
Abstract
The term congenital neutropenia encompasses a family of neutropenic disorders, both permanent and intermittent, severe (<0.5 G/l) or mild (between 0.5-1.5 G/l), which may also affect other organ systems such as the pancreas, central nervous system, heart, muscle and skin. Neutropenia can lead to life-threatening pyogenic infections, acute gingivostomatitis and chronic parodontal disease, and each successive infection may leave permanent sequelae. The risk of infection is roughly inversely proportional to the circulating polymorphonuclear neutrophil count and is particularly high at counts below 0.2 G/l.
When neutropenia is detected, an attempt should be made to establish the etiology, distinguishing between acquired forms (the most frequent, including post viral neutropenia and auto immune neutropenia) and congenital forms that may either be isolated or part of a complex genetic disease.
Except for ethnic neutropenia, which is a frequent but mild congenital form, probably with polygenic inheritance, all other forms of congenital neutropenia are extremely rare and have monogenic inheritance, which may be X-linked or autosomal, recessive or dominant.
About half the forms of congenital neutropenia with no extra-hematopoetic manifestations and normal adaptive immunity are due to neutrophil elastase (ELANE) mutations. Some patients have severe permanent neutropenia and frequent infections early in life, while others have mild intermittent neutropenia.
Congenital neutropenia may also be associated with a wide range of organ dysfunctions, as for example in Shwachman-Diamond syndrome (associated with pancreatic insufficiency) and glycogen storage disease type Ib (associated with a glycogen storage syndrome). So far, the molecular bases of 12 neutropenic disorders have been identified.
Treatment of severe chronic neutropenia should focus on prevention of infections. It includes antimicrobial prophylaxis, generally with trimethoprim-sulfamethoxazole, and also granulocyte-colony-stimulating factor (G-CSF). G-CSF has considerably improved these patients' outlook. It is usually well tolerated, but potential adverse effects include thrombocytopenia, glomerulonephritis, vasculitis and osteoporosis. Long-term treatment with G-CSF, especially at high doses, augments the spontaneous risk of leukemia in patients with congenital neutropenia.
Similar content being viewed by others


Background
Congenital neutropenia is characterized by chronic neutropenia due to a constitutional genetic defect. Since the early 1990s, and particularly during the last decade, the molecular bases of several entities have been discovered, leading to changes in the disease classification. Kostmann's syndrome is often considered as the paradigm of congenital neutropenia. This disorder, first described in a Swedish publication in 1950 [1], and subsequently in English in 1956 [2], has three main characteristics: profound neutropenia (<0.2 G/l) occurring during the first weeks of life, maturation arrest of granulopoiesis at the promyelocyte stage, and death due to bacterial infections (11 of the 14 initially reported patients died in their first year of life from bacterial infections). Nearly 50 years later, these patients' life expectancy routinely exceeds 20 years and the molecular basis of this entity has been identified [3]. It is now agreed that Kostmann's syndrome is accompanied, at least in forms due to mutations of one the two isoforms of HAX1 protein, those observed in the 'kostmann's pedigree', by neurological involvement (mental retardation and epilepsy) [4]. Thus, the "paradigm" of congenital neutropenia is a condition with early hematologic expression and later neurological involvement.
Knowledge of the molecular bases of other forms of congenital neutropenia has also modified the disease classification. Until the late 1990s, the literature distinguished cyclic neutropenia, associated with a regular pattern of change in the neutrophil count, typically every 21 days and showing autosomal dominant transmission [5], from permanent neutropenia (severe congenital neutropenia or Kostmann's syndrome). This distinction was made in publications based on the international registry of chronic neutropenia in the late 1990s [6, 7], in which cyclic neutropenia was not included among the congenital neutropenias. In 1999, M. Horwitz, analyzing 13 pedigrees of patients with cyclic neutropenia, identified mutations in the neutrophil elastase (ELANE) gene [8]. Shortly afterwards the same team found that many patients with severe congenital neutropenia also had mutations of the ELANE gene [9] This pointed to a continuum between severe congenital neutropenia and cyclic neutropenia, and showed that both could be considered "congenital".
Another example of nosologic reclassification concerns the gluco-6-phosphatase molecular complex, which is defective in glycogen storage disease Ib and also in an entity associated with cutaneous involvement, cardiac arrhythmias and malformative uropathy but not with metabolic disorders [10]
Definition: neutropenia and congenital neutropenia
General definition
Neutropenia is defined as a reduction in the absolute number of neutrophils in the blood circulation. The standard hematologic examination is microscopic cell counting, which is necessary to confirm disorders identified by automated cell counters and especially to examine the cell morphology. Neutropenia is defined by a neutrophil count below 1.5 G/l in children over 1 year, and below 2 G/l in children aged between 2 and 12 months [11–13].
The number of neutrophils is elevated during the first two months of life. The count increases during the first 72 hours, followed by a gradual decrease until the age of two months. In term neonates the neutrophil count is reported to range from 12 G/l to 15 G/l, depending on the study. Labor lasting more than 12 hours is associated with higher counts, while prematurity (<32 weeks) is associated with lower counts. Neutropenia in newborns is therefore defined by a threshold higher as in adult at least 2.5 G/l neutrophils.
Neutropenia is said to be severe when below 0.5 G/l and chronic if it lasts more than 3 months, whether it is intermittent or permanent.
It is important to stress that the neutrophil count shows physiological fluctuations [14], in a chaotic and non random manner [15]. There are also nycthemeral and seasonal variations [16–18], which persist in pathologic situations. Thus, neutropenia should ideally be confirmed on three samples per week over a 6-week period.
Neutropenia is said to be permanent when present in all samples, intermittent if there are periods of spontaneous normalization, and cyclic if episodes occur about every 21 days (perfectly sinusoidal neutropenia with a 21-day cycle is almost never seen in practice).
Only one study has focused on the periodicity in patients with a diagnosis of "cyclic" neutropenia, based on serial counts [19]. Among 10 such patients, regular periods, of 18, 20 and 30 days, were found in only three cases. The same study also showed regular variations in patients with permanent neutropenia (severe congenital and idiopathic neutropenia). Thus, it is better to use the terms "permanent neutropenia" and "intermittent neutropenia", while bearing in mind that there is a continuum between the two extremes, as the pathological processes that lead to neutropenia affect both the period of variation and the depth of the nadir.
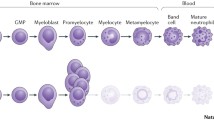
Neutropenia is said to be "central" when the bone marrow compartment is depleted, as shown by a deficiency in late maturation stages (especially <10% of mature neutrophils) and "peripheral" if bone marrow neutrophil maturation is normal (Figure 1).

Bone marrow smear differential count including % of the different granulocyte precursors. a) normal bone marrow - a regular pyramid. b) patients with severe congenital neutropenia and ELANE mutation: bone marrow myeloid arrest at promyelocyte stage with eosinophilia
Monocytosis, hypereosinophilia and polyclonal hypergammaglobulinemia are associated with neutropenia and are inversely proportional to its severity. A compensatory role of monocytes may explain the good clinical tolerance of some forms of profound constitutional neutropenia [20]
Congenital neutropenia: an evolving definition
Close examination of the literature shows that the term "congenital neutropenia" is not used homogeneously [6, 21–23]. One very restrictive definition reserves the term "congenital neutropenia" for severe forms not associated with immunological or extra-hematopoeitic abnormalities, while a broader definition includes all situations that comprise chronic neutropenia, with or without immunological or extra-hematopoeitic abnormalities. Thus, some authors but not others include glycogen storage disease Ib, Shwachman-Diamond syndrome, the WHIM syndrome, and Barth's disease in the definition of congenital neutropenia.
In this review, the term "congenital neutropenia" is not restricted to disorders in which neutropenia is the only phenotypic manifestation, but encompasses all 'congenital' disorders comprising neutropenia. We also consider "neutropenia" as a continuum, ranging from intermittent forms with various periods to permanent circulating neutrophil deficiency.
Epidemiology
The data are currently limited, owing to confusion and overlapping case definitions. Exhaustive studies are rare, and few patient registries are available[24]. General epidemiological surveys of primary immune deficiencies do not take congenital neutropenia into account [25–27], with the exception of the Iranian study [28] and a recent French study [29]. In the Iranian study, 53 cases were recorded, for a prevalence of 0.77/106. In the French registry-based study of a population of comparable size, 374 cases had been recorded in December 2006, giving a prevalence nearly 10 times higher ≈ 6.2/106. Neither study included patients with idiopathic neutropenia.
In 2003, the International Neutropenia Registry [6] reported 731 cases, of which 238 were idiopathic, recruited in a far larger geographic area than in previous studies, including the USA, Canada, Australia and Europe (excluding France), for a population close to 700 million. The prevalence was 0.7 per million inhabitants or 1 per million inhabitants when idiopathic neutropenia was included.
There are probably no major differences across countries, and the minimal prevalence of congenital neutropenia appears to be 6 cases per million inhabitants, if we take into consideration the results from the French survey - the highest rate so far described. In the French registry, 30% of patients had ELANE neutropenia (20% severe congenital neutropenia and 10% cyclic neutropenia), 30% had Shwachman-Diamond syndrome, 5% had glycogen storage disease Ib, and 35% had other disorders (1 or 2% each). However, the distribution of the different forms was influenced by the patients' geographic origin (e.g. immigrants to western countries). Some mutations are also linked to the geographic origin (HAX1 in Kurdistan and Sweden, G6PC3 in Arameans, AP14 in Mennonites), while ELANE, SBDS, SLC37A4 ( previously named G6PT1) and CXCR4 mutations appear to be universally distributed.
Clinical description
The consequences of neutropenia: infections
In vitro, the antibacterial activity of neutrophils can be represented by a simple dilution curve [30]. The risk of bacterial infection is more difficult to appreciate in vivo. Central neutropenia carries a far higher risk of bacterial and fungal infections than peripheral neutropenia. In central neutropenia the risk is low at counts above 1 G/l, increases moderately between 1 and 0.2 G/l and is very high below 0.2 G/l. The risk of infection also depends on the duration of neutropenia, with the risk of fungal infections increasing after several weeks. These data were obtained some 30 years ago in leukemic patients [31] and more recently in bone marrow graft recipients [32]. They correspond to the natural history of some constitutional forms of central neutropenia, especially that described by Kostmann [2, 33], although this has not been confirmed by other authors [34]. The preferential sites of infection are highly variable. The most frequent are the skin and mucosae, the ENT region, and the lungs. Stomatologic disorders are almost always present after age two years in patients with profound central neutropenia, and are characterized by erosive, hemorrhagic and painful gingivitis associated with papules (aphthae-like oral furuncles) of the tongue and the cheek mucosa (Additional file 1, Figure S1 Plates #1 and #2) [35]. Diffuse gastrointestinal lesions are sometimes present, leading to abdominal pain and diarrhea, and sometimes mimicking Crohn's disease on radiological studies [36]. These lesions may also be related to bacterial enteritis. It should be remembered that the symptoms of such infections may be atypical in patients with profound neutropenia, with local inflammation, the absence of pus and a necrotic tendency. One particular aspect is ecthyma gangrenosum (infectious perianal ulceration). Bacterial infections are most frequent, and generally involve Staphylococcus aureus and epidermidis, streptococci, enterococci, pneumococci, Pseudomonas aeruginosa, and Gram-negative bacilli. Most fungal infections involve Candida or Aspergillus species.
Extra-hematopoietic involvement
A variety of extra-hematopoietic involvement may be observed, contributing to the definition of several diseases or syndromes that will be examined in the Classification section, tables 1 and 2, and the Etiology/Classification section.
Physiology of myeloid differentiation
Granulopoiesis is the physiological process by which circulating neutrophils are produced and regulated. Polymorphonuclear neutrophils or granulocytes (referred to below simply as 'neutrophils') are responsible, along with monocytic cells, for innate (naïve) immunity to bacteria and fungi, based on phagocytosis and the release of proteases, antimicrobial peptides and reactive oxygen species [37]. Neutrophils also play a role in inflammation and healing. This cellular system cannot be "educated", contrary to the lymphocytic system, and emerged early in phylogenesis, being identified in mollusks, for example, as early as 1891[38].
In vitro, antibacterial activity is tightly linked to the number of neutrophils, and is absent below a critical threshold [39].
