
Abstract
Background
Sickle cell disease (SCD) is the most frequent hemoglobinopathy worldwide but remains a rare blood disorder in most western countries. Recommendations for standard of care have been produced in the United States, the United Kingdom and France, where this disease is relatively frequent because of earlier immigration from Africa. These recommendations have changed the clinical course of SCD but can be difficult to apply in other contexts. The Italian Association of Pediatric Hematology Oncology (AIEOP) decided to develop a common national response to the rising number of SCD patients in Italy with the following objectives: 1) to create a national working group focused on pediatric SCD, and 2) to develop tailored guidelines for the management of SCD that could be accessed and practiced by those involved in the care of children with SCD in Italy.
Methods
Guidelines, adapted to the Italian social context and health system, were developed by 22 pediatric hematologists representing 54 AIEOP centers across Italy. The group met five times for a total of 128 hours in 22 months; documents and opinions were circulated via web.
Results
Recommendations regarding the prevention and treatment of the most relevant complications of SCD in childhood adapted to the Italian context and health system were produced.
For each topic, a pathway of diagnosis and care is detailed, and a selection of health management issues crucial to Italy or different from other countries is described (i.e., use of alternatives for infection prophylaxis because of the lack of oral penicillin in Italy).
Conclusions
Creating a network of physicians involved in the day-to-day care of children with SCD is feasible in a country where it remains rare. Providing hematologists, primary and secondary care physicians, and caregivers across the country with web-based guidelines for the management of SCD tailored to the Italian context is the first step in building a sustainable response to a rare but emerging childhood blood disorder and in implementing the World Health Organization’s suggestion “to design (and) implement … comprehensive national integrated programs for the prevention and management of SCD".
Similar content being viewed by others


Background
Sickle cell disease (SCD) is the most common genetic disorder worldwide. Every year, an average of 300,000 children are born with sickle syndromes, and the prevalence of sickle cell among newborns ranges from 0.1/1000 in non-endemic countries to 20/1000 in several parts of Africa [1]. The United Nations and the World Health Organization (WHO) recognize inherited hemoglobin (Hb) disorders as a global public health problem [2, 3], and the WHO has urged the National Health Systems “to design, implement and reinforce … comprehensive national integrated programs for the prevention and management of SCD” [2]. In fact, although 19–27% of the African population carries the sickle cell allele, migration movements have brought SCD patients from high-frequency regions to Europe. Therefore, SCD has become the paradigm of immigration hematology in Europe [4]. Although considered a rare disease because of its global frequency in the 28 countries of the European Union, SCD is the most prevalent genetic disease in France [5] and the United Kingdom [6], and its frequency is steadily rising in many other countries of central and southern Europe [4, 7, 8]. The European Union considers SCD a rare disease and sponsored the European Network on Rare and Congenital Anemias (known as ENERCA) to facilitate collaboration among clinicians in the field of rare anemias, including SCD [9]. Several countries have developed a specific response to meet the needs of the growing number of patients with SCD. In France, national guidelines have been produced [10] and reference centers have been designated by the Health Ministry that offer focused services for SCD [11]; a newborn screening was performed at national level since 2000 for all newborns defined as being “at risk” for SCD based on ethnic origin [12]. In the United Kingdom, the development of national guidelines and the realization of a universal newborn screening program have been the first steps undertaken to offer specialized care [6]. Belgium and the Netherlands have also organized clinical services and newborn screening for hemoglobinopathies and are conducting careful investigations [13, 14].
Although the experience gained from the United States and the above mentioned European countries has forged a pathway for those involved in the care of patients with SCD, public health issues and policies associated with SCD need to be tailored according to each country’s availability of health care and the characteristics of the affected population [15–17]. Italy does not have a national hemoglobinopathy newborn screening program, and for many years, pediatric hematology services have focused primarily on beta thalassemia. A network for treatment and care of thalassemia patients has been developed in Italy since the 1970s, involving centers in the central and southern parts of the country, where the majority of thalassemia patients live. Moreover, guidelines for the management of thalassemia were implemented and are well known by physicians across the country. National public health interventions for prevention and screening of thalassemia conditions are widespread; sound research has been conducted, and patient associations continuously raise awareness of thalassemia and advocate for support.
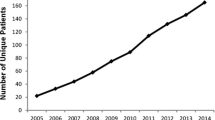
In contrast, SCD was historically a disease limited to the population of Sicily where the sickle cell allele frequency is 2–13% [18]. Recent immigration from Africa, South America, and the Balkans, mainly into the northern regions, has changed the geographic profile of SCD [7, 19, 20], however, with an increase of new diagnoses in the north, making this disease a more widespread public health issue [21]. Although the number of affected children has steadily increased over the last decade [7, 19–21], by the end of 2008, centers focused on the care of SCD were still rare, and clinical counseling for SCD was performed either by hematologists trained for thalassemia and not for SCD, or by pediatricians. No common policy of diagnosis and treatment had been developed, and the treatment was mainly case directed or locally directed. Moreover, the majority of SCD patients were African immigrants, living in northern Italy where hemoglobinopathies were less known.
