
Abstract
The diagnostic guidelines of Alzheimer’s disease (AD) have recently been updated to include brain imaging and cerebrospinal fluid (CSF) biomarkers, with the aim of increasing the certainty of whether a patient has an ongoing AD neuropathologic process or not. The CSF biomarkers total tau (T-tau), hyperphosphorylated tau (P-tau) and the 42 amino acid isoform of amyloid β (Aβ42) reflect the core pathologic features of AD, which are neuronal loss, intracellular neurofibrillary tangles and extracellular senile plaques. Since the pathologic processes of AD start decades before the first symptoms, these biomarkers may provide means of early disease detection. The updated guidelines identify three different stages of AD: preclinical AD, mild cognitive impairment (MCI) due to AD and AD with dementia. In this review, we aim to summarize the CSF biomarker data available for each of these stages. We also review results from blood biomarker studies. In summary, the core AD CSF biomarkers have high diagnostic accuracy both for AD with dementia and to predict incipient AD (MCI due to AD). Longitudinal studies on healthy elderly and recent cross-sectional studies on patients with dominantly inherited AD mutations have also found biomarker changes in cognitively normal at-risk individuals. This will be important if disease-modifying treatment becomes available, given that treatment will probably be most effective early in the disease. An important prerequisite for this is trustworthy analyses. Since measurements vary between studies and laboratories, standardization of analytical as well as pre-analytical procedures will be essential. This process is already initiated. Apart from filling diagnostic roles, biomarkers may also be utilized for prognosis, disease progression, development of new treatments, monitoring treatment effects and for increasing the knowledge about pathologic processes coupled to the disease. Hence, the search for new biomarkers continues. Several candidate biomarkers have been found in CSF, and although biomarkers in blood have been harder to find, some recent studies have presented encouraging results. But before drawing any major conclusions, these results need to be verified in independent studies.
Similar content being viewed by others

Introduction
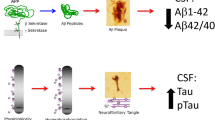
The diagnosis of Alzheimer’s disease (AD) can only be made with certainty after a patient has deceased, by histological examination of brain tissue at autopsy or, rarely, following brain biopsy. Histopathology should reveal evidence of the characteristic hallmarks of AD, which are extracellular accumulations of amyloid β (Aβ) in senile plaques and intracellular neurofibrillary tangles of hyperphosphorylated tau (P-tau) [1]. These hallmarks must be seen in sufficient numbers to confer a pathological diagnosis. This fits with the idea of a gradual progression and continuum of pathology and biomarker changes. Multiple pathologies may also be present, and the level of each can be critical.
In 1984, a working group established by the National Institute of Neurological and Communicative disorders and Stroke and the Alzheimer’s Disease and Related Disorders Association created clinical diagnostic guidelines of probable AD [2], which were validated against neuropathological diagnosis with a sensitivity and specificity of around 80 and 70%, respectively [3]. However, the criteria only allowed for making a diagnosis in the dementia stage of the disease, i.e., when the patients were so severely affected by the disease process that they could not manage daily functioning in respect to intellectual and social abilities. Due to advances in research a revision of the guidelines was made by a new working group from the National Institute of Aging in 2011 [4]. Now, imaging and cerebrospinal fluid (CSF) biomarkers have been implemented to provide evidence of an ongoing AD pathophysiological process and it is also possible to make a pre-dementia diagnosis of AD. The included CSF biomarkers are the total amount of tau (T-tau), which reflects the intensity of neuroaxonal degeneration, P-tau, which may correlate with tangle pathology, and the 42 amino acid isoform of Aβ (Aβ42), which correlates inversely with plaque pathology [5]. It is well known that the pathological processes in the brains of AD patients start more than a decade before the first symptoms are noticed [6]. The temporal dynamics of biomarker levels in relation to changes in cognition have been described in a hypothetical model on the continuum of AD [7]. In line with this, the revised diagnostic guidelines identify three different stages of AD: preclinical AD, mild cognitive impairment (MCI) due to AD and AD with dementia. In this review, we will describe the CSF biomarker characteristics of these three stages, and also give an update on the available data for biomarkers in blood. Imaging biomarkers will not be covered.
Biomarkers in preclinical AD
The working group of the National Institute of Aging was posed with the task of how to define the preclinical phase of AD [8]. They subdivided this phase into three separate stages. In the first stage, patients have amyloid pathology, as defined by positive amyloid on positron emission tomography (PET) imaging or low CSF Aβ42, but no signs of neuronal degeneration (normal volumetric magnetic resonance imaging [MRI] of the brain, normal CSF T-tau levels). In the second stage, neuronal degeneration is evident and patients have elevated CSF T-tau or signs of neuronal injury on imaging methods in addition to positive amyloid markers. To qualify in the third stage, patients have to experience subtle cognitive deficits, although not severe enough to meet the criteria for MCI. To investigate the practical applicability of these guidelines, Jack et al. applied them to a set of cognitively normal individuals from a population-based sample [9]. Only imaging, not fluid, biomarkers were used in this study. After evaluating the results, they found the need for two additional stage categories: stage 0 and Suspected non-AD pathophysiology (SNAP). Stage 0 denotes individuals with no pathological AD biomarkers and no signs of subtle cognitive impairment. Subjects with SNAP have normal amyloid biomarkers, but display signs of neurodegeneration, and could be in the preclinical stage of a non-AD neurodegenerative process. The proposed criteria are so far intended for research purposes only, and can provide a common ground to facilitate the search for biomarkers for preclinical AD. If a disease-modifying treatment for AD is found, it will most likely have the greatest effect if administered in the early disease stages [10], which underlines the importance of the work executed by this working group.
Both cross-sectional and longitudinal studies have evaluated the association between the core CSF AD biomarkers and preclinical AD. Their findings are summarized in Tables 1 and 2. The cross-sectional studies have included patients carrying familial AD (FAD)-causing mutations and determined biomarker levels long before expected disease onset. Bateman et al. evaluated biomarkers levels in subjects at risk for carrying an autosomal dominant AD mutation [11]. The expected age of symptom onset was calculated from the participants’ parents’ age at onset of AD symptoms. Linear models were then created, showing the biomarker levels as a function of time to expected onset of symptoms in mutation carriers and non-carriers. Mutation carriers had significantly elevated levels of CSF T-tau and plasma Aβ42 15 years before expected symptom onset. CSF levels of Aβ42 were significantly reduced in mutation carriers 10 years before expected onset of symptoms. Another study that included members of the Colombian Alzheimer’s Preventive Initiative Registry found that CSF and plasma Aβ42 levels in young individuals carrying a specific presenilin 1 (PS1) mutation were significantly increased compared to non-carriers more than two decades before estimated MCI onset [12]. Studies on cognitively normal mutation carriers that were closer to the expected onset of AD have found decreased levels of Aβ42 [13, 14], increased levels of T-tau and P-tau [13, 15], and reduced Aβ42:40 ratio in CSF or increased plasma Aβ42 levels [15] compared with controls. These studies are thought to constitute models that are applicable to patients with sporadic AD as well.
Longitudinal studies have correlated baseline levels of the core biomarkers in cognitively normal individuals with decrease in cognitive function or development of MCI or AD. CSF levels of Aβ42 alone [16] or in combination with T-tau [17] or P-tau [18] have been associated with future development of cognitive impairment in individuals that were cognitively normal at the time of LP. In another study, healthy older adults were examined and lumbar punctured at baseline and after a four year follow-up [19]. It was found that low levels of Aβ42 at follow-up were associated with worse performance on cognitive tests. Decreasing levels of Aβ42 and increasing levels of P-tau were also associated with worse cognitive performance. One study including healthy controls from the Alzheimer Disease Neuroimaging Initiative (ADNI) showed that individuals with low CSF Aβ42 levels displayed significantly higher rates of brain atrophy over one year than individuals with higher Aβ42 levels [23]. Increased T-tau:Aβ42 ratio [20] or low levels of CSF Aβ42 [21] have been found to predict conversion to MCI in cognitively normal or AD in non-demented elder individuals, respectively. One study on patients with subjective complaints found that CSF Aβ42 alone was superior to T-Tau, P-tau or a combination of all three biomarkers in predicting progression to MCI or AD [22]. However, there are yet no prospective long-term studies that have investigated whether CSF biomarkers can predict development of AD in healthy elderly individuals within 10–20 years. Such studies will be vital to be able to determine whether the majority of healthy individuals with low CSF Aβ42 (alone or in combination with tau) will indeed develop AD dementia within 1–2 decades.
