
Abstract
Brachydactyly (BD) refers to the shortening of the hands, feet or both. There are different types of BD; among them, type E (BDE) is a rare type that can present as an isolated feature or as part of more complex syndromes, such as: pseudohypopthyroidism (PHP), hypertension with BD or Bilginturan BD (HTNB), BD with mental retardation (BDMR) or BDE with short stature, PTHLH type. Each syndrome has characteristic patterns of skeletal involvement. However, brachydactyly is not a constant feature and shows a high degree of phenotypic variability. In addition, there are other syndromes that can be misdiagnosed as brachydactyly type E, some of which will also be discussed. The objective of this review is to describe some of the syndromes in which BDE is present, focusing on clinical, biochemical and genetic characteristics as features of differential diagnoses, with the aim of establishing an algorithm for their differential diagnosis. As in our experience many of these patients are recruited at Endocrinology and/or Pediatric Endocrinology Services due to their short stature, we have focused the algorithm in those steps that could mainly help these professionals.
Similar content being viewed by others


Brachydactyly (BD) refers to a family of limb malformations characterized by shortening of the hands, feet or both [1]. It was added to the international Nosology and Classification of Genetic Skeletal Disorders in 2001, in the group of genetically determined dysostoses [2, 3]. Different types of brachydactyly can be distinguished based on anatomic grounds, the most commonly used classification being that provided by Bell [4] and modified by Temtamy & McKusick [5]. Most types are rare, except for A3 (BDA3, OMIM#112700) and D (BDD, OMIM#113200) that have a prevalence of around 2% [1]. In this review, we focus on brachydactyly type E (BDE, OMIM#113300), which is rare and can be diagnosed as an isolated finding or as part of several genetic syndromes [1, 5, 6].
BDE encompasses variable shortening of the metacarpals/metatarsals, frequently with involvement of the phalanges [5]. Hertzog [7] classified BDE into three distinct varieties: type E1, with shortening of metacarpal IV, sometimes associated with shortening of metatarsal IV (possible involvement of an isolated metatarsal); type E2, with shortening of metacarpals IV and V (and metatarsals) associated with shortening of the distal phalanx of the thumb; and type E3, with various combinations of short metacarpals without phalangeal involvement [8]. However, these patterns are not held to exactly in all syndromes; as well as the distal phalanx of the thumb, others phalanges are often shortened.
In this review, we describe some common syndromes in which BDE is present, outlining their main clinical, biochemical and genetic characteristics (Additional file 1: Table S1), with the aim of establishing an algorithm for the accurate diagnosis of BDE in association with other features. Even if acrodysostosis and tricho-rinho-phalangeal syndrome cannot be considered as conditions with a pure brachydactyly E, as they have peculiar radiological features, sometimes they are misdiagnosed as syndromic BDE, so we will try to define the specific characteristics that distinguish them from all the other conditions.
One of these specific features is multihormonal resistance, mainly to pthyroid hormone (PTH) and thyroid hormone (TSH). We use this feature to classify the syndromes into two groups depending on the presence of multihormonal resistance as many of these patients are recruited at Endocrinology and/or Pediatric Endocrinology Services due to their short stature that leads to the analysis of hormonal profile.
Isolated brachydactyly type E: HOXD13 type (OMIM#113300)
While BDE is commonly reported as part of a syndrome, it occasionally appears as an isolated entity [9–11]. Four heterozygous mutations in the homeobox D13 gene (HOXD13, 2q31.1), two missenses and two nonsenses, have been identified as causative of isolated BDE [9–12]. In two of these four cases, an overlap between BDE and BDD (shortening of the distal phalanx of the thumb) was noted in the patients [9]. Other hand malformations that have been observed in combination with BDE are syndactyly of digits III and IV, synpolydactyly of finger IV, and long distal phalanges [9]. In addition, 21 independent mutations have been identified in HOXD13, most of them producing synpolydactyly (SPD; OMIM#186000), or in rare cases syndactyly, and other unspecified limb malformations [11].
Regarding BDE caused by mutations in HOXD13, the pattern profile showed many variations between affected patients: most of them had shortening of metacarpals III and sometimes also IV and V (Figure 1A); and in the feet, shortening of the metatarsals IV was frequently seen, sometimes in combination with that of metatarsals I, III or V and broadening of the hallux (Figure 1B). In addition, little-finger distal phalanx hypoplasia/aplasia, lateral phalangeal duplication and/or clinodactyly of finger IV, and syndactyly of fingers III/IV were frequently observed [9, 11, 13]. Finally, in general, affected individuals had normal stature and no appreciable psychomotor developmental delay (Additional file 1: Table S2) [9, 11, 13, 14].

Radiographs of hands (A) and feet (B) of a non adult individual with heterozygous mutation in HOXD13 (OMIM#113300) (courtesy of Dr. Aleksander Jamsheer). Shortening and widening of IV and V metacarpals are more evident in right; I, IV and V distal brachyphalangy, and mild clynodactyly of the distal phalanx of II and V, in left hand. In the feet, note asymmetrical shortening of the metatarsals: in right feet II, III, IV are shortened and in the left one, only III and IV. Note also, the clynodactyly of right hallux.
Brachydactyly type E as part of syndromes
BDE with multihormonal resistance
Pseudohypopthyroidism
Pseudohypopthyroidism (PHP) refers to several distinct, but related, disorders in which resistance to pthyroid hormone (PTH) is the most prominent feature [15]. However, resistance to thyroid-stimulating hormone (TSH) and several other pituitary hormones that mediate their action through G-protein-coupled receptors is often present as well. As a result of PTH resistance, patients with PHP develop hypocalcaemia and hyperphosphataemia. Resistance seems to occur only in the proximal renal tubule, while the action of PTH is unimpaired in other target tissues, such as bone and the renal thick ascending limb [16–18].
BDE is present in 70-78% of individuals with PHP [19]. There are, however, great differences in hand shortening: some patients display severe shortening of all hand bones, whereas commonly in others metacarpals and distal phalanges are more impaired than other segments. In particular, Poznanski et al. reported that the distal phalanx of the thumb and metacarpal IV were the most affected in 75% and 65% of patients respectively [20]. On the other hand, while de Sanctis et al. did not observe differences in prevalence of shortening, they reported that metacarpal V and distal phalanges I and IV were the most severely affected (85.7% < −2 SDS) (Figure 2) [19]. They also noted variations in the patterns of bone shortening between subjects with the same mutation, within the same family as well as among unrelated individuals [19]. Most publications on PHP only mention shortening of metacarpals III, IV and V or metatarsals, but it should be clarified that the shortening of these bones is usually more evident than shortening of phalanges; the latter may also occur, but is more difficult to assess because of the wide range of variation within the normal population [21].

Hand of a patient with pseudohypopthyroidism type Ia (OMIM#103580). Note the shortening of first metacarpal and proximal phalanx of the thumb and more severe shortening of metacarpals IV and V (courtesy of Dr. Beatriz Garcia-Cuartero).
PHP is associated with alterations in the GNAS locus, which maps to the telomeric end of the long arm of chromosome 20 (20q13.2-20q13.3) [22]. GNAS exemplifies a locus of high complexity [23]. One of its products is the α-subunit of the stimulatory heterotrimeric G protein (Gsα), a ubiquitous signalling protein that is essential for numerous different cellular responses. Gsα expression is biallelic in most tissues, but paternal Gsα expression is silenced in a few types of tissue (renal tubule, thyroid gland, hypophysis and gonads) [24–26] and this is important in the development of phenotypes associated with GNAS mutations. Maternal imprinting (and thus paternal expression) occurs on XLαs, the large form of Gsα, and two non-coding RNA molecules, the A/B transcript and the NESPas antisense transcript. On the other hand, paternal imprinting is seen on NESP55, a chromogranin like neuroendocrine secretory protein [27].
Heterozygous inactivating mutations within Gsα-encoding GNAS exons, leading to diminished activity, are found in patients with PHP-Ia (OMIM#103580). These patients have not only resistance to hormones but also the Albright’s hereditary osteodystrophy (AHO) phenotype (as an example, see Figure one at Miao et al. [28]) (Additional file 1: Table S3) [27]. This phenotype includes BDE (Figure 2), small stature, obesity with a rounded face, subcutaneous calcifications, and mental retardation [29]. Patients with AHO features but no evidence of hormone resistance are said to have pseudopseudohypopthyroidism (PPHP, OMIM#612463), and also carry heterozygous inactivating Gsα mutations. Maternal inheritance of such a mutation leads to PHP-Ia, AHO and hormone resistance, while paternal inheritance of the same mutation causes PPHP [27].
