
Abstract
Background
We recently described Hi-Plex, a highly multiplexed PCR-based target-enrichment system for massively parallel sequencing (MPS), which allows the uniform definition of library size so that subsequent paired-end sequencing can achieve complete overlap of read pairs. Variant calling from Hi-Plex-derived datasets can thus rely on the identification of variants appearing in both reads of read-pairs, permitting stringent filtering of sequencing chemistry-induced errors. These principles underly ROVER software (derived from Read Overlap PCR-MPS variant caller), which we have recently used to report the screening for genetic mutations in the breast cancer predisposition gene PALB2. Here, we describe the algorithms underlying ROVER and its usage.
Results
ROVER enables users to quickly and accurately identify genetic variants from PCR-targeted, overlapping paired-end MPS datasets. The open-source availability of the software and threshold tailorability enables broad access for a range of PCR-MPS users.
Methods
ROVER is implemented in Python and runs on all popular POSIX-like operating systems (Linux, OS X). The software accepts a tab-delimited text file listing the coordinates of the target-specific primers used for targeted enrichment based on a specified genome-build. It also accepts aligned sequence files resulting from mapping to the same genome-build. ROVER identifies the amplicon a given read-pair represents and removes the primer sequences by using the mapping co-ordinates and primer co-ordinates. It considers overlapping read-pairs with respect to primer-intervening sequence. Only when a variant is observed in both reads of a read-pair does the signal contribute to a tally of read-pairs containing or not containing the variant. A user-defined threshold informs the minimum number of, and proportion of, read-pairs a variant must be observed in for a ‘call’ to be made. ROVER also reports the depth of coverage across amplicons to facilitate the identification of any regions that may require further screening.
Conclusions
ROVER can facilitate rapid and accurate genetic variant calling for a broad range of PCR-MPS users.
Similar content being viewed by others

Background
Targeted massively parallel sequencing (MPS) offers significantly increased efficiencies compared with prior technologies such as Sanger sequencing and High-Resolution Melting curve analysis (HRM) for diverse genetic analysis applications [1, 2]. Such applications include disease gene discovery and mutation screening, and clinical diagnostic gene-panel testing [3, 4]. Among PCR-based approaches for MPS, we recently described a single-reaction, highly-multiplexed PCR-MPS system called Hi-Plex, designed to enable rapid and cost-effective targeted sequencing [5, 6]. Among the advantages offered by this system is the ability to define the target library size so that contemporary paired-end MPS sequencing protocols can readily result in both members of a read-pair completely covering the primer-intervening sequence of the relevant amplicon. Consequently, high-stringency filtering of sequencing chemistry errors is possible by comparing both reads of a pair over the entire read-pair length. Conceptually, this approach to PCR-MPS data handling is attractive irrespective of the upstream molecular biology. Low-level residual system noise resulting from PCR polymerase error could readily be accounted for by classical duplicate testing of specimens. This will be very useful in cases where genetic variants are present in a relatively small fraction of the whole sample, as is often the case for somatic mutations in tumour biopsies, for example.
Contemporary approaches to handling PCR-MPS data include the application of tools to ‘trim’ primer or adapter sequences or ‘merge’ read-pairs to yield ‘consensus’ read representations. For example, Trimmomatic can be used to detect and remove adapter sequences or other specified sequences at the beginning and end of a read, but discards the complementary read of the read-pair [7]. Identification of sequence to be trimmed based on anticipated adapter and/or other oligonucleotide sequences can be problematic because mismatches occur commonly due to oligonucleotide synthesis and sequencing chemistry errors. SeqPrep merges paired-end Illumina sequencing chemistry reads that are overlapping into a single longer read. Sequence identity thresholds are used to determine whether adapter sequences are present and whether read pairs overlap [8]. Trimming of reads is supported and in the context of reads-overlap having been identified, a single ‘consensus’ representation of the read-pair is produced. The accuracy of this approach depends on the relative quality measures attributed to the two reads of a pair. Incorrect consensus ‘reads’ will be derived in situations where sequencing errors are attributed with relatively high quality scores and/or the corresponding positions of the non-erroneous reads receive relatively low quality scores.