In this context, it is important to remember that several of the longitudinal studies mentioned above are in fact cross-sectional in respect to the biomarker data, with one set of baseline biomarker measures correlated to longitudinal clinical data. This is not the best study design to examine temporal dynamics in biomarker changes; the sensitivity of the analytical techniques may determine when a biomarker appears positive. A very sensitive method for one marker as compared to a less sensitive method for another marker may give a false imprecision that one pathological change precedes the other. Therefore, we now must validate the hypotheses and models mentioned above in true longitudinal studies with repeated biomarker measurements and clinical assessments over time in the same individuals.
Biomarkers in MCI
The risk for patients with MCI to develop AD during a 4.5-year follow-up has been found to be roughly tripled compared to cognitively healthy controls [24]. However, MCI is a heterogeneous disorder, and people suffering from it may progress to other dementias than AD, including vascular dementia (VAD), frontotemporal dementia (FTD) and Lewy body dementia (DLB), or remain relatively stable and decline cognitively as in normal aging [25]. The combined pattern of low levels of Aβ42 together with high levels of T-tau and P-tau in CSF can accurately discriminate incipient AD from patients with stable MCI [26]. Since the annual progression rate from MCI to AD is around 10-15% [27], it is important to have long follow up periods to identify late converters. One large study found that CSF levels of Aβ42, T-tau and P-tau among MCI patients could predict progression to AD with good accuracy after a median follow-up period of 5.2 years [28]. The combination of T-tau and Aβ42 gave a sensitivity of 95% and a specificity of 83%. The study population was evaluated again after an extended follow-up, now with a median of 9.2 years since baseline [29]. An additional set of patients from the MCI group had now converted to AD. The ratio of Aβ42:P-tau at baseline predicted development of AD within 9.2 years with sensitivity and specificity around 85-90%. Patients who converted within 5 years had significantly higher levels of T-tau and P-tau than the patients that converted within 5–10 years, while the level of Aβ42 was similar. A limitation of that particular study was however that the cutoff levels for the CSF biomarkers were established in the same cohort of patients that was then used to evaluate the diagnostic performance of the biomarkers to detect incipient AD. In some other studies, the cutoffs for Aβ42, T-tau and P-tau have been determined when comparing controls with patients with AD and then these cut offs have been applied in cohorts with MCI. Using this approach Hertze et al. found that different combinations of Aβ42, T-tau and P-tau could predict AD over 5 years with a sensitivity of 85-90% and specificity of 71-82% [30]. In a cohort from the Development of screening guidelines and criteria for predementia Alzheimer’s disease (DESCRIPA) study, 79% of the patients with amnestic MCI had an abnormal CSF Aβ42:T-tau ratio, and this profile was associated with progression to AD dementia [31]. A baseline AD-like profile of CSF T-tau:Aβ42 was present in 89% of MCI patients from ADNI that developed AD within one year [32].
A large multicenter study tested the ability of the core CSF AD biomarkers to predict incipient AD in patients with MCI [33]. The combination of Aβ42:P-tau ratio and T-Tau rendered a sensitivity of 83% and a specificity of 72% for progression to AD. The somewhat low specificity in this study may be caused by the short follow-up time of only around 2 years. Also, the biomarker cutoffs were determined in an independent cohort. This gives a lower, but truer, diagnostic accuracy. Further, lack of standardization of pre-analytical and analytical conditions between the included centers may have contributed.
The potential of the core biomarkers under standardized settings has proven to be very good. Their diagnostic performance in a homogeneous mono-center population was excellent, achieving an area under the receiver operating curve of 0.97 in the discrimination of patients with AD or patients with MCI who converted to AD against stable MCI and controls [34].
Indeed, for the biomarkers to be used clinically, standardization of pre-analytical and analytical factors is needed. This requires identification of these factors followed by harmonization between laboratories. The Alzheimer’s Association quality control program was created with the purpose of facilitating the worldwide use of CSF AD biomarkers in the clinic and research and is open for any laboratory that uses a commercially available assay for CSF Aβ42, T-tau or P-tau [35]. In the test rounds that have taken place so far, the coefficient of variation among the contributing labs has been around 15-25% [36]. Currently, only monitoring is conducted, but further on, active interventions will be crucial. To that end, several standardization efforts are in progress, e.g., the Global Biomarker Standardization Consortium, the CSF-Proteins Working Group of International Federation of Clinical Chemistry and Laboratory Medicine and the BIOMARKAPD project of the EU Joint Programme in Neurodegenerative Disease Research [36].
Biomarkers in AD with dementia
CSF levels of T-tau are about 300% higher in AD patients than control subjects [37]. The biomarker is not specific for AD and increased levels can be found in the CSF of patients who have suffered from acute stroke [38] or head trauma [39], and very high levels are seen in patients with Creutzfelt-Jakob disease [40]. Since it is a non-specific marker for axonal and neuronal degeneration, non-AD dementias may also give elevated CSF T-tau levels [41]. In the differential diagnostics against other dementias, P-tau may have a greater value since it is more AD-specific [42]. Elevated P-tau levels have been found in patients with AD compared with patients suffering from FTD and VAD [43], DLB [43, 44] and Parkinson disease with dementia [44]. P-tau differentiated between patients with AD and patients with FTD and DLB, with specificities of 92 and 64%, respectively [43]. However, a large study on patients with AD as well as DLB, FTD and VAD showed that an AD biomarker profile was present in a substantial part of the non-AD patients [45]. Potential reasons for this overlap may be misdiagnosis or the presence of mixed dementias. Future autopsy studies will hopefully shed light on this issue.
The levels of P-tau in CSF ante mortem correlate with the amount of neurofibrillary tangles and hyperphosphorylated tau in the brain post mortem[46, 47]. A study of cortical biopsies from living patients with normal pressure hydrocephalus showed a correlation between the amount of hyperphosphorylated tau in the biopsies and P-tau levels in CSF [48]. The same study also found that presence of cortical amyloid plaques was associated with lower levels of CSF Aβ42. The lower CSF levels of Aβ42 in AD patients compared with controls are believed to be caused by its accumulation in senile plaques. Indeed, autopsy studies have reported correlations between plaque load and reduced Aβ42 in lumbar CSF ante mortem[46] and ventricular CSF post mortem[49]. Using positron emission tomography several studies found an inverse correlation between brain amyloid load and CSF Aβ42 [50–52].
The combination of these biomarkers can accurately distinguish AD patients from controls, with sensitivity and specificity over 80% [26, 41]. One study that examined the performance of these biomarkers in AD patients in different ages found that although diagnostic accuracy decreased somewhat with age, the positive and negative predictive values of the biomarkers combined were stable enough for the biomarkers to be used in old patients [53]. Their longitudinal stability in AD patients has also been evaluated. Most studies have found that levels of Aβ42 and P-tau remain unaltered over time [54–57] while available data for T-tau is inconclusive [58]. Some studies report no temporal alteration in T-tau levels in AD patients, while others have found an increase. However, high levels of T-tau and P-tau seems to be associated with increased disease intensity and a more rapid disease progression [59–61].
Measurement of core AD biomarkers
There are various assays for the three core AD CSF biomarkers and it is important to bear in mind what they actually measure. One of the most common assays for Aβ42 is an enzyme-linked immunosorbent assay (ELISA) specifically constructed to measure Aβ containing both the 1st and 42nd amino acids of the protein [62], but there are also assays that are C-terminally end-specific but use N-terminal antibodies that allow for measurement of N-terminally truncated Aβ42 fragments in addition to Aβ1-42 [63]. Most data suggest that these assays measure free monomeric Aβ42 but they correlate well with a selected reaction monitoring-based mass spectrometry method for total Aβ42 [64]. Tau exists in several isoforms and may be phosphorylated at different residues. The most common ELISA for T-tau detects all isoforms of tau, independently of phosphorylation state [65]. The two most common ELISAs for P-tau measure tau that is phosphorylated at residue 181 or 231 [66][67][68], and the diagnostic performance is similar between these assays [43]. A multiparameter assay based on xMAP® technology was developed for simultaneous measurement of Aβ42, T-tau and P-tau [68]. Although it gives different absolute values than the corresponding ELISAs, correction factors can be used for direct comparisons.