On the other hand, PHP-Ib (OMIM#603233) is another form of PHP which can follow an autosomal dominant (AD) trait (AD-PHP-Ib) or occur as a sporadic disorder. These patients present with PTH and sometimes TSH resistance, but resistance for other hormones is not found. Apart from this, patients often lack AHO features and exhibit normal Gsα activity in erythrocytes and fibroblasts. Nevertheless, there have been some reports of patients with mild AHO [30–33] and these individuals have diminished Gsa activity [34]. Generally, AD-PHP-Ib is caused by specific deletions in the syntaxin 16 gene (STX16) (STX16del4-6 or STX16del2-4); which are associated with maternal inheritance and cause loss of methylation at GNAS exon A/B or 1A. Other form of AD-PHP-Ib, due to maternally inherited microdeletions of NESP55 identified within GNAS, produces a loss of all maternal GNAS methylation imprints [35, 36].
Sporadic PHP-Ib cases also display GNAS imprinting abnormalities that involve NESPas, XLαs and A/B. However, the genetic lesion, if any, underlying these epigenetic defects remains to be discovered and most of these cases could represent true stochastic errors in early embryonic maintenance of methylation [37–39]. Some of these patients have been shown to be affected by paternal uniparental isodisomy (pat20iUPD) involving part or the whole long arm of chromosome 20, which includes the GNAS locus [39–42]. A mild BDE and AHO phenotype has been reported in some studies [30, 31, 43]; in most cases, only metacarpal IV and/or V shortening was seen, although mild shortening of all metacarpals and some distal phalanges has also been described [33]. Sanchez et al. found BDE in 60% of patients with PHP-Ib in their study (no subjects presenting shortening of distal phalanx I). In two patients, BDE was combined with Madelung-like deformities (involvement of the distal ulna and radius) [43]. On the other hand, Mantovani et al. did not find any correlation between the severity of the AHO phenotype and methylation defects, their patients’ phenotype ranging from mild to severe AHO [33].
Acrodysostosis with multihormonal resistance (ACRDYS1, OMIM#101800)
pthyroid hormone-related protein and G-proteins are part of the same signalling pathway affecting cartilage differentiation and growth in the metacarpals. This could explain the similarity of PTHLH-related BDE, pseudohypopthyroidism and acrodysostosis.
Acrodysostosis is a rare skeletal dysplasia characterized by severe generalized brachydactyly of hands (Figure 3) and feet with a relatively long first thumb, dysostosis, short stature and facial abnormalities: typically a round face with maxillary and nasal hypoplasia; but sometimes also an increased mandibular angle, mandibular prognathism, epicanthal folds and hypertelorism (see Figure Three Pt.7, 8, 11 at Linglart et al. [44]). Advanced skeletal maturation, spinal stenosis, obesity, mental deficiency, notably delayed speech and impaired hearing are also frequently observed (Additional file 1: Table S4) [44–51]. Although Maroteaux & Malamut defined this pathology as a new syndrome distinct from PHP/AHO in 1968 [47], these two entities have been confused many times because they share many features [44]. One of these is multihormonal resistance to PTH or TSH [52], though it is not always present as there are several forms of acrodysostosis and the genes involved are different (see below) [44]. In the case of acrodysostosis with multihormonal resistance (ACRDYS1, OMIM#101800), the gene responsible is cAMP-dependent protein kinase A (PRKAR1A), which has been localized to 17q24.2 and is involved in the signalling pathway downstream of GNAS. This is the reason for resistance to PTH and other hormones, as in PHP [44, 48].

Hand of a patient with acrodysostosis and multihormonal resistance (OMIM#101800). Severe and generalized brachydactyly, through very short and broad tubular bones, including ulna can be observed. Metacarpals II-V are proximally pointed and cone-shaped proximal phalangeal epiphyses are prematurely fused. The general appearance of the hand is bulky and stocky (courtesy of Prof. Dr. Jesús Argente).
The characteristic feature in this syndrome is severe BDE, characterized by shortening of metacarpals/metatarsals and phalanges II-V, phalangeal epiphyses being cone-shaped and prematurely fused, and affected bones appearing bulky and stocky (Figure 4) [44, 45, 48], while thumbs and halluces tend to be less affected [44, 52, 53]. In addition, a decreased interpedicular distance has been reported in 75% of patients. These severe brachydactyly and vertebral abnormalities have not been noted in PHP patients, so they could help us to differentiate between these two syndromes [45, 52]. Finally, another type of feature that could aid the differential diagnosis is subcutaneous ossification, as this is not seen in acrodysostosis [45, 52].

Hands of a patient with Bilginturan BD or HTNB (OMIM%112410). Metacarpals are short (specially II-V) with globular ends, irregular articular surfaces and cup deformity in the epiphysis of the proximal and medial phalanges. All the phalanges of the hands are also shortened, but the proximal and middle phalanges of the III and IV digits are relatively normal (courtesy of Dr. Sylvia Bähring and Dr. Okan Toka, unpublished case).
BDE without multihormonal resistance
Short stature
Bilginturan BD or hypertension with brachydactyly syndrome (HTNB, OMIM%112410)
Bilginturan et al. were the first to describe this syndrome in a large non-consanguineous Turkish family [54]. Subsequently, other authors have also studied this family and its new generations [55–66]. Affected members of the family had severe hypertension (30 mm Hg difference between affected and unaffected family members) from childhood [54] and a stroke by the age of 50 years if untreated [56]. Almost all the affected children have systolic blood pressure values over the 99th percentile [66]. However, the renin-angiotensin-aldosterone system (supine and upright) and circulating catecholamines respond normally, patients were not salt sensitive, their renal function was normal, they had no retinopathy or cardiac enlargement and the increase in radial wall thickness was minimal [55, 56, 59]. Affected individuals also presented BDE, were on average 10 cm shorter and had lower mean birth weight than their unaffected relatives, as well as having a stocky build and rather round face (see Figures two a & three a at Bilginturan et al. [54]) (Additional file 1: Table S5) [54, 56, 66].
Brachydactyly among the affected Turkish family members was heterogeneous. In most cases, shortening of more than one metacarpal and metatarsal was present [54], digits IV and V being most frequently affected but, in some cases, shortening has been seen in all the metacarpals and metatarsals in this and other families [56, 57, 66, 67]. Further, the shortening is not always symmetrical. All the phalanges of the hand were shortened, but the proximal and middle phalanges of the III and IV digits were relatively normal. Phalanges revealed cone-shaped epiphyses at proximal ends. All the phalanges of the toes were shortened and some patients presented symmetrical fusion between the middle and distal phalanges of toes IV and V, or only V (Figure 4). Additionally, shortening of carpal bones in the axial direction was also observed [54]. Bähring et al. compared the brachydactyly pattern profile of a 5.5-year-old Japanese patient, who carried a de novo (12) (p11.21p12.2) chromosomal deletion [68], with a 6-year-old boy from the Turkish family [57]. Both boys presented almost identical brachydactylies; type 16 cone-shaped epiphyses, particularly in the proximal interpahalangeal joints of digits II and V, brachyphalangy of digits II-IV and brachymetacarpalia of digits IV and V. The Japanese patient presented brachymesophalangy and cone-shaped epiphyses in digits II-V, and the Turkish boy only in digits II and V [57]. Intellectual disability, subcutaneous calcifications, obesity or differences in body mass index have not been observed [55, 57, 66], except in an isolated case reported by Derbent et al., where the patient presented obesity and a defect in the left renal artery [69]. It is important to highlight that brachydactyly is not appreciable until around 6 years of age and it aggravates significantly during early puberty as a result of an impaired growth spurt at the prepubertal stage. Given this, it is important to pay attention to the first signs of brachydactyly when an elevated blood pressure is detected in children (systolic blood pressure values >99th percentile) [66].
Another clinical feature in the Turkish family was the presence of aberrant posterior inferior cerebellar artery (PICA) loops only in affected members. Naraghi et al.[70] reported 100% loop cosegregation with hypertension and brachydactyly in a group of 27 members of the Turkish family (15 affected and 12 unaffected). The prevalence of the loops was not influenced by age or sex. Further, a brief report described a case of a boy with the same syndrome not only with the PICA abnormality but also abnormal renal arteries [71]. Additional studies found that affected individuals showed less active sympathetic nerve traffic to muscle than unaffected family members and also could not buffer blood pressure-elevating effects of phenylephrine [60, 65]. These findings confirmed that baroreflex blood pressure buffering was impaired in affected individuals [60, 66]. The arterial baroreflex is a critical cardiovascular reflex that provides continuous buffering of acute fluctuations in arterial blood pressure due, for example, to changes in posture, exercise, and emotion [72]. PICA loops can produce neurovascular compression of the ventrolateral medulla at the root entry zone of cranial nerves IX and X [63, 70], which are very important in the baroreflex mechanism. Arterial baroreceptors are mechanoreceptors: those located in the carotid sinuses are innervated by cranial nerve IX and those in the aortic arch by cranial nerve X. When arterial pressure increases, these nerves send an input to the nucleus of the solitary tract and barosensitive neurons from here initiate a sympatho-inhibitory pathway to reduce arterial blood pressure [72].