In settings where accuracy is a high priority, we reasoned that an approach that requires agreement between both reads of a read-pair for a given position before the read-pair is allowed to contribute to variant-calling, would be desirable. Furthermore, by using co-ordinates information relating to primers used in PCR-MPS applications, read-trimming should theoretically be possible without the confounding influences of errors introduced during primer synthesis or sequencing. To realise these design features, we developed ROVER software which enables highly-accurate detection of genetic variants in PCR-MPS datasets by applying filtering based on completely overlapping reads in read-pairs. Amplicon library insert size should be shorter than the length of a sequencing read for compatibility with this software. Previously, we reported the use of ROVER in the context of screening for genetic mutations in the breast cancer predisposition gene, PALB2 [GenBank reference sequence NM_024675; MIM#610355] [9]. Here, we describe its mode of action and usage.
Implementation
ROVER is implemented in Python 2.7 and is intended to be used as a command-line application. Its two key inputs are 1) a tab-delimited file of DNA co-ordinates specifying the locations of target amplicon regions, relative to a reference sequence, in standard BED format [10]; 2) one or more files containing reads aligned to the reference sequence in the standard BAM format [11]. Its main output is a file containing a list of variants called for each input BAM file in tab-delimited format compatible with widely-used variant annotation tools such as ANNOVAR [12]. It also accepts two threshold arguments which are used to filter the list of resulting variants as described below. The types of variants considered by ROVER are single nucleotide variants (SNVs) and small insertions and deletions (indels).
The pseudocode in Figure 1 illustrates the algorithm employed by ROVER to compute variants for a single input BAM file. The algorithm is applied to each input BAM file and the results are accumulated and then saved to the output file.

Pseudocode illustrating the main variant calling algorithm employed by ROVER.
The algorithm is realised by the GET_VARIANTS function which takes four parameters: 1) the input BAM file; 2) the list of amplicon co-ordinates; 3) the absolute threshold for the number of read-pairs required for a variant to be kept; 4) the proportion threshold for the number of read-pairs required for the variant to be kept. It returns as its result a set of variants which are called for this input BAM file.
Each amplicon is processed separately by the loop from line 4 to line 29. All the read-pairs which overlap the amplicon are retrieved from the input BAM file (line 8). In practice, the fraction that a read must overlap an amplicon can be tailored by a command-line argument, defaulting to 0.9. It is important to note that we only consider reads which are part of a mapped pair. For each pair of reads, we compute the set of variants which appear in both reads (lines 9 to 15). We then discard any variants which appear on both reads but are not within the bounds of the amplicon (lines 17 to 24) (see Figure 2). For each variant which appears within the amplicon bounds, we also count the number of times that variant is seen in all the pairs which overlap the amplicon (lines 21 and 23). We then discard any variants which do not meet the threshold requirements (lines 26 to 29). The first threshold is the absolute count of read-pairs for a variant in a given amplicon. We must see a variant in at least this many read-pairs overlapping the amplicon for it to be kept in the output. The second threshold is the proportion of read-pairs containing a variant in a given amplicon. We must see a variant in at least this proportion of all the read-pairs overlapping the amplicon for it to be kept in the output. Variants which pass both of the thresholds are added to the overall set of called variants (line 29). After processing all the amplicons, the set of all called variants is returned as the result (line 31). An optional input argument allows the user to dictate that only when base changes or insertions are associated with quality scores in both reads above a selectable threshold, will a read-pair be considered in variant calling.