CSF and imaging biomarkers combined
Even though imaging biomarkers are beyond the scope of this review we would like to point out some studies where CSF and imaging biomarkers have been used together. The combination of CSF and imaging biomarkers can provide increased diagnostic accuracy compared to using either modality alone. Shaffer et al. found this to be the case when using whole-brain MRI, fluorine 18 fluorodeoxyglucose (FDG) PET and CSF T-tau, P-tau and Aβ42 to predict conversion from MCI to AD [69]. Westman et al. showed that the combination of CSF Aβ42, T-tau and P-tau with MRI-generated regional subcortical volumes and cortical thickness measures gives a better classification of MCI, AD and controls at baseline than using either biomarker alone [70]. The prediction of incipient AD in MCI subjects was also improved by using the two biomarker modalities together. Vos et al. found that the CSF Aβ42:T-tau ratio increased the predictive accuracy for MCI patients with normal and abnormal hippocampal volumes on MRI that later developed AD [71]. MRI only provided increased diagnostic accuracy in patients with a normal CSF test. Other studies have found that CSF T-tau, P-tau, Aβ40 and/or Aβ42 and atrophy of the medial temporal lobe on MRI provides independent information when discriminating between AD patients and controls [72], and between patients with stable MCI and MCI patients that develop AD [73]. Further studies need to elucidate which imaging methods that are most beneficial to use, and the correct time point to implement them, in relation to CSF biomarkers.
Candidate biomarkers
The search for new AD biomarkers may be facilitated by the core CSF AD biomarkers. By including patients with signs of an AD pathological process and ensuring that control subjects lack this profile, future biomarker studies may be more successful. Results from earlier studies may, for instance, have been clouded by the presence of non-symptomatic AD patients in the control groups. A recent study on the effect size of the major susceptibility gene for AD, APOE ϵ4, supports this reasoning; when disregarding clinical information on patients with cognitive impairment and simply grouping them on the basis of CSF tau and Aβ markers, the association of APOE ϵ4 with AD was twice as strong as compared to when classifying patients according to clinical status [74].
The potential uses for AD biomarkers are many. Besides from diagnostics, CSF biomarkers may be utilized for prognosis, assessing disease progression, developing treatments, monitoring treatment effects and studying disease mechanisms. Several studies have investigated the AD biomarker potential of various analytes related to Aβ.
BACE1
For Aβ to be produced, the amyloid precursor protein (APP) is cut by two different enzymes, β-secretase and γ-secretase. The major β-secretase in the brain is called β-site APP cleaving enzyme-1 (BACE1). Several studies have investigated the levels of CSF BACE1 activity in patients with MCI and AD, but the results are not univocal. Three small studies found that patients with AD had increased BACE1 activity compared with non-demented controls [75, 76] or patients with other dementias [77]. One study found higher activity in patients with MCI and dementia due to AD compared with controls [63]. Another study found significantly elevated BACE1 activity in patients with MCI but not with AD when comparing with controls [78]. One study failed to show any differences in BACE1 activity between MCI and AD patients compared with controls [79]. However, when patients with a pathologic profile of the core AD biomarkers were compared with controls with a normal biomarker pattern, a significant elevation of BACE1 activity was found in the patient group. The MCI patients contributed the most to this elevation. Finally, a large study did not find any differences in activity between AD patients and controls [80]. When the AD patients were stratified into mild and moderate-severe AD, an increased BACE1 activity could be seen in the group with mild AD compared the more affected AD patients and controls. These studies indicate that the activity of BACE1 may be mildly elevated in the early stages of AD, but the diagnostic usability of the biomarker seems limited. It may however prove to be valuable in clinical trials of BACE1 inhibitors.
sAPPα/sAPPβ
The levels of sAPPα and sAPPβ correlate very well in AD patients as well as controls [63]. Several studies have failed to show any differences in the levels of these biomarkers between AD patients and controls [30, 34, 63, 80, 81]. One study found higher levels of sAPPβ in MCI patients compared with controls [81], and another that MCI patients that upon follow up developed AD had higher levels of sAPPβ than patients that didn’t [82]. However, Hertze et al. found no differences in sAPP levels in MCI patients with incipient AD compared with stable MCIs or patients that developed other dementias [30]. Studies that compared patients with MCI or dementia that had a pathologic core CSF AD biomarker profile with controls with a normal profile found that the former group had increased levels of sAPPα and sAPPβ, but there were large overlaps between the groups [83–85]. The diagnostic value of sAPPα and sAPPβ appears limited. Yet, the proteins may be used for studying effects on the APP metabolism in clinical trials [5].
Aβ oligomers
Oligomers of Aβ may be more toxic than fibrillar Aβ aggregates [86]. They can inhibit hippocampal long-term potentiation in vivo[87], and cause abnormal tau phosphorylation and neuritic dystrophy [88–90]. A relatively large number of assay formats to measure Aβ oligomers in CSF have been published. These papers have shown that the CSF level of Aβ oligomers is very low, probably less than 1% of total Aβ levels, and thereby very difficult to quantify in a reliable manner.
In AD patients, elevated Aβ oligomer levels have been found in brains [91, 92] and CSF [93–96]. However, not all studies have found altered levels in AD patients versus controls [97, 98], but the latter of these found a negative correlation between oligomer levels and cognitive status, indicating a potential use for assessing disease stage in AD patients. One study found elevated levels of oligomers in human and mouse brains, but not in CSF [99]. Handoko et al. found that CSF oligomer levels increased with age and correlated with levels of T-tau in cognitively normal older adults. Also, increased levels were found in cognitively normal subjects with a biomarker profile indicating impending AD [100]. Except for technical difficulties to measure minute amounts of Aβ oligomers in CSF samples, it is also possible that different assays measure different variants of Aβ oligomers, which might explain the divergent results. The studies provide little characterization of what the various assays are even measuring.
Other candidate markers
In addition to the amyloid cascade hypothesis, genetics points to at least three other pathways that may be of relevance in AD pathogenesis: (i) the innate immune system, (ii) cholesterol metabolism and (iii) endosomal vesicle recycling. Better biomarkers for these processes may give us better tools to assess the importance of aberrations in these pathways and to subgroup AD patients according to which pathway is most abnormal.
Candidate biomarkers may help us to better characterize the ongoing pathologic processes in the AD brain. One example of this is the inflammatory biomarker YKL-40, which is secreted by activated microglia [101]. Studies have found increased levels of CSF YKL-40 in AD patients, as a sign of microglial activation [102–104]. Although Craig-Shapiro et al. suggested that YKL-40 is secreted by astrocytes, data from cell cultures and immunohistochemistry show that microglia, rather than astrocytes, are responsible for the main production of YKL-40 [101]. In an experimental setting, Simard et al. provided further insight into the role of microglia in AD by showing that blood-derived microglia can prevent the formation of or eliminate amyloid plaques in transgenic mice [105]. There are several other interesting candidate biomarkers, such as visinin-like protein-1 [106], neurogranin [107] and F2-isoprostane [108].
Blood biomarkers
Since blood is more easily accessible than CSF, finding reliable blood biomarkers for AD is desirable. Due to the blood–brain barrier, the concentration of brain-derived proteins in the blood is lower than in the CSF, which makes this task a challenge. Further, brain-derived proteins are diluted in the large plasma volume and may also get degraded. Recent studies point to important differences in the regulation of, e.g., tau concentrations in CSF as compared to blood. In patients with hypoxic brain injury following cardiac arrest, tau is rapidly released into the bloodstream but effectively (within 24 hours) cleared in patients with good neurological outcome [109]. In contrast, CSF tau levels stay elevated for many weeks following an acute neurological insult [38]. Further, tau levels are clearly elevated in CSF from patients with Alzheimer’s disease, but less so in the corresponding plasma samples; measurements of tau in these two compartments do not correlate [110].
Many studies have examined the association of plasma Aβ40 and Aβ42 levels with incipient AD, but the results are not clear-cut. Some studies have found that elevated Aβ40 [111, 112] or Aβ42 [] levels predicted development of AD, while others have found the opposite [115] or no associations [116, 117]. Some studies found that a low Aβ42:Aβ40 ratio predicted future AD [111, 118, 119] while others report an elevated ratio [112, 113] or no significant difference [116] in the group with incipient AD compared with subjects that didn’t develop AD. Recently, a meta-analysis over the literature indicated that a low Ab42:Ab40 ratio could predict development of AD, but no such association was found for the individual peptides [120]. More research in the area is needed. Cross-sectional studies that have compared Aβ levels in AD patients and controls have mostly found similar group levels [121]. In one study, high levels of Aβ42 was associated with a more rapid rate of cognitive decline among AD patients [122]. Recent studies have, by measuring the levels of several plasma analytes simultaneously, found biomarker patterns that successfully differentiated AD patients from controls [123, 124] or were associated with MCI or AD [125]. However, some of these finding have been hard to reproduce in independent studies [126, 127]. Other studies have found that plasma analytes such as desmosterol [128], transthyretin [129], clusterin [130], chitinase 3-like 1 protein [131] and matrix metalloproteinase 2 [132] may be associated with AD. This needs to be corroborated in further studies. Several of these biomarkers have also been associated with AD when measured in CSF.