The molecular basis for this syndrome is unknown, but several researchers have found evidence of linkage to chromosome 12p, specifically to 12p11.21-12p.12.2 [55, 56, 73, 74]. Surprisingly, in a genome-wide linkage analysis, chromosome 12p was also implicated in Chinese patients with primary hypertension (not brachydactyly) [75]. Bahring et al. identified a complex rearrangement involving a deletion, reinsertion and inversion of the 12p12.2-p12.1 region [64]. This region is, however, relatively gene poor, only further characterized expressed sequence tags (EST) being identified in the vicinity [64]. These authors also proposed Kir6.1, SUR2, LSOX5 and PDE3A, which are within the studied segment, as candidate genes. Although, no alterations were found in these genes, a higher mRNA expression of PDE3A was detected in older affected family members (while the younger subjects affected showed expression values in the same range as those unaffected). On this basis, they suggested that the higher vascular expression of PDE3A was probably not the cause of the hypertension, but rather the result of an increase in blood pressure with age [64]. Slightly different rearrangements in this segment were found later in other families, apart from the Turkish one [65]. Finally, in this inverted sequence, a non-protein coding exon was found only to be expressed in unaffected family members; the rearrangement could be the cause for this lack of expression. This transcript was predicted to be an miRNA expressed in primary fibroblasts [65]. The fibroblasts of affected family members showed faster cell growth, but the function of this miRNA remains to be clarified [65].
More severe cases caused by deletions in the 12p11.21-12p.12.2 region were reported by Lu et al.[74] and Nagai et al.[68]. One patient, with a deletion spanning 71 annotated genes (PDE3A→BICD1), had moderate mental retardation, short stature, borderline high blood pressure (120/80 mmHg) and characteristic brachydactyly [74]. Another patient presented a more severe phenotype, mild mental retardation, short stature, high blood pressure, brachydactyly and cone-shaped epiphyses of the hands, hypoplastic hair and skin, oligodontia, a small thoracic cage and a hypoplastic pelvis [68]. The difference in the severity between the patients might be attributable to the extent of the deletion [74].
BDE with short stature, PTHLH type (OMIM#613382)
Maass et al. were the first to implicate the pthyroid hormone-like hormone gene (PTHLH), localized to 12p11.22, in BDE, after the identification of a translocation, t(8;12) (q13;p11.2) in a family with autosomal-dominant BDE and other malformations, including short extremities, dysmorphic facies, macrocephaly, prominent forehead and a depressed nasal root [76]. Despite the PTHLH gene not being disrupted by the translocation, it produced a downregulation of the gene and consequently of its downstream targets, ADAMTS-7 and ADAMTS-12[76]. These genes are highly expressed in chondrogenically differentiated fibroblasts, so they seem to play an important role in chondrogenesis [77, 78]. In a more recent publication, Maass et al. proposed that the downregulation of PTHLH, and its targets, was due to the disruption of the interaction with a regulatory element localized 24.43 Mb downstream of PTHLH, CISTR-ACT[79]. This element is a cis-regulatory element of PTHLH and also a trans-regulatory element of SOX9 (17q) [76, 80], which is another essential gene in chondrogenesis whose haploinsufficiency causes skeletal malformations [81]. This regulation occurs by an lncRNA (DA125942), derived from the CISTR-ACT locus, that coordinates the expression of SOX9 and PTHLH generating a loop in the chromatin [79, 80]. The translocations t(8;12) (q13;p11.2) and t(4;12) (q13.2-13.3;p11.2), later identified in another family, prevent the formation of such a loop and result in overexpression of the lncRNA and misexpression of PTHLH and SOX9[79, 80].
In an independent study, a deletion and three point mutations in the coding sequence of PTHLH were found to be responsible for BDE with short stature [82]. Further, in an isolated case of symmetrical enchondromatosis caused by a de novo duplication of 12p11.23 to 12p11.22 including PTHLH, the patient showed BDE with cone-shaped phalangeal epiphyses [83].
As in the previous group, the pattern of brachydactyly varies considerably. Several authors have reported shortening of metacarpals III, IV and V in most cases, while proximal, middle and distal phalange involvement was variable and shortening and cone-shaped epiphyses on middle and distal phalanges II and V were frequently mentioned (Figure 5) [76, 82, 83]. In two cases (one patient with a deletion involving another 6 genes and the second a carrier of a small duplication), metacarpals I and II were also shortened [82, 83], and in the case of translocation t(8;12) mentioned above, shortening of metacarpals I , III, IV and V was observed (Maass et al.; IV:2 patient) [76].

Right hand in a non adult individual with heterozygous mutation in PTHLH. Severe shortening of IV and V metacarpals is clearly observed (courtesy of Dr. Cécile Teinturier-Thomas, personal collection; Dr. Caroline Silve, molecular diagnosis, unpublished case).
Apart from BDE, other features were seen in these patients (Additional file 1: Table S6): most but not all had short stature [76, 82], tooth problems were reported by Klopocki et al. in two out of five families [82], and those affected with translocation t(8;12) had dysmorphic facies with macrocephaly [76]. Neither hypertension nor mental retardation was mentioned, but learning difficulties were reported in one family [82].
AHO-like
Brachydactyly mental retardation syndrome (BDMR, OMIM#600430)
Brachydactyly mental retardation syndrome (BDMR), also referred to as Albright hereditary osteodystrophy-like syndrome, is a rare pathology characterized by variable features: short stature, obesity, developmental delay, behavioural disorders, autism spectrum disorder and craniofacial and skeletal abnormalities, like BDE (see Figure one at Fernandez-Rebollo et al. [84]). Major malformations have also been reported, including cardiac, tracheal, gastrointestinal, genitourinary, CNS and skeletal abnormalities (Additional file 1: Table S7) [85, 86].
This syndrome is caused by deletions in 2q37. Recently, the minimum critical region responsible for this syndrome has been mapped to the 200 kb which involve the histone deacetylase 4 gene (HDAC4), localized exactly to 2q37.3 [87]. In addition, point mutations in HDAC4 have been found in two patients with BDMR [88]. On the other hand, a patient with a deletion in 2q37.3, but not including HDAC4, exhibited a similar phenotype but without BDE, cardiac or other major malformations [88]. Given this, it was proposed that HDAC4 haploinsufficiency could be causative of BDE in this syndrome [88]. A recent report, of a family case with a microdeletion involving HDAC4 and other two genes, puts this statement in doubt, because none of the three affected patients showed BDE [87]. So far, we know that BDE is a variable feature and only reported in about a half of patients with 2q37 microdeletions [87].
Histone deacetylase 4 is involved in bone, muscle, neurological and cardiac development [89]. Felder et al. demonstrated that FARP2, HDLBP and PASK genes were downregulated in a patient with a 2q37.3 terminal deletion compared to levels in his healthy family members and controls [90]. These are good candidate genes because they are involved in neuronal and/or skeletal development [90], but point mutations in these genes which cause BDMR or other similar syndrome have not yet been reported in the bibliography, except for the case of a patient with autism associated with a de novo MECP2 duplication who also had a second de novo duplication of 2q37.3 material involving PASK and HDLBP. However, this patient did not have an AHO-like phenotype [91].
BDE is only penetrant in 50-60% of cases and the pattern profile is variable [88, 92]. Frequently, metacarpals III and IV and metatarsal IV are shortened, but other metacarpals (II-V) have also been found to be affected, generally sparing metacarpal/metatarsal I [86, 88, 90, 93–98]. Further, individuals with similar deletions or family members with the same deletion may present different phenotypes; this could be a result of incomplete penetrance of the haploinsufficiency genes, epigenetics or regulation by other genes [92].
It is worth mentioning that there is a very similar syndrome, but without BDE, known as Smith-Magenis syndrome (SMS; OMIM#182290) which is caused by deletions in 17p11.2 or mutations in the RAI1 gene [88]. In addition, RAI1 expression is reduced in BDMR cases [88]. Hence, when individuals are suspected of having the BDMR phenotype, but no 2q37 deletions or HDAC4 mutations are detected, deletions at 17p11.2 and the RAI1 gene should be analysed.