The Italian Association of Pediatric Hematology Oncology (AIEOP) has a membership of all 54 Pediatric Hematology Oncology Centers across Italy and has existed since the 1970s (the website is http://www.aieop.org). It has developed common protocols and a database for children with cancer and non-oncologic diseases. The AIEOP Red Cell Group, involved in non-oncologic hematology, decided to develop a common national response to the rising number of SCD patients. Its objectives are creating a national working group focused on pediatric SCD and developing tailored guidelines for the management of children with SCD in Italy that could be accessed and practiced by all those involved in the care of the pediatric SCD population in the country.
We thought that creating a network of physicians involved in the day-to-day care of children with SCD and providing physicians and caregivers across the country with guidelines for the management of SCD tailored to our specific Italian context were the first steps in building a sustainable response to this newly more widespread disorder. The purpose of this paper is not to detail the guidelines but to describe the pathway chosen by the AIEOP Group to address a rare but increasingly prevalent blood disorder in Italy and summarize the recommendations that were critical for our specific health system and national context.
Methods
All 54 AIEOP centers were invited to participate in the SCD Working Group. Issues to be addressed in the guidelines were chosen by the group considering the international recommendations and the most important clinical issues in pediatric SCD [6, 22, 23]; every topic was developed by a subgroup in a single document. Each document included a brief description of the state-of-the-art recommendations with ABC ranking of strength of the evidence and references. Statements were based on literature evidence; when available evidence was not sufficient, the working group developed specific recommendations, based on expert author opinion or existing guidelines, and such statements were clearly presented as opinion. Each document was revised by the entire group and modified accordingly after exhaustive discussion. Four members of the group completed the internal revision of the final document, and external revision was performed by three Italian expert pediatric hematologists and one patient association.
Results and discussion
The SCD working group consisted of 22 pediatric hematologists from 14 centers (north 9, central 1, south 4), who met five times during 20 months from 2008 to 2010, for a total of 120 hours of meetings. One final 8-hour meeting was held in 2012 for definitive revision and approval of the document. Specific characteristics of our health system were taken into consideration in writing the guidelines. (see below List of Characteristics of the Italian context).
Characteristics of the Italian context
No national hemoglobinopathies newborn screening program
National free health care; direct primary, secondary, and tertiary pediatric care for children
Regulation of health care provided by regional health care systems with regional differences and disparities in health care facilities and resources across the country
Specialized care delivered on a hospital-based model and not on a medical home-based model
Free vaccinations with vaccination campaigns conducted on regional basis
Care for sickle cell patients delivered in pediatric hematology oncology centers among oncology patients
-
→ lack of teams dedicated to sickle cell patients
-
→ management of pain crisis in the emergency room
No availability of oral penicillin in the country
No availability of pediatric transcranial Doppler services, but an adult stroke network exists in the country
Recommendations
The guidelines, written in Italian, consist of 27 chapters and 242 recommendations and explore the milestones of SCD diagnosis, prevention, and treatment, tailoring them to the Italian public health system organization and drug availability. Recommendations that were considered more critical in our context and deserved extensive discussion are described below.
Neonatal screening
Universal neonatal screening, or at least targeted screening, is recommended. A comprehensive program for global prix en charge should enroll children after the neonatal diagnosis, with adequate health education for parents, implementation of preventive and therapeutic measures, and appropriate follow-up. We are aware that for the present, only two pilot selective screenings are being conducted in Italy, organized on a local basis (in the Region of Friuli Venezia Giulia and in the city of Modena).
Amoxicillin prophylaxis and vaccinations
Since its introduction for children with SCD, penicillin prophylaxis has resulted in a large reduction of invasive pneumococcal infections; therefore, it is recommended starting at 2 months of age (Table 1). In Italy, the oral formulation of penicillin is not available, and a depot i.m. formulation is difficult to find; therefore, the group made an effort to search the literature and to elaborate alternative prophylaxis options. The recommendation for Italy is to use amoxicillin or, as an alternative, a macrolide (Table 2). Pneumococcus immunization with both conjugated (PVC13) and polysaccharidic (Pneumo23) vaccine is recommended. Annual influenza vaccination is also recommended to reduce possible severe complications [24].
Management of painful vaso-occlusive crisis (VOC)
VOCs are the hallmark of SCD, and extensive literature has demonstrated that the optimal management of VOC is in hematology day care centers [31, 32]. In Italy, SCD patients with VOC are usually admitted to the general emergency room (ER), however; therefore, the guidelines emphasize the need to train ER personnel in the management of VOC. Generally, pediatricians are not comfortable with the use of analgesics, especially opioids; consequently, the guidelines detail each drug, including the safety profile and practical dilution modality, to make their use easier even for pediatricians who are not familiar with them. Table 3 lists the differential diagnoses of VOC and acute emergencies to ease patient care for ER personnel who are not familiar with SCD patients.