Limitations
The major and inherent limitation of fluid biomarkers is the lack of anatomical precision in the measurements. Another limitation is the lack of assay standardization. Different assays for, e.g., Aβ42 correlate but give different absolute concentrations of the protein. This prevents from the use of global reference limits and diagnostic cut-points. Intense work is currently ongoing to achieve better assay standardization through the establishment of reference measurement procedures for tau and Aβ42 and to validate these in multi-center settings [36].
There is also a paucity of studies that connect human and experimental work. Although the biomarker CSF T-tau has been extensively studied there are some basic aspects that still are unknown. What does steady-state concentrations of tau among healthy individuals reflect; is it a low grade neurodegeneration or perhaps normal neuroaxonal plasticity? It is also not known why some tau pathologies, e.g. progressive supranuclear palsy, typically have normal levels of tau and why CSF levels of tau remain largely unaltered with progression of AD. An explanation to the latter could be that the levels of tau are more dependent on the rate of neurodegeneration than the state, but questions like these warrant translational studies spanning from models to human disease.
Conclusions
Three core CSF AD biomarkers have been evaluated in a great number of studies and may provide valuable information in all stages of AD. Due to differences in analytic results between studies and laboratories, standardization of analytic procedures is essential for the use of these biomarkers in clinics and in research. Apart from the core biomarkers, there are several candidate CSF and blood biomarkers in the pipeline, but they need verification in further studies.
References
Blennow K, de Leon MJ, Zetterberg H: Alzheimer’s disease. Lancet. 2006, 368: 387-403. 10.1016/S0140-6736(06)69113-7.
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM: Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA work group under the auspices of department of health and human services task force on Alzheimer’s disease. Neurology. 1984, 34: 939-944. 10.1212/WNL.34.7.939.
Knopman DS, DeKosky ST, Cummings JL, Chui H, Corey-Bloom J, Relkin N, Small GW, Miller B, Stevens JC: Practice parameter: diagnosis of dementia (an evidence-based review). Report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2001, 56: 1143-1153. 10.1212/WNL.56.9.1143.
Jack CR, Albert MS, Knopman DS, McKhann GM, Sperling RA, Carrillo MC, Thies B, Phelps CH: Introduction to the recommendations from the national institute on aging-Alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s dementia j Alzheimer’s Assoc. 2011, 7: 257-262. 10.1016/j.jalz.2011.03.004.
Blennow K, Hampel H, Weiner M, Zetterberg H: Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010, 6: 131-144. 10.1038/nrneurol.2010.4.
Price JL, Morris JC: Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 1999, 45: 358-368. 10.1002/1531-8249(199903)45:3<358::AID-ANA12>3.0.CO;2-X.
Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ: Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010, 9: 119-128. 10.1016/S1474-4422(09)70299-6.
Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Kaye J, Montine TJ, et al: Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the national institute on aging-Alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s dementia j Alzheimer’s Assoc. 2011, 7: 280-292. 10.1016/j.jalz.2011.03.003.
Jack CR, Knopman DS, Weigand SD, Wiste HJ, Vemuri P, Lowe V, Kantarci K, Gunter JL, Senjem ML, Ivnik RJ, et al: An operational approach to national institute on aging-Alzheimer’s association criteria for preclinical alzheimer disease. Ann Neurol. 2012, 71: 765-775. 10.1002/ana.22628.
Citron M: Alzheimer’s disease: strategies for disease modification. Nat Rev Drug Discov. 2010, 9: 387-398. 10.1038/nrd2896.
Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, et al: Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012, 367: 795-804. 10.1056/NEJMoa1202753.
Reiman EM, Quiroz YT, Fleisher AS, Chen K, Velez-Pardo C, Jimenez-Del-Rio M, Fagan AM, Shah AR, Alvarez S, Arbelaez A, et al: Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: a case–control study. Lancet Neurol. 2012, 11: 1048-1056. 10.1016/S1474-4422(12)70228-4.
Ringman JM, Coppola G, Elashoff D, Rodriguez-Agudelo Y, Medina LD, Gylys K, Cummings JL, Cole GM: Cerebrospinal fluid biomarkers and proximity to diagnosis in preclinical familial Alzheimer’s disease. Dement Geriatr Cogn Disord. 2012, 33: 1-5. 10.1159/000335729.
Moonis M, Swearer JM, Dayaw MP, St George-Hyslop P, Rogaeva E, Kawarai T, Pollen DA: Familial Alzheimer disease: decreases in CSF Abeta42 levels precede cognitive decline. Neurology. 2005, 65: 323-325. 10.1212/01.wnl.0000171397.32851.bc.
Ringman JM, Younkin SG, Pratico D, Seltzer W, Cole GM, Geschwind DH, Rodriguez-Agudelo Y, Schaffer B, Fein J, Sokolow S, et al: Biochemical markers in persons with preclinical familial Alzheimer disease. Neurology. 2008, 71: 85-92. 10.1212/01.wnl.0000303973.71803.81.
Gustafson DR, Skoog I, Rosengren L, Zetterberg H, Blennow K: Cerebrospinal fluid beta-amyloid 1–42 concentration may predict cognitive decline in older women. J Neurol Neurosurg Psychiatry. 2007, 78: 461-464.
Fagan AM, Roe CM, Xiong C, Mintun MA, Morris JC, Holtzman DM: Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 2007, 64: 343-349. 10.1001/archneur.64.3.noc60123.
Stomrud E, Hansson O, Blennow K, Minthon L, Londos E: Cerebrospinal fluid biomarkers predict decline in subjective cognitive function over 3 years in healthy elderly. Dement Geriatr Cogn Disord. 2007, 24: 118-124. 10.1159/000105017.
Stomrud E, Hansson O, Zetterberg H, Blennow K, Minthon L, Londos E: Correlation of longitudinal cerebrospinal fluid biomarkers with cognitive decline in healthy older adults. Arch Neurol. 2010, 67: 217-223. 10.1001/archneurol.2009.316.
Li G, Sokal I, Quinn JF, Leverenz JB, Brodey M, Schellenberg GD, Kaye JA, Raskind MA, Zhang J, Peskind ER, Montine TJ: CSF tau/Abeta42 ratio for increased risk of mild cognitive impairment: a follow-up study. Neurology. 2007, 69: 631-639. 10.1212/01.wnl.0000267428.62582.aa.
Skoog I, Davidsson P, Aevarsson O, Vanderstichele H, Vanmechelen E, Blennow K: Cerebrospinal fluid beta-amyloid 42 is reduced before the onset of sporadic dementia: a population-based study in 85-year-olds. Dement Geriatr Cogn Disord. 2003, 15: 169-176. 10.1159/000068478.
van Harten AC, Visser PJ, Pijnenburg YA, Teunissen CE, Blankenstein MA, Scheltens P, van der Flier WM: Cerebrospinal fluid Abeta42 is the best predictor of clinical progression in patients with subjective complaints. Alzheimer’s dementia j Alzheimer’s Assoc. 2012
Schott JM, Bartlett JW, Fox NC, Barnes J: Alzheimer’s disease neuroimaging initiative I: increased brain atrophy rates in cognitively normal older adults with low cerebrospinal fluid Abeta1-42. Ann Neurol. 2010, 68: 825-834. 10.1002/ana.22315.
Bennett DA, Wilson RS, Schneider JA, Evans DA, Beckett LA, Aggarwal NT, Barnes LL, Fox JH, Bach J: Natural history of mild cognitive impairment in older persons. Neurology. 2002, 59: 198-205. 10.1212/WNL.59.2.198.
Petersen RC: Mild cognitive impairment: aging to Alzheimer’s disease. 2003, Oxford; New York: Oxford University Press
Blennow K, Hampel H: CSF markers for incipient Alzheimer’s disease. Lancet Neurol. 2003, 2: 605-613. 10.1016/S1474-4422(03)00530-1.