Acrodysostosis without multihormonal resistance (ACRDYS2, OMIM:#614613)
In acrodysostosis without hormonal resistance, the affected gene is the phosphodiesterase 4D, cAMP-specific (PDE4D) gene, localized to 5q11.2 [44, 46, 49, 99]. Clinical features (Additional file 1: Table S4) are similar to ACRDYS1 apart from PTH resistance not generally being present [44, 99], though an exception was reported by Michot et al.[46]. Other features it has been suggested may differ in the two types are: (i) facial dysostosis, which is more evident in ACRDYS2 than in ACRDYS1; and (ii) intellectual disability, which is common in ACRDYS2, while ACRDYS1 has only been associated with some behavioural disorders and/or learning difficulties [44, 46]. On the other hand, in the study of Lee et al. all patients (three with PDE4D and two with PRKAR1A mutations) had midface hypoplasia and there were no differences in developmental disability according to the gene affected (among those with PDE4D mutations, one was not affected, a second was mildly affected and a third was significantly affected; whereas both patients with PRKAR1A mutations had mild developmental disabilities) [49]. Lynch et al. described eight patients; all of them had some degree of learning difficulties, developmental delay and intellectual disability [99]. These authors suggested that in patients with acrodysostosis associated with normal stature (only 5/15 presented short stature [46, 49, 99]), progressive obesity (frequently developing after 6 years of age), and mild intellectual disability but no PTH resistance, PDE4D rather than PRKAR1A mutations should be suspected. BDE, stenosis of the lumbar spine, nasal hypoplasia, a flat nasal bridge and short stature are features shared by both subtypes of acrodysostosis (see Figure three Pat. 15, 16 at Linglart et al. [44]). In addition, some cases of hypogonadism have been found with defects in either gene [44, 49, 99].
Thus, it is difficult to distinguish acrodysostosis cases with PDE4D mutations from those with PRKAR1A mutations on the basis of clinical observation only [49]. Therefore, biochemical studies of calcium and phosphorus metabolism are essential.
Tricho-rhino-phalangeal syndrome (TRPS)
This syndrome is classified into three types: TRPS I (OMIM#190350), TRPS II (OMIM#150230) and TRPS III (OMIM#190351). Mutations in the trichorhinophalangeal syndrome I gene (TRPS1), localized to 8q23.3, have been found in 88% of cases of TRSP I and TRSP III [100]. Lüdecke et al. described TRPS III as an extreme of the clinical spectrum of TRSP I [100]. On the other hand, TRPS II is a contiguous gene syndrome involving TRPS1 and EXT1 genes; patients develop cartilaginous exostoses in addition to the features seen in TRPS I and III [100, 101].
The striking features shared by the three types are sparse, slowly growing scalp hair, laterally sparse eyebrows, a bulbous tip of the nose (pear-shaped nose), long flat philtrum, thin upper vermillion border and protruding ears (see Figure three Momeni et al. [102]) (Additional file 1: Table S8) [100]. In radiography, the main findings are: short stature, BDE, mild metaphyseal convexity during the first year of life and premature fusion of the growth plates of the tubular bonds [100]. Additionally, hip malformations (coxa plana, coxa magna or coxa vara) have been reported in more than 70% of cases and degenerative arthrosis appears in older individuals [100]. Patients show a retarded skeletal age until puberty, and then an accelerated skeletal age, while the mean length at birth is normal [100]. That is, the growth retardation is a progressive process that can be appreciated in metacarpophalangeal pattern profile (MCPP) analysis comparing children and adults, and comparisons of their relative height [100]. Growth hormone deficiency has been reported in some cases [103–105]. Intellectual disability has also been reported in the three types, but most commonly in TRPS II [100, 101, 106, 107].
Regarding brachydactyly, shortening of the metacarpals has been reported in about half of patients and cone-shaped epiphyses of middle phalanges, type 12/12A (only appreciable after infancy in early childhood, before epiphyses fuse), in almost all patients, in most cases in mesophalanges II and III. Outcarving and deformation of the cones is another striking feature and this is more easily appreciated after epiphysis fusion (Figure 6) [108, 109]. In addition, ivory cones were reported by Giedion (again, a feature that is only appreciable after infancy in early childhood, before epiphyses fuse). Hypoplasia of the thumb, as well as shortening of metacarpals II-V and of middle phalanges II and V is frequently seen, although shortening of all middle phalanges has been also reported [101, 106, 108, 110], as have small feet and a short hallux [110]. Poznanski et al. conducted MCPP analysis comparing PHP and PPHP with TRPS and found no similarity between pattern profiles in the two syndromes [20]. These data were published before the association of the syndrome with the TRPS1 gene. More recently, Lüdecke et al. used MCPP analysis to compare various groups of TRPS patients; first of all, they concluded that like height differences, the brachydactyly was more marked in adults than in children. On the other hand, patients with missense mutations (TRPS III) had more severe brachydactyly than the patients with the nonsense ones (TRPS I). Given this, they classified the brachydactyly pattern with respect to the mean profiles for TRSP I and TRSP III, but these mean values were only intended for classification not to be used as a tool for diagnosis [100]. Additionally, patients with missense mutations were shorter than the nonsense carriers, but patients with gene contiguous syndrome (TRPS II) were the smallest [100]. Finally, the brachydactyly pattern is variable, as reflected in a lack of correlation between MCPPs in patients with the same recurrent mutation as well as the presence of asymmetric brachydactyly in some patients [100].

Hands of a patient with tricho-rhino-phalangeal syndrome I (OMIM#190350). The patient presents asymmetrical brachydactyly; metacarpals III and V are shortened on the left hand and IV and V on the right. Further, middle phalanges are shortened and cone-shaped epiphyses are shown with the typical outcarving and deformation (arrows) (courtesy of Dr. Sharona Azriel, unpublished case).
The severity of the TRPS phenotype might also depend on the size of the deletion in 8q and whether there is mosaicism for the deletion, which has been reported in four patients, two with TRPS I and two with TRPS II [101].
Turner syndrome (TS)
Turner syndrome is a chromosomal disorder with a complete or partial monosomy of X chromosome. The genetic background is variable, the most frequent karyotypes are: 45,X (~50%), karyotypes with an isochromosome of X (i(Xq) or i(Xp)), the mosaic 45,X/46XX (%30), and karyotypes containing an entire or parts of Y chromosome [111, 112].
The main features are short stature (90-100%), gonadal dysgenesis, pubertal delay, primary amenorrhea (85%), estrogens insufficiency, cardiac anomalies and/or other congenital malformations like brachydactyly E (see Figure one at Bondy [113]) [111, 112, 114–116]. Other frequent features are: lymphedema, congenital malformations of the urinary system (30-40%), abnormalities of the external ocular appendages, (epicanthal folds, ptosis, hypertelorism, upward slanting palpebral fissures), strabismus and hypermetropia (25–35%), hearing problems and ear malformations, dysmorphic craniofacial features, abnormalities in tooth development and morphology (early eruption), melanocytic nevi, normal GH secretory pattern, altered liver enzymes¸ decreased bone mineral density, etc. [111, 112, 114–117] (Additional file 1: Table S9).
Regarding brachydactyly E, the more typical brachydactyly of Turner syndrome is the shortened of the IV metacarpals, but it is variable [20, 114, 118]. Poznanski compared the brachydactyly associated with PPHP-PHP versus TS and observed that the shortening of the bones are less severe in TS than in PPHP-PHP; shortening of I distal (D1) phalanx’s is less frequent in TS (5% in contrast to the 75% of PPHP-PHP), so if a patient presents short fourth metacarpals and a normal D1, it is more probable that have Turner syndrome than PHP-PPHP. Finally, he mentions drum stick phalanges and thin bones in TS (Figure 7) [20].

Hand (A) and radius (B) of a patient with Turner syndrome. It can be observed the ulnar and palmar slant of the radial articular surfaces, with a triangular appearance of the distal radial epiphysis and slight shortening of the IV metacarpal; the radius is short and bowed.
Although TS is quite well known entity, there is often a delay in the diagnosis as over 20% of patients are diagnosed after 12 yr [115, 119]. As other syndromes mentioned in this review, it is essential to be diagnosed during infancy for proper early treatment to avoid complications linked to TS. In fact there are some publications explaining the clinical guidelines to be followed to optimize care for young woman and adults with TS [111, 112, 114–117].
Concluding remarks
The BDE pattern profile can vary considerably within a syndrome, even between unrelated patients with identical mutations and among affected family members. In addition, brachydactyly is sometimes not appreciable until 6 years of age, because it is a progressive feature and tends to become more evident during early puberty.
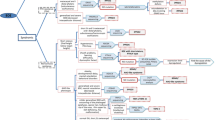
With the aim of helping to identify the most probable genetic diagnosis to inform the clinical management of the aforementioned syndromes with BDE, we have established a diagnostic algorithm including specific differential features (Figure 8). In a multidisciplinary approach for patients with BDE, there are some very subtle but helpful clues that allow us to distinguish between some of the syndromes. (i) BDE observed in individuals with acrodysostosis can be differentiated fairly clearly because of its severity: all the bones in the hand are affected and are characteristically stocky in appearance [44, 45, 48]. (ii) TRPS can also be easily recognised because the MCPP is significantly different and there are also other characteristic clinical features: sparse, slowly growing scalp hair and a bulb-shaped nose are very suggestive of this syndrome [20].

Proposed clinical algorithm to suggest the most probable genetic diagnosis. Features in brackets are not always present in the syndromes. BDE Brachydactyly type E, PHP-Ia Pseudohypopthyroidism type Ia, MLPA Multiplex ligation-dependent probe amplification, MS-MLPA Methylation specific-MLPA.