A visual analogue scale (VAS) or a numerical scale is always recommended for pain evaluation. The treatment of pain is to be established as quickly as possible, within 30 minutes from arrival in the ER, even before the identification of the cause or the possible differential diagnoses. The choice of the drug/s to be used must take into account the intensity of the pain and the drugs that have been used at home. The primary objective is to reduce the initial VAS score by 50%. Pain revaluation should be performed every 30 minutes until complete resolution. Children should receive analgesics at fixed intervals with “rescue” doses for pain that occurs in the interval between doses. The interval between doses should be determined in accordance with the intensity of pain and the duration of the analgesic effect of the drug in question. Acetaminophen, ibuprofen, ketorolac, codeine, and morphine analgesics are the recommended drugs for the treatment of pain in children.
Management of fever and infections
The management of the febrile child was the subject of intense discussion in the working group because of the disparities in health care facilities and availability of ER staff between northern and southern Italy and because of the many isolated and rural areas lying long distances from health centers in certain parts of the country. The decision was finally to define the presence of fever in a child with SCD as a medical emergency (“All those who manage children with SCD—pediatricians, hematologists, medical emergency staff, parents—should be aware that fever is a medical emergency which must be assessed and treated in the shortest possible time”), but to suggest different pathways of diagnosis and care between children at high risk of severe infection and at standard risk of severe infection (Table 4). Children at high risk of severe infection should be admitted; children at standard risk of severe infection should receive a dose of ceftriaxone and at least 3 hours of observation. The possible active involvement of the child’s pediatrician is considered.
Acute chest syndrome (ACS)
ACS is defined as the appearance of a new infiltrate on chest X-ray in association with at least one of the following: fever, dyspnea, chest pain, or desaturation. ACS is the second leading cause of hospitalization for patients with SCD and one of the leading causes of mortality. It is a potentially severe manifestation of SCD, and the patient therefore should always be hospitalized. The useful therapeutic measures are administration of O2, hydration, analgesia, early transfusion, and bronchodilators (especially if there is wheezing). Antibiotic therapy is always indicated. As a first choice, the ceftriaxone (or cefotaxime) + macrolide is preferred, considering the possibility of adding an anti-staphylococcal antibiotic. The use of “incentive spirometry” is indicated and is also effective in preventing the onset of ACS in all conditions of reduced thoracic mobility (chest pain crisis, immobilization in bed, etc.). There should be a timely transfer to the Intensive Care Unit (ICU) with sudden clinical worsening, requiring ventilatory support. The use of steroids is at present controversial, and the use of nitric oxide is still experimental. Because of the increasing recognition of this condition as a risk factor of disease severity in SCD, wheezing and asthma must be recognized and treated, as well as allergies for wheezing prevention.
Transcranial Doppler (TCD)
Early TCD screening and transfusion programs applied in patients with abnormal TCD really allows to significantly reduce stroke risk from 11 to less than 2% by age 18 and should be systematically offered to all children with SCD [33–35].
Because of the deficiency of this procedure in the Italian context, a particular emphasis has been given to this chapter, highlighting the difficulties other countries have faced in setting up a TCD program (lack of trained radiologists, missed appointments, education of patients and their parents, training of providers-pediatricians, hematologists, pediatric radiologists, service organization and patients’ access). In fact, in the USA, despite incontrovertible evidence of the value of screening for stroke risk, the adherence to the TCD surveillance programs is still less than 50% [36]. The results of a nationwide survey revealed poor compliance because of several obstacles, with the most frequent problems being poor patient attendance (as a result of the distance to the vascular screening laboratory) and the lack of personnel trained to perform TCD. The large majority of surveyed hematologists experienced difficulty in obtaining TCD assessment for their patients [37]. McCarville et al. [38] highlighted the importance of a comprehensive screening program in a single-center study in which the center achieved a 99% screening rate of the at-risk population in 3 years. The success of this comprehensive program relied on investing resources in personnel training and patient education.
TCD is recommended in children with SCD from ages 2 to 16 at least once a year It must be performed according to a specific protocol by trained staff to obtain reliable and reproducible results. If TCD is normal, a repeat screen is recommended after 12 months; if it is between 170–200 cm/s (conditional), it should be repeated within 3 months, especially if the child is under age 6 years. In individuals with TCD > 200 cm/s, a second test should be repeated within 2 months and, if confirmed as pathological, enrollment in a chronic transfusion program is recommended. In patients with abnormal TCD, another recommendation is to perform an MRI and MRA investigation for possible detection of brain injury. If blood flow is too low (TCD < 70 cm/s) or there is significant asymmetry between the two sides or if TCD cannot be performed, it is necessary to complete alternative neuroimaging studies, such as MRI and MRA. TCD imaging can be used in place of the TCD.