The overall dynamics of the neutrophil system and tissular neutrophil distribution were investigated with radiolabeling methods in the 1960-1970s. These studies show that granulopoiesis takes between 7 and 13 days, and that neutrophils have a half-life, measured after 32P labeling, of about 5.4 to 6.7 hours in peripheral blood [40, 41]. Circulating neutrophils represent only 3% to 5% of all neutrophils cells, and their total number is about 35 × 107 per kilogram. It is important to stress the highly dynamic nature of this system. In basal conditions, about 6 × 107/neutrophils/kg are replaced every hour. Thus, circulating neutrophil analyses provide only a simple "snapshot" of the situation at a given moment. The soluble mediators (cytokines) that control this process started to be identified in the 1980s and late 1990s, along with their mechanisms of action and their interactions. These discoveries led to therapeutic development of G-CSF (Granulocyte Colony-Stimulating Factor) [42], which has vastly improved the management of patients with malignancies and hematologic disorders, including congenital neutropenia.
Congenital neutropenia - classification and etiology
There is no simple consensus classification of congenital neutropenia. The genotype is the most important information for distinguishing one form of neutropenia from another, but it is not available during the initial work-up. The phenotype represents a continuum, with overlapping clinical manifestations: some important forms of organ involvement may not be present on initial examination. Table 1 shows associated disorders and likely diagnoses, while Table 2 lists the main diagnoses and affected organ systems.
Neutropenia with no extra-hematopoietic manifestations and with normal adaptive immunity
ELANE (ELA2): Permanent and cyclic neutropenia
ELANE (neutrophil Elastase) mutations are the most frequent known cause of congenital neutropenia and are observed in two subtypes: congenital or permanent severe neutropenia, and cyclic neutropenia. They are found in about 40% to 55% of patients with congenital neutropenia [43, 44].
Permanent neutropenia, usually called severe congenital neutropenia, is associated with deep-seated bacterial and fungal infections, stomatologic disorders, neutropenia usually below 0.2 G/l, monocytosis, hypereosinophilia and hypergammaglobulinemia, and sometimes with inflammatory anemia and maturation arrest of granulocytic cells at the promyelocyte stage (Additional file 1, Figure S1 Plate #3). These patients require large doses of G-CSF, both for the management of active infections and as long-term therapy. There is a high risk of leukemic transformation in this setting. Severe congenital neutropenia is usually diagnosed before age 6 months.
Cyclic neutropenia is less severe. The diagnosis is generally raised during the second year of life, or later, and the main clinical manifestation is recurrent acute stomatologic disorders (especially aphthae). The bone marrow aspect is variable over time (especially the granulocytic cell maturation pyramid), and is sometimes strictly normal.
Cyclic neutropenia nevertheless carries a risk of serious infections: the cumulative risk of experiencing at least one serious (potentially life-threatening) infection by age 20 years is similar in patients with permanent and cyclic neutropenia, although the former patients tend to have earlier manifestations.
No recurrent extra-hematopoietic disorders have been described in ELANE neutropenia.
By comparison with other forms of congenital neutropenia, neutropenia due to ELANE mutations is associated with the most severe infectious complications [43].
As the same mutations can be responsible for both types [43], and taking into account serial blood cell counts in patients with apparently cyclic or permanent neutropenia, the two subtypes can be considered as part of a continuum of the same disease. In addition, a given family may include members with very severe permanent neutropenia or more cyclic forms.
ELANE mutations were identified in 1999 by linkage analysis and positional cloning in 13 families with a long history of cyclic neutropenia with autosomal dominant transmission [8]. ELANE is a serine protease that cleaves elastin, among other proteins and its physiological inhibitor is α1-antitrypsin. ELANE is homologous to two other proteases produced by polymorphonuclear cells: proteinase 3 (the target of anti-neutrophil cytoplasm antibodies present in Wegener's disease) and azurocidin [45]. These three proteins, whose genes lie next to one another in chromosome region 19p13.3, are jointly regulated. ELANE is selectively stored in neutrophil azurophil granules, starting at the promyelocyte stage, but may also be found at the cell surface or within the cytoplasm.
Soon after the discovery of their involvement in cyclic neutropenia, ELANE mutations (about 50 listed to date) were also identified in patients with severe congenital neutropenia [9].
Some mutations creating a premature stop codon and leading to the synthesis of a truncated protein (lacking the last exon) are observed only in severe permanent congenital neutropenia. The G185R mutation is responsible for very severe phenotypes [43, 46].
The effects of these mutations on the protein are poorly documented. Mice with no ELANE gene expression or carrying mutations associated with severe congenital neutropenia in humans are not neutropenic [47]. Similarly, no correlation has been found between specific mutations and the protein's enzyme activity. In contrast, abnormal protein folding and cytoplasmic protein accumulation have been described [47–51]. Our understanding of the impact of ELANE mutations on intracellular protein trafficking, and particularly on granule packaging, has benefited from investigations of a genetic disease with very similar features and involving the gene coding for AP3 protein. This "cargo" protein is responsible for intraluminal trafficking of proteins from the Golgi apparatus to lysosomes, including neutrophil granules. Mutations of the AP3 tetramer subunit in humans are responsible for the Hermansky-Pudlak syndrome type 2, associated with partial albinism, and for cyclic neutropenia in Grey Collie dogs, considered the best animal model of cyclic neutropenia. ELANE mutations inhibit AP3 protein binding, thereby hindering its packaging [49]. This phenomenon contributes to endoplasmic reticulum stress through the unfolded protein response [48, 51]
Extracellular G-CSF receptor defects
No more than 5 cases have been reported to date. Here the clinical picture [52] is very similar to that of severe congenital neutropenia due to ELANE mutations, but this disorder is entirely unresponsive to G-CSF, even at doses up to 100 μg/kg per day. No constitutional anomaly common to all cells has so far been identified and this entity can be considered as a somatic mutant.
Congenital neutropenia with extra-hematopoietic manifestations
Kostmann's syndrome and HAX1 mutations
The disorder, described by Rolf Kostmann in 1950 and 1956 [1, 2], remains a paradigm in the field of congenital neutropenia. The term Kostmann's syndrome is sometimes used, inappropriately, for neutropenia with ELANE mutations.
The exact frequency of this entity is not precisely known but appears to be far lower than ELANE neutropenia, except in some geographic areas such as Sweden and Kurdistan.
The main clinical features are severe neutropenia with monocytosis and reactive eosinophilia and strong susceptibility to bacterial infections (11 deaths occurred before age 1 year among the 14 patients initially described). The pedigree lived in an isolated geographic area (northern Sweden) and involved consanguineous families, pointing to monogenic autosomal recessive transmission. A later publication by Kostmann, in 1975 [33], focusing on the same pedigree, showed improved survival thanks to the use of antibiotics, but also the onset of neurological disorders in the second decade, with both mental retardation and seizures. This syndrome is better described in a more recent study of the same pedigree, in which 5 of the 6 patients had neurological disorders [53]. Neurological involvement may depend on the mutation [54].
The molecular bases of this entity were discovered by classical genetic linkage analysis of three Kurdish families (two of which were consanguineous), followed by fine mapping of the region of interest on chromosome arm 1q, leading to the identification of HAX1 (HS1-associated protein X1) as the gene responsible for the disease. The mutations were different in the Kurdish families and the patients from the family described by R. Kostmann in 1956. HAX1 (35 kDa) is a ubiquitous mitochondrial protein with multiple partners. It has anti-apoptotic properties, due to mitochondrial membrane potential stabilization. These patients' neutrophils, and also their fibroblasts, are very sensitive to apoptotic stimuli, and this anomaly can be corrected in vitro by restoring a normal HAX1 protein level in CD34+ bone marrow myeloid progenitors, and in vivo through the anti-apoptotic function of G-CSF.
Shwachman-Diamond syndrome
This is quite a frequent form of congenital neutropenia, representing one-quarter of all cases of congenital neutropenia recorded in the French Congenital Neutropenia Registry.
First described by Nezelof in 1961 [55] and then by Shwachman and Diamond in 1964 [56], Shwachman-Diamond syndrome associates hematologic disorders with a malformative syndrome, the most consistent feature of which is external pancreatic insufficiency due to fatty involution, yielding a characteristic pancreatic aspect on magnetic resonance imaging [57], as well as chronic diarrhea with fat stools and low fecal elastase. Other features include cutaneous involvement (usually eczema, but sometimes icthyosis), bone involvement with metaphyseal dysplasia and narrow thorax [58], and psychomotor retardation [59]. Neutropenia is usually intermittent and moderate, with a decline in chemotactism associated with mild to moderate thrombocytopenia, moderate anemia, and a rise in fetal hemoglobin. The hematologic disorders can be complicated by bone marrow aplasia or leukemic transformation, mainly consisting of acute myeloid leukemia (FAB type 5 or 6), or a myelodysplastic syndrome with cytologic abnormalities (usually clonal), frequently affecting chromosome 7 (Additional file 1, Figure S1 Plates #4, #5, #6) [21, 60].
The predominant clinical manifestations are highly variable. Neonatal forms have been described, with respiratory distress, narrow thorax, pancytopenia [61, 62], and especially neurological involvement (mental retardation) [63], predominant gastrointestinal disorders (gluten intolerance), growth retardation in the second year of life, and predominant bone involvement suggestive of a constitutional bone disorder [64]. Depending on the presenting manifestations, differential diagnoses include Cystic fibrosis, Pearson's syndrome (characterized by cytologic abnormalities and especially mitochondrial respiratory chain defects), Fanconi anemia (distinguished by the constitutional karyotype) and gluten intolerance.
The genetic defect underlying the Shwachman Diamond syndrome has now been identified [65]. It involves the SDBS gene located on chromosome 7. This ubiquitously expressed gene encodes a ribosomal protein involved in the traduction process [66]. Nearly 98% of patients with this syndrome have mutations of the SBDS gene. Despite marked clinical polymorphism, the mutations are limited in number (practically always double heterozygous mutations) and the p.Lys62X/p.Cys84fs mutation is present in two-thirds of patients.
Glucose-6-phosphatase complex disorders: glycogen storage disease type Ib and G6PC3
Genetic studies show that the two entities are closely related, despite very different clinical phenotypes. Both feature neutropenia. Glycogen is stored in the liver and, after glycogenolysis, can yield glucose-6-phosphate, which can be used directly for energy production (glycolysis) or be dephosphorylated (by glucose 6 phosphatase) to yield glucose, which can be transported throughout the body to meet cellular energy needs.
Glucose 6 phosphatase is a complex of three proteins bound to the endoplasmic reticulum. Two of these three proteins are involved in congenital neutropenia: the translocase (SLC37A4), previously named G6PT1, transports glucose 6 phosphate between the cytoplasm and the lumen of the endoplasmic reticulum, while G6PC3 is a catalytic protein.
The most remarkable feature of the association between these molecular abnormalities and neutropenia is the fact that the glycogenolysis pathway and, more generally, the glucose 6 phosphatase metabolic pathway, is not the usual energy source in neutrophils, which mainly use the pentose pathway.
Neutropenia associated with glycogen storage disease Ib
Glycogen storage disease type Ib is characterized by metabolic disorders common to all forms of glycogen storage disease type I (hepatic glycogen accumulation, intolerance of fasting, hypoglycemic events, and hyperlactacidemia), as well as susceptibility to infections [67], and colitis resembling Crohn's disease both clinically and radiologically [36].
This susceptibility to infections is due to neutropenia and, sometimes, to neutrophil dysfunction (mainly defective chemotactism). Bone marrow smears show hyperplasia of the granulocytic lineage, without maturation arrest (Additional file 1, Figure S1 Plate #7). The origin of the neutropenia and neutrophil dysfunction is not known. It is not related to nutritional status and is not corrected by liver transplantation [68]. This, and the lack of any known role of the Gluco 6 Phosphate translocase (gene SLC37A4, previously named G6PT1), in neutrophil energy metabolism, raises the possibility that this protein has another function in neutrophils. Gene therapy in a mouse model has corrected both the metabolic and myeloid disorders [69].
Neutropenia associated with G6PC3 mutations
This entity associates severe permanent neutropenia with granulocyte maturation arrest, susceptibility to infections, and several other clinical manifestations, including thin skin with a highly visible veins, urogenital malformations, and cardiac disorders (especially arrhythmia due to defective atrioseptal conduction); some patients have a myopathic syndrome (despite a normal histologic and microscopic aspect of muscle) or perception deafness. Mutations of the G6PC3 gene are generally homozygous, but a double heterozygote has been described [10] and corresponds to an animal model [70]. Homozygous mutations have been shown to affect the endoplasmic reticulum [71].