Poznanski et al. also carried out MCPP analysis comparing isolated BDE and PHP/PPHP, but at the time these two syndromes had not been genetically well differentiated and hence the conclusions may not be reliable [20]. Indeed, patients with different pathologies could be confused if attention were to be focused on radiographic findings alone; hence, biochemical analyses should be included.
The other syndromes discussed have not yet been compared by MCPP analysis, but it seems that the pattern of brachydactyly does not differ appreciably between them. In such cases, when the BDE pattern is not sufficiently informative, there are other features which can help in the differentiation of each syndrome (Additional file 1: Table S1). These features are listed in the supplementary material (Additional file 1: Table S2–S9).
References
Temtamy SA, Aglan MS: Brachydactyly. Orphanet J Rare Dis. 2008, 3: 15. 10.1186/1750-1172-3-15.
Hall CM: International nosology and classification of constitutional disorders of bone (2001). Am J Med Genet. 2002, 113: 65-77. 10.1002/ajmg.10828.
Warman ML, Cormier-Daire V, Hall C, Krakow D, Lachman R, LeMerrer M, et al: Nosology and classification of genetic skeletal disorders: 2010 revision. Am J Med Genet A. 2011, 155A: 943-968.
Bell J: On Brachydactyly and Symphalangism. London: Cambridge University Press; 1951.
Temtamy SA, McKusick VA: The Genetics of Hand Malformations. NewYork: The National Foundation. March Dimes. Alan R Liss Inc; 1978.
Mundlos S: The brachydactylies: a molecular disease family. Clin Genet. 2009, 76: 123-136. 10.1111/j.1399-0004.2009.01238.x.
Hertzog KP: Brachydactyly and pseudo-pseudohypopthyroidism. Acta Genet Med Gemellol (Roma). 1968, 17: 428-438.
Canepa G, Maroteaux P, Pietrogrande V: Dysmorphic Syndromes and Constitutional Disease of the Skeleton. Padova, Italy: Piccin Nuova Libraria; 2001.
Johnson D, Kan SH, Oldridge M, Trembath RC, Roche P, Esnouf RM, et al: Missense mutations in the homeodomain of HOXD13 are associated with brachydactyly types D and E. Am J Hum Genet. 2003, 72: 984-997. 10.1086/374721.
Caronia G, Goodman FR, McKeown CM, Scambler PJ, Zappavigna V: An I47L substitution in the HOXD13 homeodomain causes a novel human limb malformation by producing a selective loss of function. Development. 2003, 130: 1701-1712. 10.1242/dev.00396.
Jamsheer A, Sowinska A, Kaczmarek L, Latos-Bielenska A: Isolated brachydactyly type E caused by a HOXD13 nonsense mutation: a case report. BMC Med Genet. 2012, 13: 4.
Brison N, Tylzanowski P, Debeer P: Limb skeletal malformations - what the HOX is going on?. Eur J Med Genet. 2012, 55: 1-7. 10.1016/j.ejmg.2011.06.003.
Oude Luttikhuis ME, Williams DK, Trembath RC: Isolated autosomal dominant type E brachydactyly: exclusion of linkage to candidate regions 2q37 and 20q13. J Med Genet. 1996, 33: 873-876. 10.1136/jmg.33.10.873.
Zhao X, Sun M, Zhao J, Leyva JA, Zhu H, Yang W, et al: Mutations in HOXD13 underlie syndactyly type V and a novel brachydactyly-syndactyly syndrome. Am J Hum Genet. 2007, 80: 361-371. 10.1086/511387.
Drezner MK, Neelon FA, Haussler M, McPherson HT, Lebovitz HE: 1,25-Dihydroxycholecalciferol deficiency: the probable cause of hypocalcemia and metabolic bone disease in pseudohypopthyroidism. J Clin Endocrinol Metab. 1976, 42: 621-628. 10.1210/jcem-42-4-621.
Stone MD, Hosking DJ, Garcia-Himmelstine C, White DA, Rosenblum D, Worth HG: The renal response to exogenous pthyroid hormone in treated pseudohypopthyroidism. Bone. 1993, 14: 727-735. 10.1016/8756-3282(93)90204-N.
Murray TM, Rao LG, Wong MM, Waddell JP, McBroom R, Tam CS, et al: Pseudohypopthyroidism with osteitis fibrosa cystica: direct demonstration of skeletal responsiveness to pthyroid hormone in cells cultured from bone. J Bone Miner Res. 1993, 8: 83-91.
Ish-Shalom S, Rao LG, Levine MA, Fraser D, Kooh SW, Josse RG, et al: Normal pthyroid hormone responsiveness of bone-derived cells from a patient with pseudohypopthyroidism. J Bone Miner Res. 1996, 11: 8-14.
De Sanctis L, Vai S, Andreo MR, Romagnolo D, Silvestro L, de Sanctis C: Brachydactyly in 14 genetically characterized pseudohypopthyroidism type Ia patients. J Clin Endocrinol Metab. 2004, 89: 1650-1655. 10.1210/jc.2003-030850.
Poznanski AK, Werder EA, Giedion A, Martin A, Shaw H: The pattern of shortening of the bones of the hand in PHP and PPHP–A comparison with brachydactyly E, Turner Syndrome, and acrodysostosis. Radiology. 1977, 123: 707-718.
Wilson LC, Hall CM: Albright’s hereditary osteodystrophy and pseudohypopthyroidism. Semin Musculoskelet Radiol. 2002, 6: 273-283. 10.1055/s-2002-36726.
Levine MA, Modi WS, O’Brien SJ: Mapping of the gene encoding the alpha subunit of the stimulatory G protein of adenylyl cyclase (GNAS1) to 20q13.2----q13.3 in human by in situ hybridization. Genomics. 1991, 11: 478-479. 10.1016/0888-7543(91)90164-A.
Bastepe M: The GNAS locus: quintessential complex gene encoding gsalpha, xlalphas, and other imprinted transcripts. Curr Genomics. 2007, 8: 398-414. 10.2174/138920207783406488.
Hayward BE, Barlier A, Korbonits M, Grossman AB, Jacquet P, Enjalbert A, et al: Imprinting of the G(s)alpha gene GNAS1 in the pathogenesis of acromegaly. J Clin Invest. 2001, 107: R31-R36. 10.1172/JCI11887.
Mantovani G, Ballare E, Giammona E, Beck-Peccoz P, Spada A: The gsalpha gene: predominant maternal origin of transcription in human thyroid gland and gonads. J Clin Endocrinol Metab. 2002, 87: 4736-4740. 10.1210/jc.2002-020183.
Germain-Lee EL, Ding CL, Deng Z, Crane JL, Saji M, Ringel MD, et al: Paternal imprinting of Galpha(s) in the human thyroid as the basis of TSH resistance in pseudohypopthyroidism type 1a. Biochem Biophys Res Commun. 2002, 296: 67-72. 10.1016/S0006-291X(02)00833-1.
Weinstein LS, Yu S, Warner DR, Liu J: Endocrine manifestations of stimulatory G protein alpha-subunit mutations and the role of genomic imprinting. Endocr Rev. 2001, 22: 675-705. 10.1210/er.22.5.675.
Miao ZM, Wang C, Wang BB, Meng DM, Su DM, Cheng Z, et al: Identification of a novel mutation in a pseudohypopthyroidism family. Int J Endocrinol. 2011, 201 (1): 509549.
Weinstein LS: Albright hereditary osteodystrophy, pseudohypopthyroidism, and Gs deficiency. G proteins, receptors and disease. Edited by: Spiegel AM. New Jersey: Humara Press; 1998: 23-56.
Perez de Nanclares G, Fernandez-Rebollo E, Santin I, Garcia-Cuartero B, Gaztambide S, Menendez E, et al: Epigenetic defects of GNAS in patients with pseudohypopthyroidism and mild features of Albright’s hereditary osteodystrophy. J Clin Endocrinol Metab. 2007, 92: 2370-2373. 10.1210/jc.2006-2287.
Mariot V, Maupetit-Mehouas S, Sinding C, Kottler ML, Linglart A: A maternal epimutation of GNAS leads to Albright osteodystrophy and PTH resistance. J Clin Endocrinol Metab. 2008, 93: 661-665.
Unluturk U, Harmanci A, Babaoglu M, Yasar U, Varli K, Bastepe M, et al: Molecular diagnosis and clinical characterization of pseudohypopthyroidism type-Ib in a patient with mild Albright’s hereditary osteodystrophy-like features, epileptic seizures, and defective renal handling of uric acid. Am J Med Sci. 2008, 336: 84-90. 10.1097/MAJ.0b013e31815b218f.
Mantovani G, de Sanctis L, Barbieri AM, Elli FM, Bollati V, Vaira V, et al: Pseudohypopthyroidism and GNAS epigenetic defects: clinical evaluation of albright hereditary osteodystrophy and molecular analysis in 40 patients. J Clin Endocrinol Metab. 2010, 95: 651-658. 10.1210/jc.2009-0176.
