the presence of N-{2-[1-({3-[2-(acetylamino)ethyl]-5-methoxy-1H-indol-2-yl}methyl)-5-methoxy-1H-indol-3-yl]- ethyl}acetamide: a contaminant in commercial melatonin preparations. Chemistry Central Journal 6, 412 (2012). https://doi.org/10.1186/1752-153X-6-36
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1752-153X-6-36