Cohen's syndrome
A very rare form of congenital neutropenia, this autosomal recessive syndrome associates mental retardation with a dysmorphic syndrome that includes microcephaly, facial abnormalities (moon face), myopia, pigmentary retinitis, trunk obesity, and ligament hyperlaxity [72]. Neutropenia is present in over 90% of cases and is responsible for chronic infections with gingivostomatitis. The marrow is rich, with no maturation arrest [73]. Cohen's syndrome has been linked to mutations of the VPS13B gene, located on chromosome 8 and coding for an endoplasmic reticulum protein [74].
Neutropenia associated with poikilodermia, Clericuzio type
The poikilodermia includes skin atrophy and a papular erythematous rash. Several subtypes of this genodermatosis have been described.
The Clericuzio type was first described in Navajo Indians. Onset occurs in the first year of life. The rash gradually propagates centripetally from the limbs and comprises a papular rash, followed by plaques of hypo- and hyperpigmentation and telangiectasies. The nails are affected too (pachyonychia), but no hair loss or leukoplasia is observed. Recurrent infections occur, and especially pneumonia.
The neutropenia is often severe. Granulocyte maturation arrest at the promyelocyte stage is rarely observed, but dysgranulopoiesis with late arrest is often seen [75]. An Italian linkage study [76] revealed composite mutations of the C16ORF57 gene, whose function is unknown.
Barth's disease
This X-linked syndrome combines dilated cardiomyopathy with endomyocardial fibrosis (sometimes leading to early death), myopathy and moderate or profound neutropenia, sometimes responsible for severe infections. There is also an acidopathy involving several organic acids, including 3-methylglutaconic acid. This condition is due to mutations in the G4-5 gene, which encodes tafazzin, a protein involved in phospholipid membrane homeostasis [77].
Neutropenia and albinism: AP14 deficiency
Several children of a consanguineous Mennomite family presented with partial albinism, severe neutropenia and susceptibility to pneumococcal infection. Bone marrow studies showed no maturation arrest and there were no shared morphological features with Griscelli or Chediak Higashi disease. This syndrome, which has so far been detected in only one family, is due to deficiency of a protein (AP14) involved in intracellular endosome trafficking [78].
Neutropenia and albinism: Hermansky Pudlak syndrome type 2
Hermansky Pudlak syndrome was first described in 1959, in patients with partial albinism, hemorrhagic complications and platelet granulations. In 1994, a similar syndrome associated with neutropenia was described [79]. This entity, known as Hermansky Pudlak syndrome type 2, is due to mutations in the AP3 cargo protein [80]. It is the canine equivalent of Grey Collie cyclic neutropenia [81]. To understand the packaging function of AP3, it was first necessary to elucidate the effects of neutrophil elastase mutations, as the two proteins interact during granule packaging [49, 82].
Miscellaneous malformative syndromes
Several distinct phenotypic entities combine neutropenia and a variety of other conditions, including trichothiodystrophy [83], cuti laxa, uropathy, cardiopathy [84], and Klippel Trenaumay syndrome [21]. No noteworthy genetic mutations have been found in these isolated cases.
Chronic neutropenia with defective naive/adaptive immunity, considered as congenital neutropenia
Multiple interactions take place between the innate and adaptive immune systems. Toll receptors are shared by the two systems, while some proteins expressed by the phagocyte lineage are involved in the lymphocyte lineage [85]. Some metabolic pathways and multiple effectors (e.g. interleukins) are also shared. This explains why many "lymphocyte disorders" can also be associated with neutropenia. Indeed, these associations are so frequent [86] that both adaptive immunity and other functions of the innate immune system must be investigated when chronic neutropenia is diagnosed. These morbid associations, often attributed to viral infections or autoimmunity, also involve common pathophysiologic mechanisms, as shown by studies of some extremely rare disorders like Bruton's disease [87].
Neutropenia associated with GFI1 mutations
This is an extremely rare cause of congenital neutropenia, so far described in only four patients [44, 88]. The clinical phenotype does not seem to be very homogeneous, as one patient was diagnosed with severe neutropenia at 4 months of age, together with marked monocytosis, while the father, who had the same mutation, had moderate, asymptomatic neutropenia, and the second patient, diagnosed at age 56 years with idiopathic neutropenia, had no clear susceptibility to infections. These patients all have moderate lymphopenia (CD3 cells between 1 and 1.4 G/l) with normal memory cells and humoral immunity.
GFI1, a nuclear protein, is a transcriptional repressive factor involved in T lymphomatogenesis and in the development of T cell progenitors. Its involvement in granulopoiesis and in macrophage activity has been demonstrated in knock-out mice, which also exhibit an inner-ear defect. Heterozygous GFI1 mutations, which are dominant mutations, lead to an increase in ELANE expression, in the same way as ELANE mutations.
Permanent congenital neutropenia due to Wiskott-Aldrich syndrome (WAS) gene mutation
This is also a very rare entity observed to date in 5 families. Its hematologic and infectious features resemble those of ELANE neutropenia, but with no monocytosis despite profound neutropenia [89–93]. Some cases are only diagnosed in adulthood, implying that some patients have limited infectious complications. This is an X-linked disorder. A genetic linkage study of a pedigree with suspected sex-linked genetic transmission revealed mutations in the WAS gene (encoding Wiskott-Aldrich syndrome protein) in a family with severe congenital neutropenia [92], and more recently in four other families [44, 89–91, 94].
These patients' phenotype is completely distinct from that of patients with the classical form of Wiskott-Aldrich syndrome, which comprises eczema, thrombocytopenia with small platelets, and immune deficiency.
This phenotypic difference, despite the shared involvement of the WAS gene, is due to functional differences in the respective mutations (WAS protein activation in congenital neutropenia and defective WAS protein activity in the classical syndrome).
As WAS protein is involved in intracytoplasmic actin polymerization, mutations observed in patients with neutropenia lead to an increase in actin polymerization, accompanied by an increase in the podosome level and in apoptosis.
Neutropenia associated with IRAK 4 mutations
A deficiency in interleukin 1 receptor-associated kinase 4 leads to a functional defect of innate immunity [95]. It includes marked susceptibility to pyogenic infections (especially staphylococci and pneumococci), but no other extra-hematologic or infectious manifestations. These patients have only moderate neutropenia, which tends to normalize during infections. However, functional tests, and especially the monocyte response to various stimuli, such as LPS, show defective neutrophil and monocyte mobilization [96], whereas standard immunological findings can be normal.
NK cell deficiency and neutropenia
NK cell deficiency and dysfunction have been described in four patients with chronic neutropenia and maturation arrest at the promyelocyte stage in the only relevant study. These findings were made before the main molecular abnormalities were identified [97]. It is impossible to know whether this feature is common to several forms of congenital neutropenia or whether it represents an original entity.
Wart hypogammaglobulinemia immunodeficiency myelokathexis (WHIM) syndrome
This form of constitutional neutropenia is characterized by morphological abnormalities of the rare circulating neutrophils, which are hypersegmented and contain cytoplasmic vacuoles; bone marrow cells show similar anomalies (Additional file 1, Figure S1 Plate #8). This unusual morphological aspect (kathexia meaning neutrophil retention in the bone marrow) justified the use of the initial term [98]. Later, immunological abnormalities were also reported, including lymphopenia and moderate hypogammaglobulinemia [99]. Severe papillomavirus warts are almost always present, leading to the adoption of the term "wart hypogammaglobulinemia immunodeficiency myelokathexis". Subsequent identification of the role of a chemokine receptor gene (CXCR4)[100] led to a better understanding of this disease and showed that this syndrome corresponds to the same entity, although warts may not initially be present. CXCR4 is a chemokine receptor known for its role as an HIV coreceptor [101]. This receptor and its ligand SDF1 (CXCL12) are involved in organogenesis, B lymphocyte ontogenesis, and myelopoiesis, and are required for CD34+ cell migration from bone marrow. Mutations of the CXCR4 chemokine are dominant mutations, leading to receptor hyperactivity and defective mobilization of bone marrow neutrophils (myelokathexis) and lymphocytes.
Neutropenia associated with miscellaneous constitutional disorders NOT considered as congenital neutropenia
Neutropenia is not a major clinical or biological feature of these disorders. They are not usually considered to be forms of congenital neutropenia, because the neutropenia is transient (for example in Bruton's agammaglobulinemia), or tends to occur late, or is only moderate and does not require any particular management (for example Charcot and Tooth disease with dynamin 2 mutation).
Chronic neutropenia, with defective innate/adaptive immunity NOT considered as congenital neutropenia
Humoral immune deficiencies
Bruton's agammaglobulinemia (~30% of cases), CD40 ligand deficiency (immune deficiency with IgM hypergammaglobulinemia, 50% of cases), variable hypogammaglobulinemia and unclassified hypogammaglobulinemia can be accompanied by neuropenia [86, 97, 102–105]. The neutropenia is usually detected before immunoglobulin replacement therapy and responds to immunoglobulin therapy [106]. In Bruton's agammaglobulinemia, due to BTK gene mutations, the neutropenia can be very profound at onset, with maturation arrest at the promyelocyte stage. Humoral immunity should be thoroughly investigated in patients with neutropenia.
Severe combined immune deficiency and immune deficiency syndromes
Severe combined immune deficiencies (like those associated with IL-2 receptor gamma mutation) can also include neutropenia. The lymphocyte deficit, mainly affecting T cells [107], frequently includes neutropenia, which can be severe. Other immune deficiencies that are not as severe at onset, such as defective HLA class II expression and ataxia-telangiectasia, can also include neutropenia. In Wiskott-Aldrich disease, neutropenia usually accompanies the frequent autoimmune disorders [108], through a mechanism different from that underlying X-linked neutropenia and activating WASP mutations.
Reticular dysgenesis and AK2 gene mutation
Reticular dysgenesis is an autosomal recessive form of the severe combined immune deficiency syndrome (SCID) affecting hematopoietic lineages of both the innate and adaptive immune systems. At birth, this condition is characterized by a total absence of neutrophils, T cells and NK cells, sometimes associated with anemia, thrombocytopenia and low B cell counts, while monocyte counts remain normal. This disorder also affects the inner ear, leading to profound perception deafness. Recently, the gene responsible for this form of SCID was identified by two independent teams [109, 110]. It codes for adenylate kinase 2 (AK2), a ubiquitous enzyme involved in energy metabolism and whose known function is reverse transphosphorylation of AMP and ATP into ADP.
22q11 syndrome
This is a complex malformative syndrome due to interstitial deletion of chromosome 22 at the q11 locus. Few children present all the characteristic features of this syndrome simultaneously. ENT disorders comprise velar insufficiency, facial malformation (especially of the lower face), sometimes accompanied by marked retrognatism. Other disorders include parathyroid deficiency with hypocalcemia, cardiac abnormalities (especially tetralogy of Fallot) and immunologic abnormalities, including, in the most severe cases, Di George syndrome with thymic agenesis, and T lymphocyte deficiency. Platelet disorders have been described [111, 112] and also neutropenia, sometimes of an autoimmune nature [113].
Exocytosis disorders
Neutropenia is found in several cellular exocytosis disorders [114], leading to hemophagocytic lymphohistiocytosis (HLH) but also sometimes inaugural neutropenia, as in AP14 mutation disorders and Hermansky Pudllak disease type 2. Most genetic defects associated with these disorders have now been identified, and we will only recall the main phenotypes, of which the principal extra-hematopoeitic manifestation is complete or partial albinism [115–117].
Chediak Higashi syndrome (CHS)
CHS is characterized by partial oculo-cutaneous albinism, abnormal melanosome hair repartition, giant granules in all neutrophils and in bone marrow granulocytic precursors (Additional file 1, Figure S1 Plate #9), bright red inclusions in some lymphocytes, defective bactericidal activity, and NK dysfunction. Neutropenia due to bone marrow destruction occurs early in this disorder, prior to HLH.
Griscelli syndrome type 2 (GS2)
The clinical manifestations of this disorder share many features of Chediak-Higashi syndrome, especially albinism and immune deficiency, and sometimes HLH in GS2, and not in GS1 and GS3 (Additional file 1, Figure S1 Plate #10). It differs by the absence of giant granulations in blood cells, and the microscopic aspect of the hair. Neutropenia can be present, either in isolation or during the course of a macrophage activation syndrome.