Zazo C, Thiele S, Martin C, Fernandez-Rebollo E, Martinez-Indart L, Werner R, et al: Gsalpha activity is reduced in erythrocyte membranes of patients with psedohypopthyroidism due to epigenetic alterations at the GNAS locus. J Bone Miner Res. 2011, 26: 1864-1870. 10.1002/jbmr.369.
Bastepe M, Frohlich LF, Linglart A, Abu-Zahra HS, Tojo K, Ward LM, et al: Deletion of the NESP55 differentially methylated region causes loss of maternal GNAS imprints and pseudohypopthyroidism type Ib. Nat Genet. 2005, 37: 25-27. 10.1038/ng1560.
Chillambhi S, Turan S, Hwang DY, Chen HC, Juppner H, Bastepe M: Deletion of the noncoding GNAS antisense transcript causes pseudohypopthyroidism type Ib and biparental defects of GNAS methylation in cis. J Clin Endocrinol Metab. 2010, 95: 3993-4002. 10.1210/jc.2009-2205.
Mackay DJ, Callaway JL, Marks SM, White HE, Acerini CL, Boonen SE, et al: Hypomethylation of multiple imprinted loci in individuals with transient neonatal diabetes is associated with mutations in ZFP57. Nat Genet. 2008, 40: 949-951. 10.1038/ng.187.
Meyer E, Lim D, Pasha S, Tee LJ, Rahman F, Yates JR, et al: Germline mutation in NLRP2 (NALP2) in a familial imprinting disorder (Beckwith-Wiedemann Syndrome). PLoS Genet. 2009, 5: e1000423. 10.1371/journal.pgen.1000423.
Perez-Nanclares G, Romanelli V, Mayo S, Garin I, Zazo C, Fernandez-Rebollo E, et al: Detection of hypomethylation syndrome among patients with epigenetic alterations at the GNAS Locus. J Clin Endocrinol Metab. 2012, 97: E1060-E1067. 10.1210/jc.2012-1081.
Bastepe M, Lane AH, Juppner H: Paternal uniparental isodisomy of chromosome 20q–and the resulting changes in GNAS1 methylation–as a plausible cause of pseudohypopthyroidism. Am J Hum Genet. 2001, 68: 1283-1289. 10.1086/320117.
Fernandez-Rebollo E, Lecumberri B, Garin I, Arroyo J, Bernal-Chico A, Goni F, et al: New mechanisms involved in paternal 20q disomy associated with pseudohypopthyroidism. Eur J Endocrinol. 2010, 163: 953-962. 10.1530/EJE-10-0435.
Bastepe M, Altug-Teber O, Agarwal C, Oberfield SE, Bonin M, Juppner H: Paternal uniparental isodisomy of the entire chromosome 20 as a molecular cause of pseudohypopthyroidism type Ib (PHP-Ib). Bone. 2011, 48: 659-662. 10.1016/j.bone.2010.10.168.
Sanchez J, Perera E, Jan de Beur S, Ding C, Dang A, Berkovitz GD, et al: Madelung-Like Deformity in Pseudohypopthyroidism Type 1b. J Clin Endocrinol Metab. 2011, 96: E1507-E1511. 10.1210/jc.2011-1411.
Linglart A, Fryssira H, Hiort O, Holterhus PM, Perez de Nanclares G, Argente J, et al: PRKAR1A and PDE4D mutations cause acrodysostosis but two distinct syndromes with or without GPCR-signaling hormone resistance. J Clin Endocrinol Metab. 2012, 97: E2328-E2338. 10.1210/jc.2012-2326.
Robinow M, Pfeiffer RA, Gorlin RJ, McKusick VA, Renuart AW, Johnson GF, et al: Acrodysostosis. A syndrome of peripheral dysostosis, nasal hypoplasia, and mental retardation. Am J Dis Child. 1971, 121: 195-203.
Michot C, Le GC, Goldenberg A, Abhyankar A, Klein C, Kinning E, et al: Exome sequencing identifies PDE4D mutations as another cause of acrodysostosis. Am J Hum Genet. 2012, 90: 740-745. 10.1016/j.ajhg.2012.03.003.
Maroteaux P, Malamut G: [Acrodysostosis]. Presse Med. 1968, 76: 2189-2192.
Linglart A, Menguy C, Couvineau A, Auzan C, Gunes Y, Cancel M, et al: Recurrent PRKAR1A mutation in acrodysostosis with hormone resistance. N Engl J Med. 2011, 364: 2218-2226. 10.1056/NEJMoa1012717.
Lee H, Graham JM, Rimoin DL, Lachman RS, Krejci P, Tompson SW, et al: Exome sequencing identifies PDE4D mutations in acrodysostosis. Am J Hum Genet. 2012, 90: 746-751. 10.1016/j.ajhg.2012.03.004.
Hamanishi C, Nagata Y, Nagao Y, Sohen S, Tanaka S: Acrodysostosis associated with spinal canal stenosis. Spine (Phila Pa 1976 ). 1993, 18: 1922-1925. 10.1097/00007632-199310000-00035.
Butler MG, Rames LJ, Wadlington WB: Acrodysostosis: report of a 13-year-old boy with review of literature and metacarpophalangeal pattern profile analysis. Am J Med Genet. 1988, 30: 971-980. 10.1002/ajmg.1320300416.
Silve C, Clauser E, Linglart A: Acrodysostosis. Horm Metab Res. 2012, 44: 749-758.
Graham JM, Krakow D, Tolo VT, Smith AK, Lachman RS: Radiographic findings and Gs-alpha bioactivity studies and mutation screening in acrodysostosis indicate a different etiology from pseudohypopthyroidism. Pediatr Radiol. 2001, 31: 2-9. 10.1007/s002470000355.
Bilginturan N, Zileli S, Karacadag S, Pirnar T: Hereditary brachydactyly associated with hypertension. J Med Genet. 1973, 10: 253-259. 10.1136/jmg.10.3.253.
Schuster H, Wienker TE, Bahring S, Bilginturan N, Toka HR, Neitzel H, et al: Severe autosomal dominant hypertension and brachydactyly in a unique Turkish kindred maps to human chromosome 12. Nat Genet. 1996, 13: 98-100. 10.1038/ng0596-98.
Schuster H, Wienker TF, Toka HR, Bahring S, Jeschke E, Toka O, et al: Autosomal dominant hypertension and brachydactyly in a Turkish kindred resembles essential hypertension. Hypertension. 1996, 28: 1085-1092. 10.1161/01.HYP.28.6.1085.
Bahring S, Nagai T, Toka HR, Nitz I, Toka O, Aydin A, et al: Deletion at 12p in a Japanese child with brachydactyly overlaps the assigned locus of brachydactyly with hypertension in a Turkish family. Am J Hum Genet. 1997, 60: 732-735.
Schuster H: Autosomal dominant hypertension with brachydactyly: an enigmatic form of monogenic hypertension. Nephrol Dial Transplant. 1998, 13: 1337-1340. 10.1093/ndt/13.6.1337.
Hattenbach LO, Toka HR, Toka O, Schuster H, Luft FC: Absence of hypertensive retinopathy in a Turkish kindred with autosomal dominant hypertension and brachydactyly. Br J Ophthalmol. 1998, 82: 1363-1365. 10.1136/bjo.82.12.1363.
Jordan J, Toka HR, Heusser K, Toka O, Shannon JR, Tank J, et al: Severely impaired baroreflex-buffering in patients with monogenic hypertension and neurovascular contact. Circulation. 2000, 102: 2611-2618. 10.1161/01.CIR.102.21.2611.
Luft FC: Monogenic hypertension: lessons from the genome. Kidney Int. 2001, 60: 381-390. 10.1046/j.1523-1755.2001.00810.x.
Luft FC, Toka O, Toka HR, Jordan J, Bahring S: Mendelian hypertension with brachydactyly as a molecular genetic lesson in regulatory physiology. Am J Physiol Regul Integr Comp Physiol. 2003, 285: R709-R714.
Luft FC: Mendelian forms of human hypertension and mechanisms of disease. Clin Med Res. 2003, 1: 291-300. 10.3121/cmr.1.4.291.
Bahring S, Rauch A, Toka O, Schroeder C, Hesse C, Siedler H, et al: Autosomal-dominant hypertension with type E brachydactyly is caused by rearrangement on the short arm of chromosome 12. Hypertension. 2004, 43: 471-476. 10.1161/01.HYP.0000111808.08715.ec.
Bahring S, Kann M, Neuenfeld Y, Gong M, Chitayat D, Toka HR, et al: Inversion region for hypertension and brachydactyly on chromosome 12p features multiple splicing and noncoding RNA. Hypertension. 2008, 51: 426-431. 10.1161/HYPERTENSIONAHA.107.101774.