Petersen RC: Mild cognitive impairment as a diagnostic entity. J Intern Med. 2004, 256: 183-194. 10.1111/j.1365-2796.2004.01388.x.
Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L: Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: a follow-up study. Lancet Neurol. 2006, 5: 228-234. 10.1016/S1474-4422(06)70355-6.
Buchhave P, Minthon L, Zetterberg H, Wallin AK, Blennow K, Hansson O: Cerebrospinal fluid levels of beta-amyloid 1–42, but not of tau, are fully changed already 5 to 10 years before the onset of Alzheimer dementia. Arch Gen Psychiatry. 2012, 69: 98-106. 10.1001/archgenpsychiatry.2011.155.
Hertze J, Minthon L, Zetterberg H, Vanmechelen E, Blennow K, Hansson O: Evaluation of CSF biomarkers as predictors of Alzheimer’s disease: a clinical follow-up study of 4.7 years. J Alzheimer’s disease JAD. 2010, 21: 1119-1128.
Visser PJ, Verhey F, Knol DL, Scheltens P, Wahlund LO, Freund-Levi Y, Tsolaki M, Minthon L, Wallin AK, Hampel H, et al: Prevalence and prognostic value of CSF markers of Alzheimer’s disease pathology in patients with subjective cognitive impairment or mild cognitive impairment in the DESCRIPA study: a prospective cohort study. Lancet Neurol. 2009, 8: 619-627. 10.1016/S1474-4422(09)70139-5.
Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, et al: Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects. Ann Neurol. 2009, 65: 403-413. 10.1002/ana.21610.
Mattsson N, Zetterberg H, Hansson O, Andreasen N, Parnetti L, Jonsson M, Herukka SK, van der Flier WM, Blankenstein MA, Ewers M, et al: CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA j Am Med Assoc. 2009, 302: 385-393. 10.1001/jama.2009.1064.
Johansson P, Mattsson N, Hansson O, Wallin A, Johansson JO, Andreasson U, Zetterberg H, Blennow K, Svensson J: Cerebrospinal fluid biomarkers for Alzheimer’s disease: diagnostic performance in a homogeneous mono-center population. J Alzheimer’s disease JAD. 2011, 24: 537-546.
Mattsson N, Andreasson U, Persson S, Arai H, Batish SD, Bernardini S, Bocchio-Chiavetto L, Blankenstein MA, Carrillo MC, Chalbot S, et al: The Alzheimer’s association external quality control program for cerebrospinal fluid biomarkers. Alzheimer’s dementia j Alzheimer’s Assoc. 2011, 7: 386-395. 10.1016/j.jalz.2011.05.2243. e386
Carrillo MC, Blennow K, Soares H, Lewczuk P, Mattsson N, Oberoi P, Umek R, Vandijck M, Salamone S, Bittner T, et al: Global standardization measurement of cerebral spinal fluid for Alzheimer’s disease: an update from the Alzheimer’s association global biomarkers consortium. Alzheimer’s dementia j Alzheimer’s Assoc. 2013, 9: 137-140. 10.1016/j.jalz.2012.11.003.
Blennow K, Vanmechelen E, Hampel H: CSF total tau, Abeta42 and phosphorylated tau protein as biomarkers for Alzheimer’s disease. Mol Neurobiol. 2001, 24: 87-97. 10.1385/MN:24:1-3:087.
Hesse C, Rosengren L, Andreasen N, Davidsson P, Vanderstichele H, Vanmechelen E, Blennow K: Transient increase in total tau but not phospho-tau in human cerebrospinal fluid after acute stroke. Neurosci Lett. 2001, 297: 187-190. 10.1016/S0304-3940(00)01697-9.
Zetterberg H, Hietala MA, Jonsson M, Andreasen N, Styrud E, Karlsson I, Edman A, Popa C, Rasulzada A, Wahlund LO, et al: Neurochemical aftermath of amateur boxing. Arch Neurol. 2006, 63: 1277-1280. 10.1001/archneur.63.9.1277.
Wang GR, Gao C, Shi Q, Zhou W, Chen JM, Dong CF, Shi S, Wang X, Wei Y, Jiang HY, et al: Elevated levels of Tau protein in cerebrospinal fluid of patients with probable creutzfeldt-jakob disease. Am J Med Sci. 2010, 340: 291-295. 10.1097/MAJ.0b013e3181e92a1f.
Blennow K: Cerebrospinal fluid protein biomarkers for Alzheimer’s disease. NeuroRx j Am Soc Exper NeuroTherap. 2004, 1: 213-225.
Hampel H, Blennow K, Shaw LM, Hoessler YC, Zetterberg H, Trojanowski JQ: Total and phosphorylated tau protein as biological markers of Alzheimer’s disease. Exp Gerontol. 2010, 45: 30-40. 10.1016/j.exger.2009.10.010.
Hampel H, Buerger K, Zinkowski R, Teipel SJ, Goernitz A, Andreasen N, Sjoegren M, DeBernardis J, Kerkman D, Ishiguro K, et al: Measurement of phosphorylated tau epitopes in the differential diagnosis of Alzheimer disease: a comparative cerebrospinal fluid study. Arch Gen Psychiatry. 2004, 61: 95-102. 10.1001/archpsyc.61.1.95.
Hall S, Ohrfelt A, Constantinescu R, Andreasson U, Surova Y, Bostrom F, Nilsson C, Hakan W, Decraemer H, Nagga K, et al: Accuracy of a panel of 5 cerebrospinal fluid biomarkers in the differential diagnosis of patients with dementia and/or parkinsonian disorders. Arch Neurol. 2012, 69: 1445-1452.
Schoonenboom NS, Reesink FE, Verwey NA, Kester MI, Teunissen CE, van de Ven PM, Pijnenburg YA, Blankenstein MA, Rozemuller AJ, Scheltens P, van der Flier WM: Cerebrospinal fluid markers for differential dementia diagnosis in a large memory clinic cohort. Neurology. 2012, 78: 47-54. 10.1212/WNL.0b013e31823ed0f0.
Tapiola T, Alafuzoff I, Herukka SK, Parkkinen L, Hartikainen P, Soininen H, Pirttila T: Cerebrospinal fluid {beta}-amyloid 42 and tau proteins as biomarkers of Alzheimer-type pathologic changes in the brain. Arch Neurol. 2009, 66: 382-389. 10.1001/archneurol.2008.596.
Buerger K, Ewers M, Pirttila T, Zinkowski R, Alafuzoff I, Teipel SJ, DeBernardis J, Kerkman D, McCulloch C, Soininen H, Hampel H: CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain. 2006, 129: 3035-3041. 10.1093/brain/awl269.
Seppala TT, Nerg O, Koivisto AM, Rummukainen J, Puli L, Zetterberg H, Pyykko OT, Helisalmi S, Alafuzoff I, Hiltunen M, et al: CSF biomarkers for Alzheimer disease correlate with cortical brain biopsy findings. Neurology. 2012, 78: 1568-1575. 10.1212/WNL.0b013e3182563bd0.
Strozyk D, Blennow K, White LR, Launer LJ: CSF Abeta 42 levels correlate with amyloid-neuropathology in a population-based autopsy study. Neurology. 2003, 60: 652-656. 10.1212/01.WNL.0000046581.81650.D0.
Fagan AM, Mintun MA, Mach RH, Lee SY, Dence CS, Shah AR, LaRossa GN, Spinner ML, Klunk WE, Mathis CA, et al: Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Abeta42 in humans. Ann Neurol. 2006, 59: 512-519. 10.1002/ana.20730.
Forsberg A, Engler H, Almkvist O, Blomquist G, Hagman G, Wall A, Ringheim A, Langstrom B, Nordberg A: PET imaging of amyloid deposition in patients with mild cognitive impairment. Neurobiology of aging. 2008, 29: 1456-1465. 10.1016/j.neurobiolaging.2007.03.029.
Grimmer T, Riemenschneider M, Forstl H, Henriksen G, Klunk WE, Mathis CA, Shiga T, Wester HJ, Kurz A, Drzezga A: Beta amyloid in Alzheimer’s disease: increased deposition in brain is reflected in reduced concentration in cerebrospinal fluid. Biol Psychiatry. 2009, 65: 927-934. 10.1016/j.biopsych.2009.01.027.
Mattsson N, Rosen E, Hansson O, Andreasen N, Parnetti L, Jonsson M, Herukka SK, van der Flier WM, Blankenstein MA, Ewers M, et al: Age and diagnostic performance of Alzheimer disease CSF biomarkers. Neurology. 2012, 78: 468-476. 10.1212/WNL.0b013e3182477eed.