Familial hemophagocytic lymphohistiocytosis (FHLH)
These inherited immune dysregulation syndromes are related to mutations in perforin, Munc13.4, Munc18.2 or Syntaxin11 encoding genes and are defined by early onset of severe HLH. Neutropenia is one of the diagnostic criteria of HLH, though not a major feature. Morphological anomalies are rare.
Other syndromes associated with neutropenia
Blackfan-Diamond anemia
Several years after onset, neutropenia can occur in patients with Blackfan-Diamond anemia.
Fanconi anemia and dyskeratosis congenita
Neutropenia is an integral feature of these constitutional forms of bone marrow aplasia, which are associated with complex malformations. Anemia and thrombocytopenia, but rarely neutropenia, are present at diagnosis.
Constitutional monosomy 7
Constitutional monosomy 7 has been found in several patients with sporadic or familial neutropenia. Secondary malignant transformation is the rule [118–120].
Aminoacidopathies
Neutropenia is a secondary feature of several aminoacidopathies, including hyperglycinemia, and isovaleric, propionic and methylmalonic acidemia [121]. Chronic fluctuating neutropenia is a feature of dibasic protein intolerance (also called lysinuric protein intolerance) and there is a typical cytologic aspect (Additional file 1, Figure S1 Plate #11). Other features of the macrophage activation syndrome are also present [122].
Pearson's syndrome
Pearson's syndrome associates external pancreatic insufficiency with pancytopenia. Neutropenia can be present, together with anemia and thrombocytopenia. This syndrome is due to a mitochondrial respiratory chain disorder and to mitochondrial DNA deletion [123]. The neutropenia is usually less severe than the anemia. The diagnosis is raised by sideroblastic anemia, with evocative cytologic abnormalities (Additional file 1, Figure S1 Plate #11) and unexplained acidosis.
Cartilage-hair hypoplasia
This syndrome combines dwarfism, metaphysal chondrodysplasia, sparse hair, and sometimes an immune deficiency, with lymphopenia, hypogammaglobulinemia and neutropenia [124]. This autosomal recessive disease, mainly affecting the Amish (USA) and Finnish populations, is due to mutations of the RMRP gene, coding for a ribonuclease [125].
Chronic neutropenia, recurrent fever, Behçet's disease and amyloidosis
Recurrent fevers are a set of disorders comprising recurrent fever, various inflammatory manifestations (serous and articular) and sometimes recurrent aphthosis.
Amyloidosis is a common complication of these disorders, and especially of familial mediterranean fever (FMF) [126]. Hyperleukocytososis is usually present [127], but an authentic case of FMF with neutropenia has been described [128].
Congenital neutropenia is often associated with hypergammaglobulinemia and a chronic inflammatory syndrome, but secondary amylosis (AA type) is very rare [129–131]. The particularities of these patients suggest that this is an independent entity.
Behçet's disease is distinct from recurrent fever, but the two disorders share the same geographic predominance (Mediterranean basin) and certain traits such as recurrent aphthae, as in neutropenic disorders. Polynucleosis is common, but cases with associated neutropenia have been reported [132].
Finnish nephrotic syndrome
The "Finnish" nephrotic syndrome is an autosomal recessive disorder defined by structural modification of nephrin, leading to massive renal protein leakage. An extremely severe nephrotic syndrome (albuminemia < 10 g/l) and massive proteinuria are present from birth.
Neutropenia can also occur in this setting [133, 134]. It is due to leakage of proteins, and especially ceruloplasmin (the protein responsible for copper transport), leading to very low circulating copper levels.
As shown in an animal model [135], copper deficiency can lead to severe neutropenia, with maturation arrest of granulopoeisis at the promyelocyte stage, as in typical congenital neutropenia. Copper administration suffices to correct the deficiency and to restore a normal neutrophil count [136].
Charcot-Marie-Tooth disease and dynamin 2 mutations
Charcot-Marie-Tooth disease comprises a variety of neurological disorders with hereditary sensory-motor neuropathy. Life expectancy is unaffected and there is no mental retardation.
Schematically, CMT is due to damage to the peripheral nerves connecting the spinal cord to the muscles, affecting nerve conduction. This leads to gait disorders, cramps and frequent foot deformation. CMT can occur during childhood but sometimes also in adulthood. In general, CMT deteriorates slowly, but it can also progress by exacerbations. There are several types, currently classified according to the affected part of the nerve (myelin or axon) and the mode of transmission (dominant or recessive). Type II is characterized by axonal involvement. In this form, with dominant transmission related to the mutation in the dynamin 2 gene, neurological signs are sometimes discreet and are accompanied by congenital cataract and fluctuating neutropenia; the neutropenia is usually mildly symptomatic but it may be severe and is sometimes the initial manifestation [137, 138].
Diagnosis of congenital neutropenia
Neutropenia is a relatively frequent finding, while congenital neutropenia is quite rare. Neutropenia is often well tolerated and normalizes rapidly, in which case specialized investigations are not necessary. Neutropenia is sometimes a secondary finding in a patient with far more significant disorders, who may thus be at risk of infectious complications. More rarely, neutropenia persists and/or emerges as the sole cause of a child's symptoms, in which case thorough investigations are necessary.
The interview and physical examination may reveal a particular etiology, such as a viral infection or malignant hemopathy, an iatrogenic cause, or an immune deficiency, warranting further specific investigations.
In non urgent settings, the permanent, intermittent or regressive nature of the neutropenia should be established during an observation period of a few weeks, in which the number of infections and any changes in buccal disorders (ulceration, gingivitis, etc.) should be noted, as they can help guide patient management.
Bone marrow examination is often necessary to rule out malignant hemopathies, determine cellularity, assess myeloid maturation, and detect signs of a precise etiology. Figure 1 shows a) the normal pyramid of granulocyte precursor maturation and b) early arrest at the promyelocytic stage (in a patient with ELANE mutation). Maturation arrest at the promyelocyte stage is often associated with bone marrow hypereosinophilia and monocytosis. Morphologically, few aspects are truly typical of a particular etiology. Specific hemophagocytosis of neutrophils is a sign of autoimmune neutropenia in young children (Additional file 1, Figure S1 Plate #13)[139–141], while cytoplasmic granulations are suggestive of Chediak Higashi disease (Additional file 1, Figure S1 Plate #9), hemophagocytosis points to dibasic protein intolerance(Additional file 1, Figure S1 Plate #11) and myelokathexis, defined by an increase in the granulocyte pool with hypermature and dystrophic features (Additional file 1, Figure S1 Plate #8) point to WHIM syndrome. Finally, precursor vacuolization is a sign of Pearson's syndrome (Additional file 1, Figure S1 Plate #12). The marrow smear may reveal non specific dysgranulopoeisis or be totally normal, but this does not rule out a diagnosis of congenital neutropenia. Cytogenetic bone marrow studies are now crucial when investigating isolated neutropenia that is suspected of being congenital.
Several other investigations are of interest, especially antineutrophil antibody assay, immunoglobulin assay (Ig GAM), lymphocyte immunophenotyping, pancreatic markers (serum trypsinogen and fecal elastase) and liposoluble vitamin levels (vitamins A, E and D).
The glucagon challenge test and studies of neutrophil demargination are rarely used now, as they are complex and provide little information of practical use.
The recommended diagnostic work-up for neutropenia is shown in Figure 2, while table 3 lists the main forms of acquired neutropenia.

Neutropenia: diagnostic tree.
Differential diagnosis and some frequent causes of chronic neutropenia
Allo-immune neutropenia
This form of neutropenia is present from birth and can be considered congenital.
It may be suspected following a maternofetal infection or a routine blood cell count. Initially severe (<0.1 G/l), it usually normalizes after 3-6 months. Available bone marrow studies show no maturation arrest. Neonatal allo-immune neutropenia is due to fetomaternal incompatibility for a paternally derived neutrophil antigen. By crossing the placental, these fetal neutrophils can elicit IgG antibodies in the mother, which then enter the fetal circulation. Based on fine characterization of neutrophil antigens, a new nomenclature (HNA, Human Neutrophil Antigens) has been proposed, with 5 systems. The first system, HNA1, codes for RFcγIIIb (CD16), a low-affinity IgG receptor, is polymorphous and includes three antigens: HNA1a, 1b and 1c. The genetic CD16 deficit on neutrophils and in its soluble form can lead to iso-immunization of the mother and neutropenia in the newborn. The second system, HNA-2, includes only one serologically defined antigen, HNA-2A, present on neutrophil subpopulations. The alloantibodies most frequently responsible for neonatal neutropenia are directed against the antigens of the HNA-1 system and, to a lesser extent, HNA-2. In rare cases, neonatal immune neutropenia can be due to a maternal IgG autoantibody. Diagnosis is based on the identification of a maternal antibody reacting selectively with neutrophils belonging to the panel expressing the antigen and\or with paternal neutrophils.
Autoimmune primitive neutropenia
This is the most frequent cause of chronic neutropenia in children, and is better known under the term "benign chronic neutropenia" [142–144]. This form of isolated neutropenia is usually discovered after a moderately severe infectious episode in a small child (median age 8 months). Monocytosis, eosinophilia and/or moderate splenomegaly can be found. This neutropenia is permanent, at least for several months, ordinarily very profound, but it is usually well tolerated. The marrow smear shows hyperplasia of the granulocyte lineage, sometimes with late arrest. Macrophagia of intramedullary polymorphonuclear cells is a diagnostic sign [139–141]. The detection of anti-polymorphonuclear cell antibodies necessitates repeated examinations (only about 75% of cases are positive on the first examination). Several techniques can be used (detection of circulating antibodies or antibodies adherent to polymorphonuclear cells). The autoimmune process targets the same membrane glycoproteins on polymorphonuclear cells as those involved in autoimmune neutropenia. The most frequently involved is the receptor for the gammaglobulin invariable fragment (FcRgIIIb) or CD16, that is encoded by two co-dominant alleles (HNA-1A and HNA-1B, formerly NA 1 and NA 2). The infectious consequences are limited, probably because bone marrow reserves are unaffected by the autoimmune process. The neutropenia resolves spontaneously after 12 to 24 months (36 months in a few cases). It is rarely associated with another autoimmune disease or with an immune deficiency. It can be secondary to a viral infection. The adult form differs from the childhood form by its greater severity. Cytologic studies sometimes show early maturation arrest of the granulocyte lineage. In particular, the frontiers between autoimmune neutropenia, idiopathic neutropenia, and neutropenia associated with proliferation of large granular lymphocytes (LGL) are still rather vague in adults [145–147].
Secondary autoimmune neutropenia
This form is rare in children, contrary to adults. The causes are numerous, but immune deficiencies are at the forefront. Neutropenia is generally a secondary feature, as for example in acute disseminated lupus erythematosus, and rheumatoid arthritis (especially Felty syndrome) [146, 147]. Finally, autoimmune neutropenia associated with autoimmune involvement of another blood lineage corresponds to the definition of Evans syndrome [148].
Idiopathic neutropenia
This diagnosis is generally made in adulthood [146]. Etiologic investigations are negative. The presence of anti-polymorphonuclear cell autoantibodies must be eliminated by repeated testing at intervals of several weeks, along with rare causes such as the association with a thymoma [149]. It seems that some of these neutropenias are associated restriction of T lymphocyte clonality, thus resembling hyperlymphocytosis with large granular lymphocytes [150]. Several pediatric cases of large granular lymphocytes associated neutropenia have been described, including a familial form [151].
Ethnic neutropenia
Ethnic neutropenia is the most frequent form of chronic neutropenia. It is generally isolated and moderate, and has no direct health repercussions. The mode of genetic transmission is not yet known and may be multifactorial. First described in 1941 [152], it appears to be particularly frequent in black-skinned individuals. Epidemiological studies show that the prevalence of neutropenia (<1.5 G/l) is about 4.5% in black people and 0.8% in Causasians [153]. Few data are available on other populations, but a high frequency has been noted in the Arabian peninsula [154], and the frequent mild neutropenia reported in Crete likely corresponds to the same entity [155].
Ethnic neutropenia is not associated with increased susceptibility to infections, and no symptoms have ever been reported.
Four simple but poorly specific features are classically present: moderate neutropenia (0.5 to 1.5 G/l), no infections attributable to neutropenia, no identifiable cause, and African ethnicity.
The few available studies of ethnic neutropenia have yielded strictly normal findings. In particular, the bone marrow is qualitatively and quantitatively normal. No difference in neutrophil mobilization after brief or more strenuous exercise (such as marathon running) has been observed, and this form of neutropenia does not appear to be due to excessive neutrophil margination [156]. A particular polymorphism of the Duffy antigen receptor for cytokines (DARC) [157] is associated with ethnic neutropenia in black people.