Blood transfusion
The issue of blood transfusion has been particularly focused to clearly differentiate indications for transfusion in SCD from thalassemia; the risk of alloimmunization has been stressed because of the lack of African donors in Italy and the need to avoid alloimmunization as much as possible (Table 5).
Indications for acute transfusion are acute symptomatic anemia with clinical signs of heart failure (tachycardia, tachypnea, dyspnea, fatigue) or absolute Hb ≤ 5 g/dl; it should be considered for a drop in Hb ≥ 2 g/dl from steady state or acute hepatic or splenic sequestration [1, 6]. In this last case, the values of Hb measured after transfusion are generally higher than expected; therefore, to prevent heart failure, it is better to transfuse 3.5 ml/kg of packed red blood cells, equal to half the usual dose [1] and then repeat the transfusion after a few hours. Great care must always be taken to avoid exceeding 10 g/dl Hb, above which the phenomenon of hyperviscosity may be dangerous. In severe infection, sepsis, or meningitis with significant anemia (Hb < 7 g/dl), blood transfusion should be performed because of the lower tolerance to anemia in the course of severe infection. In cases of high levels of Hb and transfusion indication, as in ACS or stroke, erythrocytapheresis is mandatory.
The indications for chronic transfusion are primary prevention of stroke in children with abnormal TCD, secondary prevention of stroke, and chronic heart failure, although there are no clinical studies that have shown its effectiveness in children. The goal of a chronic transfusion program is to maintain HbS level under 30% and Hb between 9 and 11 g/dl.
Iron overload and iron chelation
Iron overload has been a widely discussed topic, considering the lack of any official indication about its monitoring and treatment in SCD. Differences between thalassemia and SCD in the pattern of iron accumulation and complication rate have been the focus [39–43], and indications for monitoring, start of treatment, and drug choice have been addressed. Parameters for monitoring of iron overload were considered and specific recommendations made (Table 6). Recommended criteria for deciding the start of iron chelation therapy are serum ferritin level persistently ≥ 1000 ng/ml and transfused red blood cells ≥ 120 cc/kg (or after 20 transfused units) and/or a Liver Iron Concentration (LIC) ≥ 5–7 mg/g dw.
Compliance with chelation therapy affects morbidity and mortality in iron overloaded patients [44, 45], so a drug must be evaluated considering patient satisfaction, convenience, activity limitations, and preferences. Deferoxamine is an effective drug with an acceptable safety profile [46, 47], but patient adherence to this therapy is poor because of side effects and difficult route of administration (subcutaneous infusions over an 8- to 12-hour period, 5–7 days per week). Deferasirox is an oral chelator that has shown efficacy and safety in managing iron overload in SCD [48]. Patients on deferasirox have higher satisfaction and rate its convenience higher than patients do for deferoxamine [49], so deferasirox is rapidly becoming the standard care.
Hydroxyurea (HU)
Because of the reluctance of pediatricians to prescribe HU, specific indications and the safety profile have been particularly stressed in the guidelines. The clinical efficacy and low toxicity of HU have been demonstrated by the multi-center study HUG-KIDS Phase I/II [50], and recently in the randomized, double-blind, BABYHUG, which demonstrated the safety and efficacy of HU in “infants” (mean age 13.6 months) in reducing episodes of dactylitis, pain, and ACS in a population of patients not selected according to the clinical severity of the disease. The study also confirmed the safety of administration in the first few years of life [51]. Pediatric studies have highlighted other potential benefits of HU in the prevention of organ damage, the maintenance of splenic function, and improved growth [52, 53]. It is still unclear what role HU can have in primary and secondary prevention of stroke.
Conclusion and implications for public health practice
The organization of a national network and the development of national guidelines for pediatric SCD in Italy are the first steps in implementing the WHO suggestion “to design (and) implement … comprehensive national integrated programs for the prevention and management of SCD” and to address the two major priorities for the treatment of SCD, identified by patients in a questionnaire submitted in 2007 by the National Heart Lung and Blood Institute. One top priority that patients identified was the development of clear, evidence-based, local guidelines that they and their physicians could use to ensure that they were receiving optimal care, maximizing appropriate referral to hematologic specialist [54]. The second priority was appropriate management of pain. The development of national guidelines targeted to a specific national context allows addressing both priorities. The organization of the national network and the diffusion of the locally adapted guidelines in Italy were followed by several educational events and training courses on the management of SCD in childhood and TCD that involved hundreds of primary and secondary care pediatricians and pediatric hematologists across the country. Patient empowerment initiatives involving immigrant communities have begun to be developed, and every year the SCD World Day is now celebrated in most centers with open initiatives.