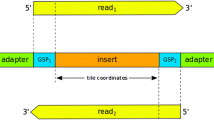
Illustration of the ROVER principles. The lower panel represents contiguous sections of a reference sequence (e.g. a specific human genome build). Numbered ‘F’ and ‘R’ pairs denote primer co-ordinates used in conjunction with mapped read starting co-ordinates to guide clipping (dotted boxes). Shaded boxes represent regions under analysis. Arrows represent reads of read-pairs. ‘X’s denote positions different from the reference sequence. Only when both reads of a read-pair concur can the read-pair contribute to genetic variant calling for a given position (circled). This contribution to a read-pair pile-up allows variant calling using thresholds for minimum number of read-pairs and the proportion of read-pairs containing the variant.
To compute the variants in a particular read we compare the aligned nucleotide bases of the read with the reference sequence. The so-called CIGAR string of mapped reads, given in the BAM file for each read, is used to relate the bases of the read to the positions of the reference sequence. Additionally, the bases from the reference sequence are deduced from the MD field of the BAM file. This allows ROVER to recover the reference sequence just from the BAM file without needing the original reference file to be specified. ROVER uses the PySam library for manipulating BAM files [13].
Results and discussion
ROVER was developed and released as open source software. ROVER performs trimming of primer sequences based on a co-ordinates file, avoiding problems associated with sequence matching in the context of errors introduced during primer synthesis, polymerase activity and sequence detection. The software considers overlapping read-pairs (as far as the start of distal primer-corresponding sequence for each read of a read-pair) and only allows read-pair concordant variants to be used for variant calling. ROVER allows thresholds for read-pair depth and read-pair proportion to be modified. The software also produces a report of mapped read-pair numbers for each amplicon, allowing the user to monitor coverage.
Using a BAM file consisting of 52,592 reads of 150 bases, derived from a Hi-Plex experiment targeting 60 amplicons,[9], ROVER took 7 seconds and required 14 MB of memory to call the variants and compute the depth of coverage across amplicons, on an Apple Mac Pro with two 2.66 GHz 6-Core processor and 64 GB RAM running OS 10.8.4. In instances where multiple BAM files are analysed, the software can be run as follows (italics indicate example parameter values):
rover –primers hiplex_primers.txt –log logfile.txt –out variant_calls.tsv
- proportionthresh 0.15 –absthresh 2 –qualthresh 20 –coverdir coverage_dir
sampleX.bam sampleY.bam sampleZ.bam
In the era of MPS-based high-throughput sequencing both in research and clinical settings, one of the biggest challenges is the generation of false-positive signals. The requirement by ROVER for read-pair concordance essentially restricts chemistry-induced error to polymerase misincorporation during PCR. Using germline DNA as input material, this source of error can readily be filtered out by application of depth and proportion of read-pairs thresholds, since many copies of PCR template (typically hundreds or thousands) are generally present at the outset of a PCR reaction and it is highly unlikely for a significant proportion of early amplification events to result in identical polymerase errors. In more challenging settings, for example the identification of rare somatic genetic variants in tumour biopsies, duplicate testing in conjunction with ROVER will afford highly accurate variant calling. Other sources of error, such as alignment errors arising from segmental duplication, can be accounted for by comparison across specimens treated using the same chemistry and bioinformatic analysis pipeline, using principles we have described previously [14].
As reported in [9], application of ROVER to a high-throughput (96-well format) Hi-Plex-based dataset enabled rapid and accurate identification of genetic variants in the coding regions of PALB2. In this blinded study, 99.93% (5,696/5,700) of amplicons were represented at ≥10× coverage, across samples. We accurately detected all 56 variant calls identified through previous mutation screening and genotyping. Heterozygous variants were observed in 37.23% (35/94) to 62.33% (513/823) of read-pairs (median = 51.23%). No false positive calls were assigned.
Conclusions
ROVER is a freely available open-source tool that enables accurate genetic variant calling from paired-end PCR-MPS datasets in which reads completely overlap for the regions under analysis.
Availability and requirements
Project name: ROVER
Project home page: https://github.com/bjpop/rover
Operating systems: POSIX-like operating systems (OS X, Linux)
Programming language: Python
Other requirements: PySam library
License: BSD
Any restrictions to use by non-academic: None
Abbreviations
- MPS:
-
Massively parallel sequencing; derived from Read Overlap PCR-MPS variant caller, ROVER software
- HRM:
-
High Resolution Melting-curve analysis
- IGV:
-
Integrative genome viewer.