Antenatal diagnosis
Monogenic congenital neutropenias are severe diseases with a sometimes major impact on the patient's daily life; antenatal diagnosis is therefore warranted, taking into account the mode of transmission [158].
Antenatal diagnosis has already been performed for the following disorders: severe congenital neutropenia with pathogenic ELANE mutations, Shwachman-Diamond syndrome, WHIM syndrome and glycogen storage disease type Ib and may be offered for many other entities, such as HAX1 and G6PC3.
Antenatal diagnosis must be preceded by a familial survey. Neutropenia due to mutation of the ELANE gene, which shows autosomal dominant transmission, can exist in the mosaic state in one of the parents, with a unusually high frequency compared with other genetic abnormalities [159].
The decision to implement antenatal diagnosis and, possibly, pregnancy termination, must be taken within a precise ethical framework, after providing the family with thorough information and obtaining medical expert opinion in each individual case.
Treatment and Patient Management
Historical perspective
Until the 1960s, children with severe congenital neutropenia had a grim prognosis [2, 30]. Survival started to improve markedly in the 1970s, thanks to progress in curative parenteral antimicrobial chemotherapy and to more general use of antibiotic prophylaxis. However, quality of life remained mediocre because of recurrent infections and permanent stomatitis.
With the exception of bone marrow transplantation[160], no other treatment (steroids, levamisole, lithium, etc.) was able to correct the neutropenia. Hematopoietic growth factors (G-CSF and GM-CSF) started to be used in 1988 [161], and immediately proved capable of correcting both the neutropenia and susceptibility to infections. The arrival of these drugs also kindled interest in these disorders, leading to the creation of registries in several western countries.
Management of acute infectious episodes
It is important to assess the potential gravity of each infectious episode by means of a thorough physical examination. Precious information on the risk associated with bacterial infections can be gleaned from experience in cancer chemotherapy, which shows the importance of body temperature (>39°C) and the decline in the monocyte count (<0.1 G/l) as gravity factors [162, 163].
In moderate neutropenia complicated by superficial or ENT infections, oral antibiotic therapy is adequate, with close ambulatory monitoring, if inflammatory markers are absent (e.g. CRP < 15 mg/L).
In contrast, patients with severe neutropenia and sepsis require immediate hospitalization [164]. After bacteriological examinations (blood culture, urinalysis, local samples, etc.) and chest radiography, parenteral antibiotic therapy should be started rapidly, with a combination of a third-generation cephalosporin and an aminoside by IV route. The place of glycopeptides (vancomycin and teicoplanin) in first-line therapy is controversial. In fever persists beyond 48 hours, antifungal treatment should be added.
If the child's condition is worrisome from the outset, G-CSF administration should be started, either at a dose to which the patient is known to respond, or at the standard dose of 5 μg/kg/d. The dose should be increased if no improvement is observed. This is justified even when the precise etiology of the neutropenia is not known. There is no reason to suspect that temporary G-CSF administration would hinder subsequent investigations.
The utility of leukocyte concentrates should be underlined, even if their use is now generally limited to cellulitis and documented bacterial or fungal infections showing no clinical response to appropriate antibiotic therapy.
Prevention of infections
It is crucial to prevent recurrent infections. The use of antimicrobial prophylaxis depends on the individual risk, history, and severity of neutropenia.
Antimicrobial prophylaxis
The principal measure is antimicrobial chemoprophylaxis. The ideal antimicrobial chemotherapy regimen will be effective against the pathogens most frequently encountered in this setting, well tolerated, and not select resistant strains. On this basis the best antibiotic is the oral sulfamethoxazole/trimethoprim combination (Bactrim®), at a daily dose of 50 mg/kg per day. Its value in constitutional neutropenia has not been studied, but it is reasonable to extrapolate data from patients with leukemia [165] and chronic granulomatous disease (CGD) [166]. The use of this drug in chronic neutropenia sometimes appears paradoxical, because it can occasionally cause neutropenia [167]. However, the risk-benefit ratio remains favorable. Sulfamethoxazole/trimethoprim only partially prevents these patients' gingivostomatitis, and concurrent therapy with an agent active on the oral saprophytic flora, and especially anaerobes (metronidazole), is therefore warranted.
Use of cytokines in constitutional neutropenia
The second possibility is to correct the neutropenia by using hematopoietic growth factors (G-CSF and GM-CSF) produced by genetic engineering.
In practice, only G-CSF is used in this indication [42]. Indeed, GM-CSF has several disadvantages, with lesser efficacy in these indications and poorer immediate tolerability ('flu-like syndrome, marked eosinophilia) [168–170]. G-CSF is currently available in two forms: filgrastim (Neupogen, in vials containing 480 μg or 330 μg) and lenograstim (Granocyte, in vials containing 340 μg or 130 μg). These two molecules are nearly identical, lenograstim being the glycosylated form of G-CSF. Their biological effects are also practically the same, but one study suggests that filgrastim yields a slightly larger increase in the neutrophil count compared with the same dose of lenograstim [171]. It is important to underline that the pegylated form of G-CSF (PegFilgrastim Neulasta®) is not registered for patients with congenital neutropenia. Pegfilgrastim -- a combination of filgrastim and polyethylene glycol -- has a half-life of 15 to 80 hours, reducing the required number of injections [172]. However, pending specific pharmacokinetic data in congenital neutropenia, its use carries a risk of overdose and potentially severe adverse effects [173], or, on the contrary, a lack of efficacy [174].
Treatment schedule
In severe congenital neutropenia, the time and the dose required to increase the neutrophil count cannot be predicted. In other indications the schedule is generally simpler. Long-term treatment takes place in two phases [175]: an induction phase and a maintenance phase. The aim of the induction phase is to characterize the individual response to G-CSF. The response is evaluated in terms of the increase in the neutrophil count (> 1.5 G/l) and the clinical improvement, after 10 to 15 days. Serial blood cell counts are useful for following changes in the neutrophil count during this period.
The recommended initial daily dose is 5 μg/kg subcutaneously, with no particular constraints regarding the timing of injections during the day. If no response is seen after 15 days, the daily dose is increased in steps of 5 μg/kg. On the contrary, if the response is rapid or even excessive (PN > 5 G/l), the dose should be halved. Short-term tolerability is also assessed during the induction phase, including dose-dependent adverse effects that will have to be taken into account during long-term treatment. Once the minimal daily dose has been determined, the maintenance phase can begin, which aims to determine the minimal dose and rhythm of injection to sustain a clinical response. In the maintenance phase it is of course possible to modulate the dose and sometimes to attempt a dose reduction or a longer dosing interval. But it may be necessary to increase the daily dose, especially for a growing child. Unnecessary blood counts should be avoided during this period: unless clinical problems arise, a monitoring interval of 4 to 6 months is acceptable.
Efficacy
Severe congenital neutropenia
Between 1988 and 2010, the international and French registries collated data on G-CSF therapy in no more than 500 patients with severe congenital neutropenia [6, 21]. Long-term follow-up confirms the results of short phase I/II trials in small groups of patients [175, 176]. The efficacy of G-CSF is based first on the neutrophil count. It is now clear that the response does not wane with time. However, the most important criterion for efficacy is the reduction in infectious complications, including stomatologic status, although there are no well-established criteria for this endpoint. During G-CSF development only one randomized study, involving 36 patients, focused on infections [176]. In this study, some patients received G-CSF routinely and others only after a 4-month observation period. This study demonstrated a benefit in terms of both infections and quality of life. No long-term randomized trials have been published. The dose required to obtain a response varies widely from one patient to another. Almost two-thirds of patients respond to a daily dose of between 2 and 10 μg/kg, while nearly 20% respond to 10-20 μg/kg. A small number of patients require even higher doses of up to 100 μg/kg. Only 13 cases of complete G-CSF treatment failure have been reported [6, 21]
The neutrophil count increment is dose-dependent beyond a minimum threshold, but it fluctuates over time on a stable dose, with no identifiable pattern.
There are no clinical or biological features predictive of the dose to which a given patient will respond.
Cyclic neutropenia
G-CSF is always effective in this setting, avoiding the neutrophil nadir. In contrast, it does not abolish the cyclic nature of granulopoeisis, and the oscillations tend to be more unstable, with peak counts sometimes exceed 30 G/l. Despite several attempts, no cyclic schedule of G-CSF administration, such as once every third week, has proven effective. In contrast, the dose required to correct the nadir is generally below 5 μg/kg/d and the injections can be given intermittently, for example once every three days.
Tolerability of G-CSF
Short-term adverse effects
G-CSF has been used briefly (<15 days), at a dose of 1 to 5 μg/kg/d, in more than a million cancer patients (children and adults) receiving chemotherapy. Tolerability during such short-term use is good or excellent. Both intravenous and subcutaneous injections provoke only occasional immediate (<1 per 100) and local reactions. 'Flu-like reactions, as observed with other cytokines, are also infrequent.
Bone pain is more frequent, affecting 2 to 5% of subjects. It rapidly subsides on treatment cessation (within less than 24 hours) and generally does not recur when a lower dose is adopted.
Long-term tolerability
Few situations necessitate long-term G-CSF therapy. In addition to chronic neutropenia, G-CSF is sometimes administered in the long term for aplastic anemia. Published reports of long-term G-CSF safety concern fewer than 1500 patients, with variable levels of drug exposure [6, 21, 177]. Although the action of G-CSF is, in principle, limited to the granulocyte lineage, various hematologic abnormalities can be present or occur transiently during treatment. Monocytosis beyond 1.5 G/l is frequent. Eosinophilia, frequent at diagnosis, can be amplified by G-CSF. Lymphocytosis is unaffected, as is the hemoglobin level in most patients. However, reticulocytosis occasionally increases, along with the hemoglobin level, especially if inflammatory anemia is present at the outset of treatment. Thrombocytopenia seems to be the most common hematologic adverse effect. However, it is generally moderate and regresses when the G-CSF dose is reduced. Thrombocytopenia can also be due to hypersplenism [177]. The spleen almost always enlarges (on imaging studies) at the outset of treatment. Clinical confirmation of this splenomegaly is rarely obtained, except in glycogen storage disease Ib, in which this complication is very frequent. Spleen rupture necessitating splenectomy can occasionally occur [177]. The uricemia rises during long-term treatment but this has no clinical consequences. Exacerbation of long-standing gout has been observed during short-course therapy [178]. The first cases of leukocytoclasic vascularitis, corresponding to Sweet's syndrome, were observed after short-term treatment (<1 month)[179]. The G-CSF-induced increase in neutrophil adhesion molecule expression seems to be responsible. These cutaneous manifestations always regress after a dose reduction or treatment cessation. Two cases of mesangioproliferative glomerulonephritis have been reported during long-term treatment, both resolving after a dose reduction or treatment cessation. Osteoporosis occurs in nearly one-quarter of patients with severe congenital neutropenia who receive chronic G-CSF therapy [180]. Two cases of pathological fracture have been reported. But, severe congenital neutropenia itself seems to be associated with osteopenia, which is often present before treatment begins. Somatic development is unaffected by G-CSF and so is puberty.
Multidisciplinary management of multisystem disorders
Glycogen storage disease Ib
Glycogen storage disease Ib patients must be managed by a multidisciplinary team including experienced specialists in pediatric nutrition and hematology. The carbohydrate balance is very fragile in these patients, who must be fed day and night. Severe carbohydrate instability may necessitate liver transplantation [68]. Low doses of G-CSF (< 5 μg/kg/d) are usually necessary both to correct the neutropenia and to obtain a clinical improvement [21, 181]. The response is obtained within 48 hours, which is compatible with neutrophil release from the bone marrow compartment and with the absence of maturation arrest in these patients. Tolerability is generally good, although G-CSF-induced thrombocytopenia and splenomegalia are relatively frequent.
Shwachman-Diamond syndrome
These patients too must be managed by a multidisciplinary team. Their external pancreatic insufficiency leads to nutritional deficiency, which may be severe or simply consist of liposoluble vitamin deficiencies, requiring supplementation. Attention must be paid to the accompanying bone disorders, and counseling is needed because of these patients' frequently abnormal mental development. The use of G-CSF is far less frequent in patients with permanent ELANE neutropenia.
Hematopoietic stem cell transplantation (HSCT)
HSCT can permanently correct the neutropenia in these patients, and is the only option for patients who continue to experience severe infections despite G-CSF therapy.