A better and standardized management of this relatively rare but complex disease implies that health systems will waste less money to treat acute or chronic complications and will optimize patient management. Children with SCD are generally admitted to ER and hospitals much more than their peers [7, 16, 17]. Moreover, SCD is in fifth place among the top 10 diagnoses with the highest readmission frequency, having the highest percentage of readmission (79.4%) [55]. The yearly cost of care for a pediatric patient 1–9 years of age with SCD is 7948 euro in countries where neonatal screening allows early diagnosis and comprehensive care is organized, assuring a holistic approach to health care; the morbidity and mortality and, therefore, the costs are much higher in cases of late diagnosis or no access to SCD specialized centers [16, 17, 56–60]. A preliminary single-center experience even in a low-prevalence country such as Italy has shown that comprehensive specialized care for a vulnerable group of immigrant children with SCD produces lower admission rates and lower use of the ER [61]. Detailed cost analysis is available for the USA but scarce for European countries; therefore, research in this field in Europe is needed.
In conclusion, creating a network of physicians involved with the day-to-day care of children with SCD and providing physicians and caregivers across the country with guidelines for the management of SCD tailored to the specific Italian context can be the first steps in building a sustainable response to a rare but nationally emerging blood disorder in childhood.
Authors informations
Members of the AIEOP Sickle Cell Disease Working Group
Andrea Ciliberti (San Giovanni Rotondo, Italy), Gian Carlo Del Vecchio (Bari, Italy), Domenico De Mattia (Bari, Italy), Benedetta Fabrizzi (Ancona, Italy), Cinzia Favara Scacco (Catania, Italy), Paola Giordano (Bari, Italy), Valentina Kiren (Triseste, Italy), Saverio Ladogana (San Giovanni Rotondo, Italy), Agostino Nocerino (Udine, Italy), Lucia Dora Notarangelo (Brescia, Italy), Anna Pusiol (Udine, Italy), Anita Regalia (Monza, Italy), Paola Saracco (Torino, Italy), Marco Zecca (Pavia, Italy).
References
Weatherall DJ, Clegg JB: Inherited haemoglobin disorders: an increasing global health problem. Bull World Health Organ. 2001, 79: 704-712.
WHO Report A59/9 on Sickle Cell Anemia. 2006, Available from: http://www.who.int/gb/ebwha/pdf_files/WHA59-REC1/e/WHA59_2006_REC1-en.pdf.
UN Resolution A/63/L.63 “Recognition of Sickle-Cell Anaemia as a Public Health Problem”. 2008, Available from: http://www.un.org/News/Press/docs/2008/ga10803.doc.htm.
Roberts I, de Montalambert M: Sickle Cell disease as a paradigm of immigration haematology: new challenges for immigration hematologists in Europe. Haematologica. 2007, 92: 865-871. 10.3324/haematol.11474.
de Montalembert M, Girot R, Galactéros F: Sickle cell disease in France in 2006: results and challenges. Arch Pediatr. 2006, 13: 1191-1194. 10.1016/j.arcped.2006.06.006.
NHS Standard and Guidelines for Clinical Care.http://www.sct.screening.nhs.uk/standardsandguidelines.
Colombatti R, Dalla Pozza LV, Mazzucato M, Sainati L, Pierobon M, Facchin P: Hospitalization of children with sickle cell disease in a region with increasing immigration rates. Haematologica. 2008, 93: 463-464. 10.3324/haematol.11766.
Mañú Pereira M, Corrons JL: Neonatal haemoglobinopathy screening in Spain. J Clin Pathol. 2009, 62: 22-25. 10.1136/jcp.2008.058834.
European Network on Rare and Congenital Anemias.http://www.enerca.org/.
http://www.has-sante.fr/portail/upload/docs/application/pdf/Drepanocytose_reco.pdf. Last accessed October 10th 2013.
Plan National Maladies Rares 2005-2008: Assurer l’équité pour l’accès au diagnostic, au traitement et à la prise en charge. http://asso.orpha.net/ORPHAWEB/cgi-bin/file/Plan_Maladies_Rares.pdf. Last accessed October 10th 2013.
Bardakdjian-Michau J, Bahuau M, Hurtrel D, Godart C, Riou J, Mathis M, Goossens M, Badens C, Ducrocq R, Elion J, Perini JM: Neonatal screening for sickle cell disease in France. J Clin Pathol. 2009, 62: 31-33. 10.1136/jcp.2008.058867.
Gulbis B, Cotton F, Ferster A, Ketelslegers O, Dresse MF, Rongé-Collard E, Minon JM, Lé PQ, Vertongen F: Neonatal haemoglobinopathy screening in Belgium. J Clin Pathol. 2009, 62: 49-52. 10.1136/jcp.2008.060517.
Bouva MJ, Mohrmann K, Brinkman HB, Kemper-Proper EA, Elvers B, Loeber JG, Verheul FE, Giordano PC: Implementing neonatal screening for haemoglobinopathies in the Netherlands. J Med Screen. 2010, 17: 58-65. 10.1258/jms.2010.009075.