Blennow K, Zetterberg H, Minthon L, Lannfelt L, Strid S, Annas P, Basun H, Andreasen N: Longitudinal stability of CSF biomarkers in Alzheimer’s disease. Neurosci Lett. 2007, 419: 18-22. 10.1016/j.neulet.2007.03.064.
Zetterberg H, Pedersen M, Lind K, Svensson M, Rolstad S, Eckerstrom C, Syversen S, Mattsson UB, Ysander C, Mattsson N, et al: Intra-individual stability of CSF biomarkers for Alzheimer’s disease over two years. J Alzheimer’s dis JAD. 2007, 12: 255-260.
Mattsson N, Portelius E, Rolstad S, Gustavsson M, Andreasson U, Stridsberg M, Wallin A, Blennow K, Zetterberg H: Longitudinal cerebrospinal fluid biomarkers over four years in mild cognitive impairment. J Alzheimer’s dis JAD. 2012, 30: 767-778.
Buchhave P, Blennow K, Zetterberg H, Stomrud E, Londos E, Andreasen N, Minthon L, Hansson O: Longitudinal study of CSF biomarkers in patients with Alzheimer’s disease. PLoS One. 2009, 4: e6294-10.1371/journal.pone.0006294.
Le Bastard N, Aerts L, Sleegers K, Martin JJ, Van Broeckhoven C, De Deyn PP, Engelborghs S: Longitudinal stability of cerebrospinal fluid biomarker levels: fulfilled requirement for pharmacodynamic markers in Alzheimer’s disease. J Alzheimer’s dis JAD. 2013, 33: 807-822.
Samgard K, Zetterberg H, Blennow K, Hansson O, Minthon L, Londos E: Cerebrospinal fluid total tau as a marker of Alzheimer’s disease intensity. Int J Geriatr Psychiatry. 2010, 25: 403-410. 10.1002/gps.2353.
Wallin AK, Blennow K, Zetterberg H, Londos E, Minthon L, Hansson O: CSF biomarkers predict a more malignant outcome in Alzheimer disease. Neurology. 2010, 74: 1531-1537. 10.1212/WNL.0b013e3181dd4dd8.
van Rossum IA, Vos SJ, Burns L, Knol DL, Scheltens P, Soininen H, Wahlund LO, Hampel H, Tsolaki M, Minthon L, et al: Injury markers predict time to dementia in subjects with MCI and amyloid pathology. Neurology. 2012, 79: 1809-1816. 10.1212/WNL.0b013e3182704056.
Andreasen N, Hesse C, Davidsson P, Minthon L, Wallin A, Winblad B, Vanderstichele H, Vanmechelen E, Blennow K: Cerebrospinal fluid beta-amyloid(1–42) in Alzheimer disease: differences between early- and late-onset Alzheimer disease and stability during the course of disease. Arch Neurol. 1999, 56: 673-680. 10.1001/archneur.56.6.673.
Zetterberg H, Andreasson U, Hansson O, Wu G, Sankaranarayanan S, Andersson ME, Buchhave P, Londos E, Umek RM, Minthon L, et al: Elevated cerebrospinal fluid BACE1 activity in incipient Alzheimer disease. Arch Neurol. 2008, 65: 1102-1107. 10.1001/archneur.65.8.1102.
Pannee J, Portelius E, Oppermann M, Atkins A, Hornshaw M, Zegers I, Hojrup P, Minthon L, Hansson O, Zetterberg H, et al: A selected reaction monitoring (SRM)-based method for absolute quantification of Abeta38, Abeta40, and Abeta42 in cerebrospinal fluid of Alzheimer’s disease patients and healthy controls. J Alzheimer’s dis JAD. 2013, 33: 1021-1032.
Blennow K, Wallin A, Agren H, Spenger C, Siegfried J, Vanmechelen E: Tau protein in cerebrospinal fluid: a biochemical marker for axonal degeneration in Alzheimer disease?. Mol chem neuropathol/spons Int Soc Neurochem World Feder Neurol res groups neurochem cerebrospinal fluid. 1995, 26: 231-245.
Vanmechelen E, Vanderstichele H, Davidsson P, Van Kerschaver E, Van Der Perre B, Sjogren M, Andreasen N, Blennow K: Quantification of tau phosphorylated at threonine 181 in human cerebrospinal fluid: a sandwich ELISA with a synthetic phosphopeptide for standardization. Neurosci Lett. 2000, 285: 49-52. 10.1016/S0304-3940(00)01036-3.
Kohnken R, Buerger K, Zinkowski R, Miller C, Kerkman D, DeBernardis J, Shen J, Moller HJ, Davies P, Hampel H: Detection of tau phosphorylated at threonine 231 in cerebrospinal fluid of Alzheimer’s disease patients. Neurosci Lett. 2000, 287: 187-190. 10.1016/S0304-3940(00)01178-2.
Olsson A, Vanderstichele H, Andreasen N, De Meyer G, Wallin A, Holmberg B, Rosengren L, Vanmechelen E, Blennow K: Simultaneous measurement of beta-amyloid(1–42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin Chem. 2005, 51: 336-345. 10.1373/clinchem.2004.039347.
Shaffer JL, Petrella JR, Sheldon FC, Choudhury KR, Calhoun VD, Coleman RE, Doraiswamy PM: Alzheimer’s Disease Neuroimaging I: Predicting cognitive decline in subjects at risk for Alzheimer disease by using combined cerebrospinal fluid, MR imaging, and PET biomarkers. Radiology. 2013, 266: 583-591. 10.1148/radiol.12120010.
Westman E, Muehlboeck JS, Simmons A: Combining MRI and CSF measures for classification of Alzheimer’s disease and prediction of mild cognitive impairment conversion. NeuroImage. 2012, 62: 229-238. 10.1016/j.neuroimage.2012.04.056.
Vos S, van Rossum I, Burns L, Knol D, Scheltens P, Soininen H, Wahlund LO, Hampel H, Tsolaki M, Minthon L, et al: Test sequence of CSF and MRI biomarkers for prediction of AD in subjects with MCI. Neurobiol aging. 2012, 33: 2272-2281. 10.1016/j.neurobiolaging.2011.12.017.
Schoonenboom NS, van der Flier WM, Blankenstein MA, Bouwman FH, Van Kamp GJ, Barkhof F, Scheltens P: CSF and MRI markers independently contribute to the diagnosis of Alzheimer’s disease. Neurobiol aging. 2008, 29: 669-675. 10.1016/j.neurobiolaging.2006.11.018.
Brys M, Glodzik L, Mosconi L, Switalski R, De Santi S, Pirraglia E, Rich K, Kim BC, Mehta P, Zinkowski R, et al: Magnetic resonance imaging improves cerebrospinal fluid biomarkers in the early detection of Alzheimer’s disease. J Alzheimer’s dis JAD. 2009, 16: 351-362.
Andreasson U, Lautner R, Schott JM, Mattsson N, Hansson O, Herukka SK, Helisalmi S, Ewers M, Hampel H, Wallin A, et al: CSF biomarkers for Alzheimer’s pathology and the effect size of APOE varepsilon4. Mol Psychiatry. 2013
Holsinger RM, McLean CA, Collins SJ, Masters CL, Evin G: Increased beta-Secretase activity in cerebrospinal fluid of Alzheimer’s disease subjects. Ann Neurol. 2004, 55: 898-899. 10.1002/ana.20144.
Verheijen JH, Huisman LG, van Lent N, Neumann U, Paganetti P, Hack CE, Bouwman F, Lindeman J, Bollen EL, Hanemaaijer R: Detection of a soluble form of BACE-1 in human cerebrospinal fluid by a sensitive activity assay. Clin Chem. 2006, 52: 1168-1174. 10.1373/clinchem.2006.066720.
Holsinger RM, Lee JS, Boyd A, Masters CL, Collins SJ: CSF BACE1 activity is increased in CJD and Alzheimer disease versus [corrected] other dementias. Neurology. 2006, 67: 710-712. 10.1212/01.wnl.0000229925.52203.4c.
Zhong Z, Ewers M, Teipel S, Burger K, Wallin A, Blennow K, He P, McAllister C, Hampel H, Shen Y: Levels of beta-secretase (BACE1) in cerebrospinal fluid as a predictor of risk in mild cognitive impairment. Arch Gen Psychiatry. 2007, 64: 718-726. 10.1001/archpsyc.64.6.718.