References
Mardis ER: The impact of next-generation sequencing technology on genetics. Trends Genet. 2008, 24: 133-141. 10.1016/j.tig.2007.12.007.
Moorthie S, Mattocks CJ, Wright CF: Review of massively parallel DNA sequencing technologies. The HUGO Journal. 2011, 5: 1-12.
Morgan JE, Carr IM, Sheridan E, Chu CE, Hayward B, Camm N, Lindsay HA, Mattocks CJ, Markham AF, Bonthron DT, Taylor GR: Genetic diagnosis of familial breast cancer using clonal sequencing. Hum Mutat. 2010, 31: 484-491. 10.1002/humu.21216.
Meldrum C, Doyle MA, Tothill RW: Next-generation sequencing for cancer diagnostics: a practical perspective. Clin Biochem Rev. 2011, 32: 177-195.
Nguyen-Dumont T, Pope BJ, Hammet F, Southey MC, Park DJ: A high-plex PCR approach for massively parallel sequencing. Biotechniques. 2013, 55: 69-74.
Nguyen-Dumont T, Pope BJ, Hammet F, Mahmoodi M, Tsimiklis H, Southey MC, Park DJ: Cross-platform compatibility of Hi-Plex, a streamlined approach for targeted massively parallel sequencing. Anal Biochem. 2013, 442 (2): 127-129. 10.1016/j.ab.2013.07.046.
Trimmomatic: a flexible read trimming tool for Illumina NGS data.http://www.usadellab.org/cms/index.php?page=trimmomatic.
SeqPrep.https://github.com/jstjohn/SeqPrep.
Nguyen-Dumont T, Teo ZL, Pope BJ, Hammet F, Mahmoodi M, Tsimiklis H, Sabbaghian N, Tischkowitz M, Foulkes WD, Giles GG, et al: Hi-Plex for high-throughput mutation screening: application to the breast cancer susceptibility gene PALB2. BMC Med Genomics. 2013, 6: 48-10.1186/1755-8794-6-48.
Quinlan AR, Hall IM: BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010, 26: 841-842. 10.1093/bioinformatics/btq033.
Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup: The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009, 25: 2078-2079. 10.1093/bioinformatics/btp352.
Wang K, Li M, Hakonarson H: ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010, 38: e164-10.1093/nar/gkq603.
PySam: Python interface for the SAM/BAM sequence alignment and mapping format.http://code.google.com/p/pysam/.
Pope BJ, Nguyen-Dumont T, Odefrey F, Hammet F, Bell R, Tao K, Tavtigian SV, Goldgar DE, Lonie A, Southey MC, Park DJ: FAVR (Filtering and Annotation of Variants that are Rare): methods to facilitate the analysis of rare germline genetic variants from massively parallel sequencing datasets. BMC Bioinforma. 2013, 14: 65-10.1186/1471-2105-14-65.
Acknowledgements
This work was supported by the Australian National Health and Medical Research Council (APP1025879 and APP1029974), the National Institute of Health, USA (RO1CA155767) and by a Victorian Life Sciences Computation Initiative (VLSCI) grant (number VR0182) on its Peak Computing Facility at the University of Melbourne, an initiative of the Victorian Government. TN-D is a Susan G. Komen for the Cure Postdoctoral Fellow.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
BJP and DJP conceived the design of the software. BJP performed the coding. FH and TN-D conducted testing of the software. BJP, TN-D, FH and DJP contributed to the writing of the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Pope, B.J., Nguyen-Dumont, T., Hammet, F. et al. ROVER variant caller: read-pair overlap considerate variant-calling software applied to PCR-based massively parallel sequencing datasets. Source Code Biol Med 9, 3 (2014). https://doi.org/10.1186/1751-0473-9-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1751-0473-9-3