Validated indications of bone marrow grafting include G-CSF resistance (> 50 μg/kg/day) and myelodysplasia/leukemic transformation, in which case it is the only therapeutic option [182–184].
Patients with malignant transformation (with the exception of frank leukemia) should not receive chemotherapy before the bone marrow graft.
In patients with neutropenia dependent on chronically high doses of G-CSF (at least 20 μg/kg per injection at least three months a year), given the high risk of leukemic transformation, bone marrow grafting should be considered on a case by case basis, taking into account the possibility of finding a related donor.
Standard HSCT procedures can be used, with myeloablative conditioning. Even in patients with malignant transformation survival now exceeds 70%, with the exception of patients with Schwachman's syndrome.
The second disease in which HSCT may be indicated is Shwachman-Diamond syndrome. Schematically, there are two distinct indications for HSCT in this setting: pancytopenia with no detectable malignant clone, and myelodysplasia/leukemic transformation [185]. The results are very different in the two indications but tend to be good (favorable outcome in >80% of cases) in the absence of a malignant clone, while they are very mediocre in case of leukemic transformation (<35% survival). In the absence of clonal progression, the so-called 'reduced intensity' conditioning regimen (especially fludarabine and Campath) appears promising [186, 187].
Daily life
It should be remembered that intramuscular injections and rectal temperature measurement may be harmful. Most vaccines can be administered, including live viral vaccines. However, BCG vaccine should be avoided. Pneumococcal and influenza vaccination is recommended. No dietary restrictions are necessary in neutropenic children. They are not unusually susceptible to viral epidemics and there is therefore no reason to deprive them of opportunities for social interaction.
Prognosis and Outcome
Several complications can occur in patients with congenital neutropenia, including infectious complications, complications related to extra-hematological involvement, and the risk of leukemia related both to the disease and its treatment.
Outcome - the infectious risk
Bacterial infections represent the main risk. Infections can be life-threatening or otherwise impair quality of life. This is particularly the case of chronic oral infections, leading to recurrent aphthosis, paradontopathy and tooth loss.
The natural risk of life-threatening invasive infections is very high. In the 1950s, almost all patients with the most severe form of the disease, with permanent and profound neutropenia, died in the first 2 years of life from sepsis, cellulitis or pneumonia; this was the case of 11 of the 14 patients in Kostmann's pedigree [2]. Two deaths from pneumonia were reported among 16 patients with cyclic neutropenia [188], while no deaths were reported among patients with chronic benign neutropenia [34, 142].
In the sixties and seventies, with more extensive used of antibiotic therapy, lethal sepsis became less frequent even in the most severe forms of congenital neutropenia. The report of Kostmann's pedigree in 1975 showed long-term survival [33] and prior the G-CSF period (since the ninety's) death from infections is already exceptional in such category of patients but occasionally is described even cyclic neutropenia [5].
Chronic infections remain very frequent, and especially stomatologic infections with painful gingivitis associated with papules (aphthae-like oral furuncles) of the tongue, and parodontopathies [35]. Diffuse gastrointestinal lesions are sometimes present, leading to abdominal pain and diarrhea, and sometimes mimicking Crohn's disease on radiological studies [36]. This complication is frequent in glycogen storage disease type Ib.
The availability of G-CSF since 1988 dramatically changed these patients' medical management, but lethal bacterial infections are still reported [21, 189], especially in patients with a poor response to G-CSF, or with poor compliance. However, chronic stomatologic infection remains very difficult to manage, even with G-CSF and neutrophil recovery, leading to tooth loss [190]. Finally, the infections risk may not be related only to the neutropenia: the best example is the WHIM syndrome, which combines lymphopenia, hypogammaglobulinemia and very high susceptibility to human papillovirus infections [191].
Outcome: morbidity related to extra-hematopoietic involvement
Extra-hematopoetic involvement may have a very strong impact on these patients' lives, such as the neurodevelopmental disorders observed in Kostmann's disease, Shwachman Diamond syndrome, and Cohen's disease. Cardiac dysfunction may be very severe in Shwachman Diamond syndrome and is almost always observed in Barth's syndrome.
Malignant transformation: Risk factors and possible role of G-CSF
First introduced in the late 1980s [42], growth factors have vastly improved the management of chronic neutropenia. Once their efficacy on the neutropenia associated with cancer chemotherapy had been demonstrated [161] and the need for long-term administration in some cases had emerged [176], questions of safety were raised, especially regarding the risk of malignant transformation.
Although congenital neutropenias are preleukemic states, the risk of malignant transformation is difficult to evaluate in isolation, as the spontaneous risk and the potential role of G-CSF must both be taken into account.
The main question is the risk-benefit ratio, as mentioned in the very first article reporting the effect of G-CSF in this setting [161], particularly as leukemias had been observed in the rare patients with congenital neutropenia who survived beyond their first decade of life [192–194].
In 1993 international teams opted to create a patient registry to examine this issue. The data confirmed the marked increase in the risk of leukemia in these patients. The cumulative incidence of leukemia among patients with severe congenital neutropenia is about 15% at age 20 years [21].
Leukemic transformation has been observed in patients with mutations in ELANE[21, 195], HAX1[196], WASP[91], SBDS[21, 197] and G6PC3 or SLC37A4[198, 199].
These leukemias have a number of particularities. They usually involve proliferation of poorly differentiated cells, and the most consistent cytogenetic anomaly is monosomy 7. They are often preceded by the emergence of an acquired somatic anomaly of the intracytoplasmic part of the G-CSF receptor CSF3R [200]. These mutations are not found in de novo acute myeloblastic or lymphoblastic leukemia [201] and have never been found in Shwachman Diamond disease, even though this entity is associated with a high risk of leukemic transformation.
Studies of patients with ELANE mutations show that the main risk factor for leukemic onset is the severity of neutropenia and not the nature of the ELANE mutation. Thus, the leukemic risk is very low or inexistent in cyclic neutropenia, while it is maximal in patients with permanent neutropenia below 0.1 G/l. However, several other factors are associated with severe neutropenia, such as infections and the use of G-CSF, especially at high doses (> 15 μg/kg/day) and for long periods[21].
The link between intracytoplasmic G-CSF receptor defects, often observed in these patients before and during malignant transformation[202, 203], and monosomy 7 is not known. In contrast, blast cell lineages harboring monosomy 7 are particularly sensitive to G-CSF, which selects cells carrying this anomaly [204]. The absence of any direct oncogenic effect of ELANE mutations and the impact of both the severity of neutropenia (that strongly modifies bone marrow homeostasis and leads to compensatory hyperstimulation of the monocyte lineage) and high doses of G-CSF suggests that long-term bone marrow stimulation can be responsible for leukemic transformation. The leukemic risk in patients receiving the highest doses of G-CSF may warrant HSCT [21, 182, 189].
Evaluation of the risk factors of secondary leukemia with patient registries
The number of reported cases of leukemia and myelodysplasia in patients with severe congenital neutropenia has increased markedly in recent years, since the advent of G-CSF [6, 7, 205–210] relative to the pre-G-CSF era [192–194]. Numerous cases have been described in patients with Shwachman-Diamond syndrome, both before the advent of G-CSF and also in patients not receiving this drug [60, 211–213], whereas few have been reported in patients receiving G-CSF. However, this syndrome is rarely treated with G-CSF.
Reviews of the literature provide less reliable information on this adverse effect than patient registries, which can be used to calculate and to compare the risk.
The risk of leukemia and myelodysplasia has been studied in the French registry [21]. Factors favoring malignant transformation included disease-related factors and G-CSF exposure. Disease-related factors comprise the type of neutropenia as myelodysplasia and leukemia are observed only in patients with severe congenital neutropenia or Shwachman Diamond syndrome, the severity of neutropenia (i.e. the number and severity of infections), the degree of neutropenia, and the level of bone marrow myeloid arrest. Two characteristics of G-CSF exposure are significantly linked to the risk of leukemic transformation: the cumulative dose and the mean dose per injection. The cumulative duration of G-CSF exposure and the length of post-treatment follow-up are not associated with an increased risk. No threshold of exposure below which G-CSF does not increase the leukemic risk has been identified. In addition, the small size of this sample rules out firm conclusions, but patients requiring more than 10 μg/kg per injection and who receive a cumulative dose of more than 10 000 μg/kg, clearly have an increased risk of malignant transformation.
Initial publications from the international registry did not examine the link between the intensity of G-CSF exposure and the leukemic risk [6, 7], but more recent analyses have shown an increase in the risk of leukemia among patients receiving the highest doses [189, 214].
A relation between G-CSF exposure and secondary myelodysplasia and leukemia have been obtained in cohort studies of patients with bone marrow aplasia [215, 216] and breast cancer [217–219], showing that G-CSF has a leukemogenic effect in situations clearly distinct from congenital neutropenia.
Monitoring of the leukemic risk
This leukemic risk warrants close patient monitoring, especially when high doses of G-CSF are used. Repeated blood cell counts are required to detect anemia or thrombocytopenia, which may necessitate bone marrow sampling.
The place of routine bone marrow sampling is more controversial. Intracytoplasmic G-CSF- receptor mutations correlate with the appearance of leukemic clones, which can also be detected by cytogenetic examination with the FISH technique.
Conclusion on the leukemic risk of G-CSF therapy
Although the data are somewhat fragmentary and heterogeneous, several clinical studies suggest that G-CSF exposure beyond a certain threshold can be leukemogenic in patents with disorders known to favor leukemic transformation. The precise threshold dose at which this effect emerges cannot be determined, as it is not mentioned in most studies.
Lessons from congenital neutropenia and perspectives
Knowledge of the molecular bases of congenital neutropenia provides important information on two aspects of myeloid differentiation.
How congenital neutropenia contribute to understand dynamics of granulopoeisis?
The link between permanent neutropenia and defective neutrophil production or excessive apoptosis of neutrophil precursors is clear: a decrease in the production of cells or shorter half-life results in a lower number in the periphery. The cyclic aspect of the peripheral neutrophil count is more difficult to analyze and suggests the existence of a cryptic biological clock that regulates granulopoiesis. This putative clock might be revealed by particular mutations. To unify cyclic and permanent neutropenia, older notions on the dynamics of granulopoiesis can be helpful. First, the neutrophil count in healthy subjects varies markedly (between 1.8 and 4.5 G/l), in an unpredictable and chaotic manner [220]. In addition, 'extrinsic' factors that affect the neutrophil count also modify the cyclic variations in circulating neutrophil numbers relative to healthy subjects. Thus, G-CSF not only increases the number of circulating neutrophils but also leads to pseudo-cyclic irregularities [161]. In contrast, a cytostatic drug such as cyclophosphamide, administered at low doses, transforms chaotic variations in the neutrophil count into pseudo-cyclic changes[221], while high doses cause profound permanent neutropenia. These phenomena correspond to a non linear mathematical model [222]. Such models can describe temporal variations in the size of a population as a chaotic variation, with no precise cycle but between permanent extremes, up to total abolition of the population, via quasi-sinusoidal variations, from variations in a single coefficient corresponding to the reproductive rate of the population, i.e. the relationship between the production and death of individuals composing the population. In the case of the neutrophil population, excessive apoptosis, that cannot be precisely quantified and that can be influenced by precise mutations and the epigenetic context [223], contributes to excessive cell death [224]. This model remains theoretical but can be used to integrate physiological and pathological situations affecting granulopoiesis and is the only way to unify the different situations observed in congenital neutropenia[19].
The fate of stem cells from immature myeloid cells to mature polymorphonuclear neutrophils. lessons from congenital neutropenia
Congenital neutropenia represents a physiological model for studying granulopoeisis. In the past 10 years, 12 genes responsible for congenital neutropenia have been identified. Each mutation is responsible for a very peculiar molecular defect. Surprisingly, most known molecular abnormalities responsible for neutropenia do not involve genes with a transcriptional role in granulopoeisis, but rather genes involved in endoplasmic reticulum functions, like granule stability or intracytoplasmic granule trafficking or protein packaging.
Defective packaging of cellular enzymes in granules (due to ELANE mutations) or cytoskeleton changes (WASP and dynamin 2 mutations) modify intracytoplasmic trafficking and result in neutropenia, possibly through an excess of apoptosis or defective maturation. This is similar to the situation in several clinical disorders comprising albinism and neutropenia and characteristic of the Hermansky-Pudlak syndrome type 2 (AP3 defect), AP14 deficiency [78] (AP14 is a protein with similar functions to AP3), Chediak-Higashi syndrome and Griscelli's syndrome (the latter two entities also involve a macrophage activation syndrome in addition to neutropenia) [114]. A transmembrane protein of the Golgi apparatus is also involved in other disorders comprising neutropenia, such as glycogen storage disease Ib (SLC37A4), G6PC3 and Cohen's disease, but whose phenotypic expression also involves other systems.