Raphael JL, Kavanagh PL, Wang CJ, Mueller BU, Zuckerman B: Translating scientific advances to improved outcomes for children with sickle cell disease: a timely opportunity. Pediatr Blood Cancer. 2011, 56: 1005-1008. 10.1002/pbc.23059.
Olney RS: Preventing morbidity and mortality from sickle cell disease. A public health perspective. Am J Prev Med. 1999, 16: 116-121. 10.1016/S0749-3797(98)00140-8.
Grosse SD, James AH, Lloyd-Puryear MA, Atrash HK: A public health framework for rare blood disorders. Am J Prev Med. 2011, 41 (Suppl 4): S319-S323.
Schiliro G: Sicily: the world reservoir for thalassemias and haemoglobinopathies. Nature. 1978, 276: 761. 10.1038/276761a0.
Russo-Mancuso G, Romeo MA, Guardabasso V, Schilirò G: Survey of sickle cell disease in Italy. Haematologica. 1998, 83: 875-881.
Russo-Mancuso G, La Spina M, Schiliro G: The changing pattern of sickle cell disease in Italy. Eur J Epidemiol. 2003, 18: 923-924. 10.1023/A:1025635118305.
Cataldo F: Immigration and changes in the epidemiology of hemoglobin disorders in Italy : an emerging public health burden. Ital J Pediatr. 2012, 38: 32. 10.1186/1824-7288-38-32.
American Academy of Pediatrics: Health supervision for children with sickle cell disease. Pediatrics. 2002, 109: 526-535.
National Institute of Health, National Heart, Lung and Blood Institute: The Management of Sickle Cell Disease.http://www.nhlbi.nih.gov/health/prof/blood/sickle/sc_mngt.pdf.
Colombatti R, Perrotta S, Masera N, Palazzi G, Notarangelo LD, Pusiol A, Bonetto E, De Zen L, Nocerino A, Samperi P, Russo-Mancuso G, Sainati L: Lessons learned from the H1N1 pandemic: the need to improve systematic vaccination in sickle cell disease children. A multi Center survey in Italy. Vaccine. 2011, 29: 1126-1128. 10.1016/j.vaccine.2010.11.089.
Working Party of the British Committee for Standards in Haematology Clinical Haematology Task Force: Guidelines for the prevention and the treatment of infection in patients with an absent or dysfunctional spleen. BMJ. 1996, 312: 430-434.
Davies JM, Barnes R, Milligan D: Update of guidelines for the prevention and treatment of infection in patients with an absent or dysfunctional spleen. Clin Med. 2002, 2: 440-443. 10.7861/clinmedicine.2-5-440.
Price VE, Dutta S, Blanchette VS, Butchart S, Kirby M, Langer JC, Ford-Jones EL: The prevention and treatment of bacterial infections in children with asplenia or hyposplenia: practice considerations at the hospital for sick children, Toronto. Pediatr Blood Cancer. 2006, 46 (5): 597-603. 10.1002/pbc.20477.
Price VE, Blanchette VS, Ford-Jones EL: The prevention and management of infections in children with asplenia or hyposplenia. Infect Dis Clin N Am. 2007, 697-710.
Infectious Diseases and Immunization Committee Cps: Prevention and therapy of bacterial infections foe children with asplenia or hyposplenia. Paediatr Child Health. 1999, 4: 417-421.
Spelman D, Buttery J, Daley A, Isaacs D, Jennens I, Kakakios A, Lawrence R, Roberts S, Torda A, Watson DA, Woolley I, Anderson T, Street A: Australasian society for infectious diseases: guidelines for the prevention of sepsis in asplenic and hyposplenic patients. Int Med J. 2008, 38: 349-356. 10.1111/j.1445-5994.2007.01579.x.
Benjamin LJ, Swinson GI, Nagel RL: Sickle cell anemia day hospital: an approach for the management of uncomplicated painful crises. Blood. 2000, 95: 1130-1136.
Wright J, Bareford D, Wright C, Augustine G, Olley K, Musamadi L, Dhanda C, Knight C: Day case management of sickle pain: 3 years experience in a UK sickle cell unit. Br J Haematol. 2004, 126: 87.
Adams R, McKie V, Nichols F, Carl E, Zhang DL, McKie K, Figueroa R, Litaker M, Thompson W, Hess D: The use of transcranial ultrasonography to predict stroke in sickle cell disease. N Engl J Med. 1992, 326: 605-610. 10.1056/NEJM199202273260905.
Adams RJ, Brambilla D: Optimizing primary stroke prevention in sickle cell anemia (STOP 2) trial investigators: discontinuing prophylactic transfusions used to prevent stroke in sickle cell disease. N Engl J Med. 2005, 353: 2769-2778.