Mulder SD, van der Flier WM, Verheijen JH, Mulder C, Scheltens P, Blankenstein MA, Hack CE, Veerhuis R: BACE1 activity in cerebrospinal fluid and its relation to markers of AD pathology. J Alzheimer’s dis JAD. 2010, 20: 253-260.
Rosen C, Andreasson U, Mattsson N, Marcusson J, Minthon L, Andreasen N, Blennow K, Zetterberg H: Cerebrospinal fluid profiles of amyloid beta-related biomarkers in Alzheimer’s disease. Neuromolecular Med. 2012, 14: 65-73. 10.1007/s12017-012-8171-4.
Olsson A, Hoglund K, Sjogren M, Andreasen N, Minthon L, Lannfelt L, Buerger K, Moller HJ, Hampel H, Davidsson P, Blennow K: Measurement of alpha- and beta-secretase cleaved amyloid precursor protein in cerebrospinal fluid from Alzheimer patients. Exp Neurol. 2003, 183: 74-80. 10.1016/S0014-4886(03)00027-X.
Perneczky R, Tsolakidou A, Arnold A, Diehl-Schmid J, Grimmer T, Forstl H, Kurz A, Alexopoulos P: CSF soluble amyloid precursor proteins in the diagnosis of incipient Alzheimer disease. Neurology. 2011, 77: 35-38. 10.1212/WNL.0b013e318221ad47.
Lewczuk P, Kamrowski-Kruck H, Peters O, Heuser I, Jessen F, Popp J, Burger K, Hampel H, Frolich L, Wolf S, et al: Soluble amyloid precursor proteins in the cerebrospinal fluid as novel potential biomarkers of Alzheimer’s disease: a multicenter study. Mol psychiatry. 2010, 15: 138-145. 10.1038/mp.2008.84.
Lewczuk P, Popp J, Lelental N, Kolsch H, Maier W, Kornhuber J, Jessen F: Cerebrospinal fluid soluble amyloid-beta protein precursor as a potential novel biomarkers of Alzheimer’s disease. J Alzheimer’s dis JAD. 2012, 28: 119-125.
Gabelle A, Roche S, Geny C, Bennys K, Labauge P, Tholance Y, Quadrio I, Tiers L, Gor B, Chaulet C, et al: Correlations between soluble alpha/beta forms of amyloid precursor protein and Abeta38, 40, and 42 in human cerebrospinal fluid. Brain res. 2010, 1357: 175-183.
Sakono M, Zako T: Amyloid oligomers: formation and toxicity of Abeta oligomers. FEBS J. 2010, 277: 1348-1358. 10.1111/j.1742-4658.2010.07568.x.
Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ: Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002, 416: 535-539. 10.1038/416535a.
Zempel H, Thies E, Mandelkow E, Mandelkow EM: Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines. J Neurosci. 2010, 30: 11938-11950. 10.1523/JNEUROSCI.2357-10.2010.
Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ: Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A. 2011, 108: 5819-5824. 10.1073/pnas.1017033108.
De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, Bigio EH, Jerecic J, Acton PJ, Shughrue PJ, et al: Alzheimer’s Disease-type neuronal tau hyperphosphorylation induced by a beta oligomers. Neurobiol aging. 2008, 29: 1334-1347. 10.1016/j.neurobiolaging.2007.02.029.
Bruggink KA, Jongbloed W, Biemans EA, Veerhuis R, Claassen JA, Kuiperij HB, Verbeek MM: Amyloid-beta oligomer detection by ELISA in cerebrospinal fluid and brain tissue. Anal Biochem. 2013, 433: 112-120. 10.1016/j.ab.2012.09.014.
Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, et al: Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nat Med. 2008, 14: 837-842. 10.1038/nm1782.
Gao CM, Yam AY, Wang X, Magdangal E, Salisbury C, Peretz D, Zuckermann RN, Connolly MD, Hansson O, Minthon L, et al: Abeta40 oligomers identified as a potential biomarker for the diagnosis of Alzheimer’s disease. PLoS One. 2010, 5: e15725-10.1371/journal.pone.0015725.
Fukumoto H, Tokuda T, Kasai T, Ishigami N, Hidaka H, Kondo M, Allsop D, Nakagawa M: High-molecular-weight beta-amyloid oligomers are elevated in cerebrospinal fluid of Alzheimer patients. FASEB J. 2010, 24: 2716-2726. 10.1096/fj.09-150359.
Georganopoulou DG, Chang L, Nam JM, Thaxton CS, Mufson EJ, Klein WL, Mirkin CA: Nanoparticle-based detection in cerebral spinal fluid of a soluble pathogenic biomarker for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2005, 102: 2273-2276. 10.1073/pnas.0409336102.
Pitschke M, Prior R, Haupt M, Riesner D: Detection of single amyloid beta-protein aggregates in the cerebrospinal fluid of Alzheimer’s patients by fluorescence correlation spectroscopy. Nat Med. 1998, 4: 832-834. 10.1038/nm0798-832.
Bruggink KA, Jongbloed W, Biemans EA, Veerhuis R, Claassen JA, Kuiperij HB, Verbeek MM: Amyloid-beta oligomer detection by enzyme-linked immunosorbent assay in cerebrospinal fluid and brain tissue. Anal Biochem. 2012, 433: 112-120.
Santos AN, Ewers M, Minthon L, Simm A, Silber RE, Blennow K, Prvulovic D, Hansson O, Hampel H: Amyloid-beta oligomers in cerebrospinal fluid are associated with cognitive decline in patients with Alzheimer’s disease. J Alzheimer’s dis JAD. 2012, 29: 171-176.
Yang T, Hong S, O’Malley T, Sperling RA, Walsh DM, Selkoe DJ: New ELISAs with high specificity for soluble oligomers of amyloid beta-protein detect natural Abeta oligomers in human brain but not CSF. Alzheimers Dement. 2013, 9: 99-112. 10.1016/j.jalz.2012.11.005.
Handoko M, Grant M, Kuskowski M, Zahs KR, Wallin A, Blennow K, Ashe KH: Correlation of specific amyloid-beta oligomers with Tau in cerebrospinal fluid from cognitively normal older adults. JAMA Neurol. 2013, 70: 597-599.
Bonneh-Barkay D, Bissel SJ, Wang G, Fish KN, Nicholl GC, Darko SW, Medina-Flores R, Murphey-Corb M, Rajakumar PA, Nyaundi J, et al: YKL-40, a marker of simian immunodeficiency virus encephalitis, modulates the biological activity of basic fibroblast growth factor. The American journal of pathology. 2008, 173: 130-143. 10.2353/ajpath.2008.080045.
Craig-Schapiro R, Perrin RJ, Roe CM, Xiong C, Carter D, Cairns NJ, Mintun MA, Peskind ER, Li G, Galasko DR, et al: YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biol Psychiatry. 2010, 68: 903-912. 10.1016/j.biopsych.2010.08.025.
Olsson B, Hertze J, Lautner R, Zetterberg H, Nagga K, Hoglund K, Basun H, Annas P, Lannfelt L, Andreasen N, et al: Microglial markers are elevated in the prodromal phase of Alzheimer’s disease and vascular dementia. J Alzheimer’s dis JAD. 2013, 33: 45-53.
Perrin RJ, Craig-Schapiro R, Malone JP, Shah AR, Gilmore P, Davis AE, Roe CM, Peskind ER, Li G, Galasko DR, et al: Identification and validation of novel cerebrospinal fluid biomarkers for staging early Alzheimer’s disease. PLoS One. 2011, 6: e16032-10.1371/journal.pone.0016032.
Simard AR, Soulet D, Gowing G, Julien JP, Rivest S: Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron. 2006, 49: 489-502. 10.1016/j.neuron.2006.01.022.
Tarawneh R, D’Angelo G, Macy E, Xiong C, Carter D, Cairns NJ, Fagan AM, Head D, Mintun MA, Ladenson JH, et al: Visinin-like protein-1: diagnostic and prognostic biomarker in Alzheimer disease. Ann Neurol. 2011, 70: 274-285. 10.1002/ana.22448.
Thorsell A, Bjerke M, Gobom J, Brunhage E, Vanmechelen E, Andreasen N, Hansson O, Minthon L, Zetterberg H, Blennow K: Neurogranin in cerebrospinal fluid as a marker of synaptic degeneration in Alzheimer’s disease. Brain research. 2010, 1362: 13-22.