The involvement of endoplasmic reticulum (ER) proteins or ER packaging processes in these forms of neutropenia shows the importance of ER stress. Increased ER stress elicits a cellular response known as the unfolded protein response (UPR). The UPR is activated in response to an accumulation of unfolded or misfolded proteins in the lumen of the ER. In this scenario, the UPR has two primary aims: to restore normal cell function by halting protein translation and to activate the signaling pathways that lead to increased production of molecular chaperones involved in protein folding. If these objectives are not achieved within a certain time lapse or if the disruption is prolonged, the UPR initiates programmed cell death (apoptosis). Three ER-localized protein sensors are known: IRE1alpha (inositol-requiring 1alpha), PERK (Protein kinase (PKR)-like ER kinase), and activating transcription factor 6 (ATF6). In cases of ER stress, these sensors are activated and trigger a complex series of events designed to maintain ER homeostasis and to promote protein folding, maturation, secretion, and ER-associated protein degradation. Changes in the shape of the protein, or a change in its function - such as a gain of function - may be one trigger of ER stress as shown for ELANE mutations [48, 51]. No comprehensive mechanism has been proposed for the impact of several different mutations, such as SBDS[225]. Indeed, mutation of the only transcription factor (GFI1) known to be involved in congenital neutropenia may not directly affect the transcription process but interfere with interaction with ELANE protein [88, 226].
Lymphoid enhancer factor 1 (LEF1) is a 48-kD nuclear protein expressed in pre-B and T cells and in myeloid cells. A low level of this factor has been observed in the myeloid cells of patients with congenital neutropenia with maturation arrest at the promyelocyte level [227]. The decrease in transcription factor expression is difficult to interpret both because LEF1 gene is normal and because LEF1 expression is depending of type of congenital neutropenia. This suggests that the decreased expression of LEF1 is more a proteic consequence to some mutations causing congenital neutropenia. Indeed Horwicz's team, who initially showed the involvement of LEF1 in this pathway, have shown that LEF-1 cooperates with Core-binding Factor α to activate ELANE in vivo [228]. They also raised the possibility that up regulating promoter mutations may contribute to SCN.
Pro LL37 is a antibacterial peptide usually packaged in neutrophil granules and its level is low, whatever the genetic backgrounds of the congenital neutropenia [229]. Low pro LL37 level may be responsible for the persistence of parondothopathy in patients with CN treated with G-CSF. Interestingly, vitamin D is able to correct the pro LL37 level [230].
Vitamin B3 (nicotamide) participates in a regulatory loop controlling the transcriptional expression of G-CSF. Vitamin B3 induces a peripheral increase in neutrophils [231].
References
Kostmann R: Hereditär reticulos - en ny systemsjukdom. Svenska Läkartideningen. 1950, 47: 2861-2868.
Kostmann R: Infantile genetic agranulocytosis; agranulocytosis infantilis hereditaria. Acta Paediatr Suppl. 1956, 45: 1-78. 10.1111/j.1651-2227.1956.tb17668.x.
Klein C, Grudzien M, Appaswamy G, Germeshausen M, Sandrock I, Schaffer AA, Rathinam C, Boztug K, Schwinzer B, Rezaei N: HAX1 deficiency causes autosomal recessive severe congenital neutropenia (Kostmann disease). Nat Genet. 2007, 39: 86-92. 10.1038/ng1940.
Carlsson G, van't HI, Melin M, Entesarian M, Laurencikas E, Nennesmo I, Trebinska A, Grzybowska E, Palmblad J, Dahl N: Central nervous system involvement in severe congenital neutropenia: neurological and neuropsychological abnormalities associated with specific HAX1 mutations. J Intern Med. 2008, 264: 388-400. 10.1111/j.1365-2796.2008.01982.x.
Palmer SE, Stephens K, Dale DC: Genetics, phenotype, and natural history of autosomal dominant cyclic hematopoiesis. American Journal of Medical Genetics. 1996, 66: 413-22. 10.1002/(SICI)1096-8628(19961230)66:4<413::AID-AJMG5>3.0.CO;2-L.
Dale DC, Cottle TE, Fier CJ, Bolyard AA, Bonilla MA, Boxer LA, Cham B, Freedman MH, Kannourakis G, Kinsey SE: Severe chronic neutropenia: Treatment and follow-up of patients in the Severe Chronic Neutropenia International Registry. Am J Hematol. 2003, 72: 82-93. 10.1002/ajh.10255.
Freedman MH, Bonilla MA, Fier C, Bolyard AA, Scarlata D, Boxer LA, Brown S, Cham B, Kannourakis G, Kinsey SE: Myelodysplasia syndrome and acute myeloid leukemia in patients with congenital neutropenia receiving G-CSF therapy. Blood. 2000, 96: 429-436.
Horwitz M, Benson KF, Person RE, Aprikyan AG, Dale DC: Mutations in ELA2, encoding neutrophil elastase, define a 21-day biological clock in cyclic haematopoiesis. Nat Genet. 1999, 23: 433-436. 10.1038/70544.
Dale DC, Person RE, Bolyard AA, Aprikyan AG, Bos C, Bonilla MA, Boxer LA, Kannourakis G, Zeidler C, Welte K: Mutations in the gene encoding neutrophil elastase in congenital and cyclic neutropenia. Blood. 2000, 96: 2317-2322.
Boztug K, Appaswamy G, Ashikov A, Schaffer AA, Salzer U, Diestelhorst J, Germeshausen M, Brandes G, Lee-Gossler J, Noyan F: A syndrome with congenital neutropenia and mutations in G6PC3. N Engl J Med. 2009, 360: 32-43. 10.1056/NEJMoa0805051.
Manroe BL, Weinberg AG, Rosenfeld CR, Browne R: The neonatal blood count in health and disease. I. Reference values for neutrophilic cells. J Pediatr. 1979, 95: 89-98. 10.1016/S0022-3476(79)80096-7.
Schelonka RL, Yoder BA, desJardins SE, Hall RB, Butler J: Peripheral leukocyte count and leukocyte indexes in healthy newborn term infants. J Pediatr. 1994, 125: 603-606. 10.1016/S0022-3476(94)70018-4.
Schelonka RL, Yoder BA, Hall RB, Trippett TM, Louder DS, Hickman JR, Guerra CG: Differentiation of segmented and band neutrophils during the early newborn period. J Pediatr. 1995, 127: 298-300. 10.1016/S0022-3476(95)70314-4.
Maughan WZ, Bishop CR, Pryor TA, Athens JW: The question of cycling of the blood neutrophil concentrations and pitfalls in the statistical analysis of sampled data. Blood. 1973, 41: 85-91.
Mackey MC: Cell kinetic status of haematopoietic stem cells. Cell Prolif. 2001, 34: 71-83. 10.1046/j.1365-2184.2001.00195.x.
Sletvold O, Smaaland R, Laerum OD: Cytometry and time-dependent variations in peripheral blood and bone marrow cells: a literature review and relevance to the chronotherapy of cancer. Chronobiol Int. 1991, 8: 235-250. 10.3109/07420529109063929.
Smaaland R, Sothern RB, Lote K, Sandberg S, Aakvaag A, Laerum OD: Circadian phase relationships between peripheral blood variables and bone marrow proliferative activity in clinical health. In Vivo. 1995, 9: 379-389.
Smaaland R, Sothern RB, Laerum OD, Abrahamsen JF: Rhythms in human bone marrow and blood cells. Chronobiol Int. 2002, 19: 101-127. 10.1081/CBI-120002594.
Haurie C, Dale DC, Mackey MC: Occurrence of periodic oscillations in the differential blood counts of congenital, idiopathic, and cyclical neutropenic patients before and during treatment with G-CSF. Experimental Hematology. 1999, 27: 401-9. 10.1016/S0301-472X(98)00061-7.
Baehner RL, Johnston RB: Monocyte function in children with neutropenia and chronic infections. Blood. 1972, 40: 31-41.
Donadieu J, Leblanc T, Bader MB, Barkaoui M, Fenneteau O, Bertrand Y, Maier-Redelsperger M, Micheau M, Stephan JL, Phillipe N: Analysis of risk factors for myelodysplasias, leukemias and death from infection among patients with congenital neutropenia. Experience of the French Severe Chronic Neutropenia Study Group. Haematologica. 2005, 90: 45-53.
Notarangelo LD, Fischer A, Geha RS, Casanova JL, Chapel H, Conley ME, Cunningham-Rundles C, Etzioni A, Hammartrom L, Nonoyama S: Primary immunodeficiencies: 2009 update. J Allergy Clin Immunol. 2009, 124: 1161-1178. 10.1016/j.jaci.2009.10.013.
Zeidler C, Germeshausen M, Klein C, Welte K: Clinical implications of ELA2-, HAX1-, and G-CSF-receptor (CSF3R) mutations in severe congenital neutropenia. Br J Haematol. 2009, 144: 459-467. 10.1111/j.1365-2141.2008.07425.x.
Dreyer NA, Garner S: Registries for robust evidence. Jama. 2009, 302: 790-791. 10.1001/jama.2009.1092.
Abuzakouk M, Feighery C: Primary immunodeficiency disorders in the Republic of Ireland: first report of the national registry in children and adults. J Clin Immunol. 2005, 25: 73-77. 10.1007/s10875-005-0360-9.
Golan H, Dalal I, Garty BZ, Schlesinger M, Levy J, Handzel Z, Wolach B, Rottem M, Goldberg A, Tamir R: The incidence of primary immunodeficiency syndromes in Israel. Isr Med Assoc J. 2002, 4: 868-871.
Matamoros FN, Mila LJ, Espanol BT, Raga BS, Fontan CG: Primary immunodeficiency syndrome in Spain: first report of the National Registry in Children and Adults. J Clin Immunol. 1997, 17: 333-339. 10.1023/A:1027382916924.
Rezaei N, Aghamohammadi A, Moin M, Pourpak Z, Movahedi M, Gharagozlou M, Atarod L, Ghazi BM, Isaeian A, Mahmoudi M: Frequency and clinical manifestations of patients with primary immunodeficiency disorders in Iran: update from the Iranian Primary Immunodeficiency Registry. J Clin Immunol. 2006, 26: 519-532. 10.1007/s10875-006-9047-x.
The French national registry of primary immunodeficiency diseases. Clin Immunol. 2010, 135: 264-272.
Li Y, Karlin A, Loike JD, Silverstein SC: A critical concentration of neutrophils is required for effective bacterial killing in suspension. Proc Natl Acad Sci USA. 2002, 99: 8289-8294. 10.1073/pnas.122244799.
Bodey GP, Buckley M, Sathe YS, Freireich EJ: Quantitative relationships between circulating leukocytes and infection in patients with acute leukemia. Ann Intern Med. 1966, 64: 328-340.
Meyers JD, Atkinson K: Infection in bone marrow transplantation. Clin Haematol. 1983, 12: 791-811.
Kostmann R: Infantile genetic agranulocytosis. Acta Paediatrica Scandinavica. 1975, 64: 362-368. 10.1111/j.1651-2227.1975.tb03847.x.
Pincus SH, Boxer LA, Stossel TP: Chronic neutropenia in childhood. Analysis of 16 cases and a review of the literature. American Journal of Medicine. 1976, 61: 849-61. 10.1016/0002-9343(76)90409-5.
Kalkwarf KL, Gutz DP: Periodontal changes associated with chronic idiopathic neutropenia. Pediatr Dent. 1981, 3: 189-195.
Roe TF, Coates TD, Thomas DW, Miller JH, Gilsanz V: Brief report: treatment of chronic inflammatory bowel disease in glycogen storage disease type Ib with colony-stimulating factors. N Engl J Med. 1992, 326: 1666-1669. 10.1056/NEJM199206183262504.
Borregaard N, Sorensen OE, Theilgaard-Monch K: Neutrophil granules: a library of innate immunity proteins. Trends Immunol. 2007, 28: 340-345. 10.1016/j.it.2007.06.002.
metchnikof E: Leçons sur la pathologie comparée de l'inflammation, faites à l'Institut Pasteur en avril et mai 1891. Masson. 1892.