Bernaudin F, Verlhac S, Arnaud C, Kamdem A, Chevret S, Hau I, Coïc L, Leveillé E, Lemarchand E, Lesprit E, Abadie I, Medejel N, Madhi F, Lemerle S, Biscardi S, Bardakdjian J, Galactéros F, Torres M, Kuentz M, Ferry C, Socié G, Reinert P, Delacourt C: Impact of early transcranial Doppler screening and intensive therapy on cerebral vasculopathy outcome in a newborn sickle cell anemia cohort. Blood. 2011, 117: 1130-1140. 10.1182/blood-2010-06-293514.
Raphael JL, Shetty PB, Liu H, Mahoney DH, Mueller BU: A critical assessment of transcranial Doppler screening rates in a large pediatric sickle cell center: opportunities to improve healthcare quality. Pediatr Blood Cancer. 2008, 5: 647-651.
Fullerton HJ, Gardner M, Adams RJ, Lo LC, Johnston SC: Obstacles to primary stroke prevention in children with sickle cell disease. Neurology. 2006, 67: 1098-1099. 10.1212/01.wnl.0000237398.13545.86.
McCarville MB, Goodin GS, Fortner G, Li CS, Smeltzer MP, Adams R, Wang W: Evaluation of a comprehensive transcranial doppler screening program for children with sickle cell anemia. Pediatr Blood Cancer. 2008, 50: 818-821. 10.1002/pbc.21430.
Vichinsky E, Butensky E, Fung E, Hudes M, Theil E, Ferrell L, Williams R, Louie L, Lee PD, Harmatz P: Comparison of organ dysfunction in transfused patients with SCD or b Thalassemia. Am J Hematol. 2005, 80: 70-74. 10.1002/ajh.20402.
Wood JC, Tyszka JM, Carson S, Nelson MD, Coates TD: Myocardial iron loading in transfusion-dependent thalassemia and sickle cell disease. Blood. 2004, 103: 1934-1936. 10.1182/blood-2003-06-1919.
Wood JC: Cardiac iron across different transfusion-dependent diseases. Blood Rev. 2008, 22 (2): S14-S21.
Fung EB, Harmatz PR, Lee PD, Milet M, Bellevue R, Jeng MR, Kalinyak KA, Hudes M, Bhatia S, Vichinsky EP: Multi-centre study of iron overload research group: increased prevalence of iron-overload associated endocrinopathy in thalassaemia versus sickle-cell disease. Br J Haemat. 2006, 135: 574-582. 10.1111/j.1365-2141.2006.06332.x.
Fung EB, Harmatz PR, Milet M, Coates TD, Thompson AA, Ranalli M, Mignaca R, Scher C, Giardina P, Robertson S, Neumayr L, Vichinsky EP: Multi-center iron overload study group:fracture prevalence and relationship to endocrinopathy in iron overloaded patients with sickle cell disease and thalassemia. Bone. 2008, 43: 162-168. 10.1016/j.bone.2008.03.003.
Cohen AR, Martin MB: Iron chelation therapy in sickle cell disease. Semin Hematol. 2001, 38 (suppl 1): 69-72.
Silliman CC, Peterson VM, Mellman DL, Dixon DJ, Hambidge KM, Lane PA: Iron chelation by deferoxamine in sickle cell patients with severe transfusion-induced hemosiderosis: a randomized, double-blind study of the dose–response relationship. J Lab Clin Med. 1993, 122: 48-54.
Lucania G, Vitrano A, Filosa A, Maggio A: Chelation treatment in sickle-cell-anaemia: much ado about nothing?. Br J Haematol. 2011, 154: 545-555. 10.1111/j.1365-2141.2011.08769.x.
Vichinsky E, Onyekwere O, Porter J, Swerdlow P, Eckman J, Lane P, Files B, Hassell K, Kelly P, Wilson F, Bernaudin F, Forni GL, Okpala I, Ressayre-Djaffer C, Alberti D, Holland J, Marks P, Fung E, Fischer R, Mueller BU, Coates T: Deferasirox in sickle cell investigators: a randomized comparison of Deferasirox versus Deferoxamine for the treatment of transfusional iron overload in sickle cell disease. Br J Haematol. 2007, 136: 501-508. 10.1111/j.1365-2141.2006.06455.x.
Vichinsky E, Bernaudin F, Forni GL, Gardner R, Hassell K, Heeney MM, Inusa B, Kutlar A, Lane P, Mathias L, Porter J, Tebbi C, Wilson F, Griffel L, Deng W, Giannone V, Coates T: Long-term safety and efficacy of deferasirox (Exjade) for up to 5 years in transfusional iron-overloaded patients with sickle cell disease. Br J Haematol. 2011, 154: 387-397. 10.1111/j.1365-2141.2011.08720.x.
Vichinsky E, Pakbaz Z, Onyekwere O, Porter J, Swerdlow P, Coates T, Lane P, Files B, Mueller BU, Coïc L, Forni GL, Fischer R, Marks P, Rofail D, Abetz L, Baladi JF: Patient-reported outcomes of deferasirox (Exjade, ICL670) versus deferoxamine in sickle cell disease patients with transfusional hemosiderosis. Substudy of a randomized open-label phase II trial. Acta Haematol. 2008, 119: 133-141. 10.1159/000125550.