Montine TJ, Markesbery WR, Morrow JD, Roberts LJ: Cerebrospinal fluid F2-isoprostane levels are increased in Alzheimer’s disease. Ann Neurol. 1998, 44: 410-413. 10.1002/ana.410440322.
Randall J, Mortberg E, Provuncher GK, Fournier DR, Duffy DC, Rubertsson S, Blennow K, Zetterberg H, Wilson DH: Tau proteins in serum predict neurological outcome after hypoxic brain injury from cardiac arrest: results of a pilot study. Resuscitation. 2013, 84: 351-356. 10.1016/j.resuscitation.2012.07.027.
Mehta PD, Pirttila T, Mehta SP, Sersen EA, Aisen PS, Wisniewski HM: Plasma and cerebrospinal fluid levels of amyloid beta proteins 1–40 and 1–42 in Alzheimer disease. Arch Neurol. 2000, 57: 100-105. 10.1001/archneur.57.1.100.
van Oijen M, Hofman A, Soares HD, Koudstaal PJ, Breteler MM: Plasma Abeta(1–40) and Abeta(1–42) and the risk of dementia: a prospective case-cohort study. Lancet Neurol. 2006, 5: 655-660. 10.1016/S1474-4422(06)70501-4.
Mayeux R, Honig LS, Tang MX, Manly J, Stern Y, Schupf N, Mehta PD: Plasma A[beta]40 and A[beta]42 and Alzheimer’s disease: relation to age, mortality, and risk. Neurology. 2003, 61: 1185-1190. 10.1212/01.WNL.0000091890.32140.8F.
Mayeux R, Tang MX, Jacobs DM, Manly J, Bell K, Merchant C, Small SA, Stern Y, Wisniewski HM, Mehta PD: Plasma amyloid beta-peptide 1–42 and incipient Alzheimer’s disease. Ann Neurol. 1999, 46: 412-416. 10.1002/1531-8249(199909)46:3<412::AID-ANA19>3.0.CO;2-A.
Schupf N, Tang MX, Fukuyama H, Manly J, Andrews H, Mehta P, Ravetch J, Mayeux R: Peripheral Abeta subspecies as risk biomarkers of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2008, 105: 14052-14057. 10.1073/pnas.0805902105.
Sundelof J, Giedraitis V, Irizarry MC, Sundstrom J, Ingelsson E, Ronnemaa E, Arnlov J, Gunnarsson MD, Hyman BT, Basun H, et al: Plasma beta amyloid and the risk of Alzheimer disease and dementia in elderly men: a prospective, population-based cohort study. Arch Neurol. 2008, 65: 256-263. 10.1001/archneurol.2007.57.
Hansson O, Zetterberg H, Vanmechelen E, Vanderstichele H, Andreasson U, Londos E, Wallin A, Minthon L, Blennow K: Evaluation of plasma Abeta(40) and Abeta(42) as predictors of conversion to Alzheimer’s disease in patients with mild cognitive impairment. Neurobiol aging. 2010, 31: 357-367. 10.1016/j.neurobiolaging.2008.03.027.
Lopez OL, Kuller LH, Mehta PD, Becker JT, Gach HM, Sweet RA, Chang YF, Tracy R, DeKosky ST: Plasma amyloid levels and the risk of AD in normal subjects in the cardiovascular health study. Neurology. 2008, 70: 1664-1671. 10.1212/01.wnl.0000306696.82017.66.
Graff-Radford NR, Crook JE, Lucas J, Boeve BF, Knopman DS, Ivnik RJ, Smith GE, Younkin LH, Petersen RC, Younkin SG: Association of low plasma Abeta42/Abeta40 ratios with increased imminent risk for mild cognitive impairment and Alzheimer disease. Arch Neurol. 2007, 64: 354-362. 10.1001/archneur.64.3.354.
Yaffe K, Weston A, Graff-Radford NR, Satterfield S, Simonsick EM, Younkin SG, Younkin LH, Kuller L, Ayonayon HN, Ding J, Harris TB: Association of plasma beta-amyloid level and cognitive reserve with subsequent cognitive decline. JAMA j Am Med Assoc. 2011, 305: 261-266. 10.1001/jama.2010.1995.
Koyama A, Okereke OI, Yang T, Blacker D, Selkoe DJ, Grodstein F: Plasma amyloid-beta as a predictor of dementia and cognitive decline: a systematic review and meta-analysis. Arch Neurol. 2012, 69: 824-831. 10.1001/archneurol.2011.1841.
Irizarry MC: Biomarkers of Alzheimer disease in plasma. NeuroRx j Am Soc Exper NeuroTherap. 2004, 1: 226-234.
Laske C, Sopova K, Gkotsis C, Eschweiler GW, Straten G, Gawaz M, Leyhe T, Stellos K: Amyloid-beta peptides in plasma and cognitive decline after 1 year follow-up in Alzheimer’s disease patients. J Alzheimer’s dis JAD. 2010, 21: 1263-1269.
Doecke JD, Laws SM, Faux NG, Wilson W, Burnham SC, Lam CP, Mondal A, Bedo J, Bush AI, Brown B, et al: Blood-based protein biomarkers for diagnosis of alzheimer disease. Arch Neurol. 2012, 69: 1318-1325. 10.1001/archneurol.2012.1282.
Ray S, Britschgi M, Herbert C, Takeda-Uchimura Y, Boxer A, Blennow K, Friedman LF, Galasko DR, Jutel M, Karydas A, et al: Classification and prediction of clinical Alzheimer’s diagnosis based on plasma signaling proteins. Nat Med. 2007, 13: 1359-1362. 10.1038/nm1653.
Hu WT, Holtzman DM, Fagan AM, Shaw LM, Perrin R, Arnold SE, Grossman M, Xiong C, Craig-Schapiro R, Clark CM, et al: Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease. Neurology. 2012, 79: 897-905. 10.1212/WNL.0b013e318266fa70.
Bjorkqvist M, Ohlsson M, Minthon L, Hansson O: Evaluation of a previously suggested plasma biomarker panel to identify Alzheimer’s disease. PLoS One. 2012, 7: e29868-10.1371/journal.pone.0029868.
Soares HD, Chen Y, Sabbagh M, Roher A, Schrijvers E, Breteler M: Identifying early markers of Alzheimer’s disease using quantitative multiplex proteomic immunoassay panels. Ann N Y Acad Sci. 2009, 1180: 56-67. 10.1111/j.1749-6632.2009.05066.x.
Sato Y, Suzuki I, Nakamura T, Bernier F, Aoshima K, Oda Y: Identification of a new plasma biomarker of Alzheimer’s disease using metabolomics technology. J Lipid Res. 2012, 53: 567-576. 10.1194/jlr.M022376.
Velayudhan L, Killick R, Hye A, Kinsey A, Guntert A, Lynham S, Ward M, Leung R, Lourdusamy A, To AW, et al: Plasma transthyretin as a candidate marker for Alzheimer’s disease. J Alzheimer’s dis JAD. 2012, 28: 369-375.
Thambisetty M, An Y, Kinsey A, Koka D, Saleem M, Guntert A, Kraut M, Ferrucci L, Davatzikos C, Lovestone S, Resnick SM: Plasma clusterin concentration is associated with longitudinal brain atrophy in mild cognitive impairment. NeuroImage. 2012, 59: 212-217. 10.1016/j.neuroimage.2011.07.056.
Choi J, Lee HW, Suk K: Plasma level of chitinase 3-like 1 protein increases in patients with early Alzheimer’s disease. J Neurol. 2011, 258: 2181-2185. 10.1007/s00415-011-6087-9.
Lim NK, Villemagne VL, Soon CP, Laughton KM, Rowe CC, McLean CA, Masters CL, Evin G, Li QX: Investigation of matrix metalloproteinases, MMP-2 and MMP-9, in plasma reveals a decrease of MMP-2 in Alzheimer’s disease. J Alzheimer’s dis JAD. 2011, 26: 779-786.
Acknowledgements
Work in the authors’ laboratories is supported by the Swedish Research Council, Swedish State Support for Clinical Research and Alzheimer’s Association.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
CR drafted the manuscript. OH, KB and HZ revised the manuscript for important intellectual content. All authors read and approved the final manuscript.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Rosén, C., Hansson, O., Blennow, K. et al. Fluid biomarkers in Alzheimer’s disease – current concepts. Mol Neurodegeneration 8, 20 (2013). https://doi.org/10.1186/1750-1326-8-20
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1326-8-20