Toka O, Maass PG, Aydin A, Toka H, Hubner N, Ruschendorf F, et al: Childhood hypertension in autosomal-dominant hypertension with brachydactyly. Hypertension. 2010, 56: 988-994. 10.1161/HYPERTENSIONAHA.110.156620.
Chitayat D, Grix A, Balfe JW, Abramowicz JS, Garza J, Fong CT, et al: Brachydactyly-short stature-hypertension (Bilginturan) syndrome: report on two families. Am J Med Genet. 1997, 73: 279-285. 10.1002/(SICI)1096-8628(19971219)73:3<279::AID-AJMG10>3.0.CO;2-G.
Nagai T, Nishimura G, Kato R, Hasegawa T, Ohashi H, Fukushima Y: Del(12)(p11.21p12.2) associated with an asphyxiating thoracic dystrophy or chondroectodermal dysplasia-like syndrome. Am J Med Genet. 1995, 55: 16-18. 10.1002/ajmg.1320550106.
Derbent M, Baskin E, Agildere M, Agras PI, Saatci U: Brachydactyly short-stature hypertension syndrome: a case with associated vascular malformations. Pediatr Nephrol. 2006, 21: 390-393. 10.1007/s00467-005-2111-6.
Naraghi R, Schuster H, Toka HR, Bahring S, Toka O, Oztekin O, et al: Neurovascular compression at the ventrolateral medulla in autosomal dominant hypertension and brachydactyly. Stroke. 1997, 28: 1749-1754. 10.1161/01.STR.28.9.1749.
Litwin M, Jurkiewicz E, Nowak K, Kosciesza A, Grenda R, Malczyk K, et al: Arterial hypertension with brachydactyly in a 15-year-old boy. Pediatr Nephrol. 2003, 18: 814-819. 10.1007/s00467-003-1169-2.
Benarroch EE: The arterial baroreflex: functional organization and involvement in neurologic disease. Neurology. 2008, 71: 1733-1738. 10.1212/01.wnl.0000335246.93495.92.
Toka HR, Bahring S, Chitayat D, Melby JC, Whitehead R, Jeschke E, et al: Families with autosomal dominant brachydactyly type E, short stature, and severe hypertension. Ann Intern Med. 1998, 129: 204-208. 10.7326/0003-4819-129-3-199808010-00008.
Lu HY, Cui YX, Shi YC, Xia XY, Liang Q, Yao B, et al: A girl with distinctive features of borderline high blood pressure, short stature, characteristic brachydactyly, and 11.47 Mb deletion in 12p11.21-12p12.2 by oligonucleotide array CGH. Am J Med Genet A. 2009, 149A: 2321-2323. 10.1002/ajmg.a.33030.
Gong M, Zhang H, Schulz H, Lee YA, Sun K, Bahring S, et al: Genome-wide linkage reveals a locus for human essential (primary) hypertension on chromosome 12p. Hum Mol Genet. 2003, 12: 1273-1277. 10.1093/hmg/ddg135.
Maass PG, Wirth J, Aydin A, Rump A, Stricker S, Tinschert S, et al: A cis-regulatory site downregulates PTHLH in translocation t(8;12)(q13;p11.2) and leads to Brachydactyly Type E. Hum Mol Genet. 2010, 19: 848-860. 10.1093/hmg/ddp553.
Bai XH, Wang DW, Kong L, Zhang Y, Luan Y, Kobayashi T, et al: ADAMTS-7, a direct target of PTHrP, adversely regulates endochondral bone growth by associating with and inactivating GEP growth factor. Mol Cell Biol. 2009, 29: 4201-4219. 10.1128/MCB.00056-09.
Bai XH, Wang DW, Luan Y, Yu XP, Liu CJ: Regulation of chondrocyte differentiation by ADAMTS-12 metalloproteinase depends on its enzymatic activity. Cell Mol Life Sci. 2009, 66: 667-680. 10.1007/s00018-008-8633-x.
Maass PG, Rump A, Schulz H, Stricker S, Schulze L, Platzer K, et al: A misplaced lncRNA causes brachydactyly in humans. J Clin Invest. 2012, 122: 3990-4002. 10.1172/JCI65508.
Troy A, Sharpless NE: Genetic “lnc”-age of noncoding RNAs to human disease. J Clin Invest. 2012, 122: 3837-3840. 10.1172/JCI66645.
Stricker S, Mundlos S: Mechanisms of digit formation: human malformation syndromes tell the story. Dev Dyn. 2011, 240: 990-1004. 10.1002/dvdy.22565.
Klopocki E, Hennig BP, Dathe K, Koll R, de Ravel T, Baten E, et al: Deletion and point mutations of PTHLH cause brachydactyly type E. Am J Hum Genet. 2010, 86: 434-439. 10.1016/j.ajhg.2010.01.023.
Collinson M, Leonard SJ, Charlton J, Crolla JA, Silve C, Hall CM, et al: Symmetrical enchondromatosis is associated with duplication of 12p11.23 to 12p11.22 including PTHLH. Am J Med Genet A. 2010, 152A: 3124-3128. 10.1002/ajmg.a.33567.
Fernandez-Rebollo E, Perez O, Martinez-Bouzas C, Cotarelo-Perez MC, Garin I, Ruibal JL, et al: Two cases of deletion 2q37 associated with segregation of an unbalanced translocation 2;21: choanal atresia leading to misdiagnosis of CHARGE syndrome. Eur J Endocrinol. 2009, 160: 711-717.
Wilson LC, Leverton K, Oude Luttikhuis ME, Oley CA, Flint J, Wolstenholme J, et al: Brachydactyly and mental retardation: an Albright hereditary osteodystrophy-like syndrome localized to 2q37. Am J Hum Genet. 1995, 56: 400-407.
Falk RE, Casas KA: Chromosome 2q37 deletion: clinical and molecular aspects. Am J Med Genet C Semin Med Genet. 2007, 145C: 357-371. 10.1002/ajmg.c.30153.
Villavicencio-Lorini P, Klopocki E, Trimborn M, Koll R, Mundlos S, Horn D: Phenotypic variant of Brachydactyly-mental retardation syndrome in a family with an inherited interstitial 2q37.3 microdeletion including HDAC4. Eur J Hum Genet. 2013, 21: 743-748. 10.1038/ejhg.2012.240.
Williams SR, Aldred MA, Der Kaloustian VM, Halal F, Gowans G, McLeod DR, et al: Haploinsufficiency of HDAC4 causes brachydactyly mental retardation syndrome, with brachydactyly type E, developmental delays, and behavioral problems. Am J Hum Genet. 2010, 87: 219-228. 10.1016/j.ajhg.2010.07.011.
Tammachote R, Kingsuwannapong N, Tongkobpetch S, Srichomthong C, Yeetong P, Kingwatanakul P, et al: Primary hyperoxaluria type 1 and brachydactyly mental retardation syndrome caused by a novel mutation in AGXT and a terminal deletion of chromosome 2. Am J Med Genet A. 2012, 158A: 2124-2130. 10.1002/ajmg.a.35495.
Felder B, Radlwimmer B, Benner A, Mincheva A, Todt G, Beyer KS, et al: FARP2, HDLBP and PASK are downregulated in a patient with autism and 2q37.3 deletion syndrome. Am J Med Genet A. 2009, 149A: 952-959. 10.1002/ajmg.a.32779.
Grasshoff U, Bonin M, Goehring I, Ekici A, Dufke A, Cremer K, et al: De novo MECP2 duplication in two females with random X-inactivation and moderate mental retardation. Eur J Hum Genet. 2011, 19: 507-512. 10.1038/ejhg.2010.226.
Aldred MA, Sanford RO, Thomas NS, Barrow MA, Wilson LC, Brueton LA, et al: Molecular analysis of 20 patients with 2q37.3 monosomy: definition of minimum deletion intervals for key phenotypes. J Med Genet. 2004, 41: 433-439. 10.1136/jmg.2003.017202.
Shrimpton AE, Braddock BR, Thomson LL, Stein CK, Hoo JJ: Molecular delineation of deletions on 2q37.3 in three cases with an Albright hereditary osteodystrophy-like phenotype. Clin Genet. 2004, 66: 537-544. 10.1111/j.1399-0004.2004.00363.x.
Power MM, James RS, Barber JC, Fisher AM, Wood PJ, Leatherdale BA, et al: RDCI, the vasoactive intestinal peptide receptor: a candidate gene for the features of Albright hereditary osteodystrophy associated with deletion of 2q37. J Med Genet. 1997, 34: 287-290. 10.1136/jmg.34.4.287.
Chaabouni M, Le MM, Raoul O, Prieur M, de Blois MC, Philippe A, et al: Molecular cytogenetic analysis of five 2q37 deletions: refining the brachydactyly candidate region. Eur J Med Genet. 2006, 49: 255-263. 10.1016/j.ejmg.2005.07.001.