Kinney TR, Helms RW, O’Branski EE, Ohene Frempong K, Wang W, Daeschner C, Vichinsky E, Redding Lallinger R, Gee B, Platt OS, Ware RE, Pediatric Hydroxyurea Group: Safety of hydroxyurea in children with sickle cell anemia: results of the HUG,KIDS study, a phase I/II trial. Blood. 1999, 94: 1550-1554.
Wang WC, Ware RE, Miller ST, Iyer RV, Casella JF, Minniti CP, Rana S, Thornburg CD, Rogers ZR, Kalpatthi RV, Barredo JC, Brown RC, Sarnaik SA, Howard TH, Wynn LW, Kutlar A, Armstrong FD, Files BA, Goldsmith JC, Waclawiw MA, Huang X, Thompson BW, BABY HUG investigators: Hydroxycarbamide in very young children with sickle,cell anaemia: a multicentre, randomised, controlled trial (BABY HUG). Lancet. 2011, 377: 1663-1672. 10.1016/S0140-6736(11)60355-3.
Hankins JS, Helton KJ, McCarville MB, Li CS, Wang WC, Ware RE: Preservation of spleen and brain function in children with sickle cell anemia treated with hydroxyurea. Pediatr Blood Cancer. 2007, 50: 293-297.
Ferster A, Vermylen C, Cornu G, Buyse M, Corazza F, Devalck C, Fondu P, Toppet M, Sariban E: Hydroxyurea for treatment of severe sickle cell anemia: a pediatric clinical trial. Blood. 1996, 88: 1960-1964.
Hoots WK, Shurin SB: Future directions of sickle cell disease research: the NIH perspective. Pediatr Blood Cancer. 2012, 59: 353-357. 10.1002/pbc.24180.
Berry JG, Toomey SL, Zaslavsky AM, Jha AK, Nakamura MM, Klein DJ, Feng JY, Shulman S, Chiang VW, Kaplan W, Hall M, Schuster MA: Pediatric readmission prevalence and variability across hospitals. JAMA. 2013, 309: 372-380. 10.1001/jama.2012.188351.
Vichinsky EP: Comprehensive care in sickle cell disease: its impact on morbidity and mortality. Semin Hematol. 1991, 28: 220-226.
Yang YM, Shah AK, Watson M, Mankad VN: Comparison of costs to the health sector of comprehensive and episodic health care for sickle cell disease patients. Public Health Rep. 1995, 110: 80-86.
Okpala I, Thomas V, Westerdale N, Jegede T, Raj K, Daley S, Costello-Binger H, Mullen J, Rochester-Peart C, Helps S, Tulloch E, Akpala M, Dick M, Bewley S, Davies M, Abbs I: The comprehensiveness care of sickle cell disease. Eur J Haematol. 2002, 68: 157-162. 10.1034/j.1600-0609.2002.01523.x.
Amendah DD, Mvundura M, Kavanagh PL, Sprinz PG, Grosse SD: Sickle cell disease-related pediatric medical expenditures in the U.S. Am J Prev Med. 2010, 38 (4): S550-S556. 10.1016/j.amepre.2010.01.004.
Kauf TL, Coates TD, Huazhi L, Mody-Patel N, Hartzema AG: The cost of health care for children and adults with sickle cell disease. Am J Hematol. 2009, 84 (6): 323-327. 10.1002/ajh.21408.
Colombatti R, Montanaro M, Guasti F, Rampazzo P, Meneghetti G, Giordan M, Basso G, Sainati L: Comprehensive care for sickle cell disease immigrant patients: a reproducible model achieving high adherence to minimum standards of care. Pediatr Blood Cancer. 2012, 59 (7): 1275-1279. 10.1002/pbc.24110.
Acknowledgments
The authors thank the external reviewers of the guidelines: Loris Brunetta, Gianluca Forni, Antonio Piga, and especially the late Renzo Galanello.
The research was partially supported by Grant 04/11 of the Fondazione Città della Speranza and by a Grant from the Pediatric Department of the University of Naples.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
RC, SP, PS, MC, GP, NM, LS, GR participated in the meetings and wrote the guidelines; RC, PS, LS and GR internally reviewed the guidelines; RC, SP drafted the manuscript; LS and GR reviewed the manuscript. All authors have seen and approved the manuscript.
Raffaella Colombatti, Silverio Perrotta, Laura Sainati and Giovanna Russo contributed equally to this work.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an open access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Colombatti, R., Perrotta, S., Samperi, P. et al. Organizing national responses for rare blood disorders: the Italian experience with sickle cell disease in childhood. Orphanet J Rare Dis 8, 169 (2013). https://doi.org/10.1186/1750-1172-8-169
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1172-8-169