Morris B, Etoubleau C, Bourthoumieu S, Reynaud-Perrine S, Laroche C, Lebbar A, et al: Dose dependent expression of HDAC4 causes variable expressivity in a novel inherited case of brachydactyly mental retardation syndrome. Am J Med Genet A. 2012, 158A: 2015-2020. 10.1002/ajmg.a.35463.
Casas KA, Mononen TK, Mikail CN, Hassed SJ, Li S, Mulvihill JJ, et al: Chromosome 2q terminal deletion: report of 6 new patients and review of phenotype-breakpoint correlations in 66 individuals. Am J Med Genet A. 2004, 130A: 331-339. 10.1002/ajmg.a.30156.
Giardino D, Finelli P, Gottardi G, De CG, Della MM, Lonardo F, et al: Narrowing the candidate region of Albright hereditary osteodystrophy-like syndrome by deletion mapping in a patient with an unbalanced cryptic translocation t(2;6)(q37.3;q26). Am J Med Genet A. 2003, 122A: 261-265. 10.1002/ajmg.a.20287.
Lynch DC, Dyment DA, Huang L, Nikkel SM, Lacombe D, Campeau PM, et al: Identification of novel mutations confirms PDE4D as a major gene causing acrodysostosis. Hum Mutat. 2012, 34: 97-102.
Ludecke HJ, Schaper J, Meinecke P, Momeni P, Gross S, von Holtum D, et al: Genotypic and phenotypic spectrum in tricho-rhino-phalangeal syndrome types I and III. Am J Hum Genet. 2001, 68: 81-91. 10.1086/316926.
Shanske AL, Patel A, Saukam S, Levy B, Ludecke HJ: Clinical and molecular characterization of a patient with Langer-Giedion syndrome and mosaic del(8)(q22.3q24.13). Am J Med Genet A. 2008, 146A: 3211-3216. 10.1002/ajmg.a.32615.
Momeni P, Glockner G, Schmidt O, von Holtum D, Albrecht B, Gillessen-Kaesbach G, et al: Mutations in a new gene, encoding a zinc-finger protein, cause tricho-rhino-phalangeal syndrome type I. Nat Genet. 2000, 24: 71-74. 10.1038/71717.
Sohn YB, Ki CS, Park SW, Cho SY, Ko AR, Kwon MJ, et al: Clinical, biochemical, and genetic analysis of two korean patients with trichorhinophalangeal syndrome type I and growth hormone deficiency. Ann Clin Lab Sci. 2012, 42: 307-312.
Riedl S, Giedion A, Schweitzer K, Mullner-Eidenbock A, Grill F, Frisch H, et al: Pronounced short stature in a girl with tricho-rhino-phalangeal syndrome II (TRPS II, Langer-Giedion syndrome) and growth hormone deficiency. Am J Med Genet A. 2004, 131: 200-203.
Stagi S, Bindi G, Galluzzi F, Lapi E, Salti R, Chiarelli F: Partial growth hormone deficiency and changed bone quality and mass in type I trichorhinophalangeal syndrome. Am J Med Genet A. 2008, 146A: 1598-1604. 10.1002/ajmg.a.32348.
Weaver DD, Cohen MM, Smith DW: The tricho-rhino-phalangeal syndrome. J Med Genet. 1974, 11: 312-314. 10.1136/jmg.11.3.312.
Sidler JA, Filges I, Boesch N, Ramelli GP, Rothlisberger B, Huber AR, et al: TRPS1 codon 952 constitutes a mutational hot spot in trichorhinophalangeal syndrome type I and could be associated with intellectual disability. Clin Dysmorphol. 2012, 21: 87-90. 10.1097/MCD.0b013e32834e9248.
Giedion A, Burdea M, Fruchter Z, Meloni T, Trosc V: Autosomal-dominant transmission of the tricho-rhino-phalangeal syndrome. Report of 4 unrelated families, review of 60 cases. Helv Paediatr Acta. 1973, 28: 249-259.
Giedion A: Phalangeal cone-shaped epiphyses of the hand: their natural history, diagnostic sensitivity, and specificity in cartilage hair hypoplasia and the trichorhinophalangeal syndromes I and II. Pediatr Radiol. 1998, 28: 751-758. 10.1007/s002470050460.
Noltorp S, Kristoffersson UL, Mandahl N, Stigsson L, Svensson B, Werner CO: Trichorhinophalangeal syndrome type I: symptoms and signs, radiology and genetics. Ann Rheum Dis. 1986, 45: 31-36. 10.1136/ard.45.1.31.
Akierman SV, Skappak CD, Girgis R, Ho J: Turner syndrome and apparent absent uterus: a case report and review of the literature. J Pediatr Endocrinol Metab. 2013, 26: 587-589.
Improda N, Rezzuto M, Alfano S, Parenti G, Vajro P, Pignata C, et al: Precocious puberty in Turner syndrome: report of a case and review of the literature. Ital J Pediatr. 2012, 38: 54. 10.1186/1824-7288-38-54.
Bondy CA: New issues in the diagnosis and management of Turner syndrome. Rev Endocr Metab Disord. 2005, 6: 269-280. 10.1007/s11154-005-6185-z.
Stochholm K, Juul S, Juel K, Naeraa RW, Gravholt CH: Prevalence, incidence, diagnostic delay, and mortality in Turner syndrome. J Clin Endocrinol Metab. 2006, 91: 3897-3902. 10.1210/jc.2006-0558.
Pinsker JE: Clinical review: Turner syndrome: updating the pdigm of clinical care. J Clin Endocrinol Metab. 2012, 97: E994-E1003. 10.1210/jc.2012-1245.
Liu XY, Zhang HG, Chen S, Wang RX, Zhang ZH, Liu RZ: 45, X mosaicism in northeast China: a clinical report and review of the literature. J Assist Reprod Genet. 2013, 30: 407-412. 10.1007/s10815-012-9927-3.
Bondy CA: Care of girls and women with Turner syndrome: a guideline of the Turner Syndrome Study Group. J Clin Endocrinol Metab. 2007, 92: 10-25.
Archibald RM, FinbY N, De Vito F: Endocrine significance of short metacarpals. J Clin Endocrinol Metab. 1959, 19: 1312-1322. 10.1210/jcem-19-10-1312.
Massa G, Verlinde F, De Schepper J, Thomas M, Bourguignon JP, Craen M, et al: Trends in age at diagnosis of Turner syndrome. Arch Dis Child. 2005, 90: 267-268. 10.1136/adc.2004.049817.
Acknowledgements
We thank all the members of the Spanish PHP group for their clinical advice and all the people that provide us with clinical radiographies to complete our review: Dr. Aleksander Jamsheer, Prof. Dr. Jesus Argente, Dr. Beatriz Garcia-Cuartero, Dr. Sylvia Bähring, Dr. Okan Toka, Dr. Cécile Teinturier-Thomas, Dr. Caroline Silve and Dr. Sharona Azriel.
This work was partially supported by the Carlos III Health Institute (PI10/0148). Mrs Arrate Pereda is funded by the Spanish Ministry of Health (FI11/00309), Dr Intza Garin by a Spanish fund for health research (FIS) programme (I3SNSCA10/01056), Dr Elena Beristain by a grant from the Spanish Ministry of Science and Innovation (MICINN; PTA2009-2147), and Dr Guiomar Perez de Nanclares in part by the I3SNS Programme of the Spanish Ministry of Health (CP03/0064; SIVI 1395/09).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
AP and GPN designed the review. IG and EB participated in the molecular description of the syndromes. MGB and BG participated in the clinical description of the syndromes. MGB and AMI participated in the description of the radiological findings. All authors read and approved the final manuscript.
Electronic supplementary material
13023_2013_642_MOESM1_ESM.doc
Additional file 1: Table S1: Summary of description of each syndrome. ACRDYS: acrodysostosis; BDE: brachydactyly type E; BDMR: Brachydactyly mental retardation syndrome; PHP: pseudohypopthyrodism; TRPS: Tricho-rhino-phalangeal syndrome. Table S2. Summary of the phenotype associated with isolated BDE HOXD13 type [9–12]. Table S3. Summary of the phenotype associated with PHP-Ia [27, 29]. Table S4. Summary of the phenotype of acrodysostosis [46, 52, 99]. Table S5. Summary of the phenotype associated with Bilginturan BD or hypertension with brachydactyly syndrome [54–66]. Table S6. Summary of the phenotype associated with BDE with short stature, PTHLH type [76, 82]. Table S7. Summary of the phenotype associated with Brachydactyly mental retardation syndrome [86, 88, 90, 92]. Table S8. Summary of the phenotype associated with TRPS [100, 108]. Table S9. Summary of the phenotype associated with Turner syndrome [111, 112, 114–117]. (DOC 134 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Pereda, A., Garin, I., Garcia-Barcina, M. et al. Brachydactyly E: isolated or as a feature of a syndrome. Orphanet J Rare Dis 8, 141 (2013). https://doi.org/10.1186/1750-1172-8-141
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1172-8-141