Li Y, Karlin A, Loike JD, Silverstein SC: Determination of the critical concentration of neutrophils required to block bacterial growth in tissues. J Exp Med. 2004, 200: 613-622. 10.1084/jem.20040725.
Cartwright GE, Athens JW, Wintrobe MM: The kinetics of granulopoiesis in normal man. Blood. 1964, 24: 780-803.
Dresch C, Najean Y, Bauchet J: Kinetic studies of 51Cr and DF32P labelled granulocytes. Br J Haematol. 1975, 29: 67-80. 10.1111/j.1365-2141.1975.tb01800.x.
Dale DC: The discovery, development and clinical applications of granulocyte colony-stimulating factor. Transactions of the American Clinical & Climatological Association. 1998, 109: 27-36.
Bellanne-Chantelot C, Clauin S, Leblanc T, Cassinat B, Rodrigues-Lima F, Beaufils S, Vaury C, Barkaoui M, Fenneteau O, Maier-Redelsperger M: Mutations in the ELA2 gene correlate with more severe expression of neutropenia: a study of 81 patients from the French Neutropenia Register. Blood. 2004, 103: 4119-4125. 10.1182/blood-2003-10-3518.
Xia J, Bolyard AA, Rodger E, Stein S, Aprikyan AA, Dale DC, Link DC: Prevalence of mutations in ELANE, GFI1, HAX1, SBDS, WAS and G6PC3 in patients with severe congenital neutropenia. Br J Haematol. 2009, 147: 535-542. 10.1111/j.1365-2141.2009.07888.x.
Korkmaz B, Horwitz MS, Jenne DE, Gauthier F: Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases 2. Pharmacol Rev. 2010, 62: 726-759. 10.1124/pr.110.002733.
Massullo P, Druhan LJ, Bunnell BA, Hunter MG, Robinson JM, Marsh CB, Avalos BR: Aberrant subcellular targeting of the G185R neutrophil elastase mutant associated with severe congenital neutropenia induces premature apoptosis of differentiating promyelocytes. Blood. 2005, 105: 3397-3404. 10.1182/blood-2004-07-2618.
Grenda DS, Johnson SE, Mayer JR, McLemore ML, Benson KF, Horwitz M, Link DC: Mice expressing a neutrophil elastase mutation derived from patients with severe congenital neutropenia have normal granulopoiesis. Blood. 2002, 100: 3221-3228. 10.1182/blood-2002-05-1372.
Grenda DS, Murakami M, Ghatak J, Xia J, Boxer LA, Dale D, Dinauer MC, Link DC: Mutations of the ELA2 gene found in patients with severe congenital neutropenia induce the unfolded protein response and cellular apoptosis. Blood. 2007, 110: 4179-4187. 10.1182/blood-2006-11-057299.
Horwitz M, Benson KF, Duan Z, Li FQ, Person RE: Hereditary neutropenia: dogs explain human neutrophil elastase mutations. Trends Mol Med. 2004, 10: 163-170. 10.1016/j.molmed.2004.02.002.
Horwitz MS, Duan Z, Korkmaz B, Lee HH, Mealiffe ME, Salipante SJ: Neutrophil elastase in cyclic and severe congenital neutropenia. Blood. 2007, 109: 1817-1824. 10.1182/blood-2006-08-019166.
Kollner I, Sodeik B, Schreek S, Heyn H, von NN, Germeshausen M, Zeidler C, Kruger M, Schlegelberger B, Welte K: Mutations in neutrophil elastase causing congenital neutropenia lead to cytoplasmic protein accumulation and induction of the unfolded protein response. Blood. 2006, 108: 493-500. 10.1182/blood-2005-11-4689.
Druhan LJ, Ai J, Massullo P, Kindwall-Keller T, Ranalli MA, Avalos BR: Novel mechanism of G-CSF refractoriness in patients with severe congenital neutropenia. Blood. 2005, 105: 584-591. 10.1182/blood-2004-07-2613.
Carlsson G, Fasth A: Infantile genetic agranulocytosis, morbus Kostmann: presentation of six cases from the original "Kostmann family" and a review. Acta Paediatr. 2001, 90: 757-764.
Germeshausen M, Grudzien M, Zeidler C, Abdollahpour H, Yetgin S, Rezaei N, Ballmaier M, Grimbacher B, Welte K, Klein C: Novel HAX1 mutations in patients with severe congenital neutropenia reveal isoform-dependent genotype-phenotype associations. Blood. 2008, 111: 4954-4957. 10.1182/blood-2007-11-120667.
Nezelof C, Watchi M: L'hypoplasie congénitale lipomateuse du pancréas du pancréas exocrine chez l'enfant (2 observations et revue de la littérature). Arch Fr Pediatr. 1961, 18: 1135-1172.
Shwachman H, Diamond LK, Oski FA, Khaw KT: The syndrome of pancreatic insufficiency and bone marrow dysfunction. J Pediatr. 1964, 65: 645-663. 10.1016/S0022-3476(64)80150-5.
Lacaille F, Mani TM, Brunelle F, Lallemand D, Schmitz J: Magnetic resonance imaging for diagnosis of Shwachman's syndrome. Journal of Pediatric Gastroenterology & Nutrition. 1996, 23: 599-603. 10.1097/00005176-199612000-00015.
Dror Y, Freedman MH: Shwachman-diamond syndrome. Br J Haematol. 2002, 118: 701-713.
Kerr EN, Ellis L, Dupuis A, Rommens JM, Durie PR: The behavioral phenotype of school-age children with shwachman diamond syndrome indicates neurocognitive dysfunction with loss of Shwachman-Bodian-Diamond syndrome gene function. J Pediatr. 2010, 156: 433-438. 10.1016/j.jpeds.2009.09.026.
Dror Y, Freedman MH: Shwachman-Diamond syndrome: An inherited preleukemic bone marrow failure disorder with aberrant hematopoietic progenitors and faulty marrow microenvironment. Blood. 1999, 94: 3048-3054.
Black LV, Soltau T, Kelly DR, Berkow RL: Shwachman-Diamond syndrome presenting in a premature infant as pancytopenia. Pediatr Blood Cancer. 2008, 51: 123-124. 10.1002/pbc.21550.
Kuijpers TW, Nannenberg E, Alders M, Bredius R, Hennekam RC: Congenital aplastic anemia caused by mutations in the SBDS gene: a rare presentation of Shwachman-Diamond syndrome. Pediatrics. 2004, 114: e387-e391. 10.1542/peds.2003-0651-F.
Toiviainen-Salo S, Makitie O, Mannerkoski M, Hamalainen J, Valanne L, Autti T: Shwachman-Diamond syndrome is associated with structural brain alterations on MRI. Am J Med Genet A. 2008, 146A: 1558-1564. 10.1002/ajmg.a.32354.
Nishimura G, Nakashima E, Hirose Y, Cole T, Cox P, Cohn DH, Rimoin DL, Lachman RS, Miyamoto Y, Kerr B: The Shwachman-Bodian-Diamond syndrome gene mutations cause a neonatal form of spondylometaphysial dysplasia (SMD) resembling SMD Sedaghatian type. J Med Genet. 2007, 44: e73. 10.1136/jmg.2006.043869.
Boocock GR, Morrison JA, Popovic M, Richards N, Ellis L, Durie PR, Rommens JM: Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet. 2003, 33: 97-101. 10.1038/ng1062.
Finch AJ, Hilcenko C, Basse N, Drynan LF, Goyenechea B, Menne TF, Gonzalez FA, Simpson P, D'Santos CS, Arends MJ: Uncoupling of GTP hydrolysis from eIF6 release on the ribosome causes Shwachman-Diamond syndrome. Genes Dev. 2011, 25: 917-929. 10.1101/gad.623011.
Ambruso DR, McCabe ER, Anderson DC, Beaudet A, Ballas LM, Brandt IK: Infectious and bleeding complications in patients with glycogen Ib. Am J Dis Child. 2003, 139: 691-697.
Lachaux A, Boillot O, Stamm D, Canterino I, Dumontet C, Regnier F, Floret D, Hermier M: Treatment with lenograstim (glycosylated recombinant human granulocyte colony-stimulating factor) and orthotopic liver transplantation for glycogen storage disease type Ib. J Pediatr. 1993, 123: 1005-1008. 10.1016/S0022-3476(05)80403-2.
Yiu WH, Pan CJ, Allamarvdasht M, Kim SY, Chou JY: Glucose-6-phosphate transporter gene therapy corrects metabolic and myeloid abnormalities in glycogen storage disease type Ib mice. Gene Ther. 2007, 14: 219-226. 10.1038/sj.gt.3302869.
Cheung YY, Kim SY, Yiu WH, Pan CJ, Jun HS, Ruef RA, Lee EJ, Westphal H, Mansfield BC, Chou JY: Impaired neutrophil activity and increased susceptibility to bacterial infection in mice lacking glucose-6-phosphatase-beta. J Clin Invest. 2007, 117: 784-793. 10.1172/JCI30443.
Jun HS, Lee YM, Cheung YY, McDermott DH, Murphy PM, De Ravin SS, Mansfield BC, Chou JY: Lack of glucose recycling between endoplasmic reticulum and cytoplasm underlies cellular dysfunction in glucose-6-phosphatase-beta-deficient neutrophils in a congenital neutropenia syndrome. Blood. 2010, 116: 2783-2792. 10.1182/blood-2009-12-258491.
Chandler KE, Kidd A, Al-Gazali L, Kolehmainen J, Lehesjoki AE, Black GC, Clayton-Smith J: Diagnostic criteria, clinical characteristics, and natural history of Cohen syndrome. J Med Genet. 2003, 40: 233-241. 10.1136/jmg.40.4.233.
Kivitie-Kallio S, Rajantie J, Juvonen E, Norio R: Granulocytopenia in Cohen syndrome. Br J Haematol. 1997, 98: 308-311. 10.1046/j.1365-2141.1997.2323049.x.
Kolehmainen J, Black GC, Saarinen A, Chandler K, Clayton-Smith J, Traskelin AL, Perveen R, Kivitie-Kallio S, Norio R, Warburg M: Cohen syndrome is caused by mutations in a novel gene, COH1, encoding a transmembrane protein with a presumed role in vesicle-mediated sorting and intracellular protein transport. Am J Hum Genet. 2003, 72: 1359-1369. 10.1086/375454.
Mostefai R, Morice-Picard F, Boralevi F, Sautarel M, Lacombe D, Stasia MJ, McGrath J, Taieb A: Poikiloderma with neutropenia, Clericuzio type, in a family from Morocco. Am J Med Genet A. 2008, 146A: 2762-2769. 10.1002/ajmg.a.32524.
Volpi L, Roversi G, Colombo EA, Leijsten N, Concolino D, Calabria A, Mencarelli MA, Fimiani M, Macciardi F, Pfundt R: Targeted next-generation sequencing appoints c16orf57 as clericuzio-type poikiloderma with neutropenia gene. Am J Hum Genet. 2010, 86: 72-76. 10.1016/j.ajhg.2009.11.014.
Barth PG, Wanders RJ, Vreken P, Janssen EA, Lam J, Baas F: X-linked cardioskeletal myopathy and neutropenia (Barth syndrome) (MIM 302060). Journal of Inherited Metabolic Disease. 1999, 22: 555-67. 10.1023/A:1005568609936.
Bohn G, Allroth A, Brandes G, Thiel J, Glocker E, Schaffer AA, Rathinam C, Taub N, Teis D, Zeidler C: A novel human primary immunodeficiency syndrome caused by deficiency of the endosomal adaptor protein p14. Nat Med. 2007, 13: 38-45. 10.1038/nm1528.
Kotzot D, Richter K, Gierth-Fiebig K: Oculocutaneous albinism, immunodeficiency, hematological disorders, and minor anomalies: a new autosomal recessive syndrome?. Am J Med Genet. 1994, 50: 224-227. 10.1002/ajmg.1320500303.
Huizing M, Scher CD, Strovel E, Fitzpatrick DL, Hartnell LM, Anikster Y, Gahl WA: Nonsense mutations in ADTB3A cause complete deficiency of the beta3A subunit of adaptor complex-3 and severe Hermansky-Pudlak syndrome type 2. Pediatr Res. 2002, 51: 150-158. 10.1203/00006450-200202000-00006.
Dale DC, Rodger E, Cebon J, Ramesh N, Hammond WP, Zsebo KM: Long-term treatment of canine cyclic hematopoiesis with recombinant canine stem cell factor. Blood. 1995, 85: 74-9.
Meng R, Bridgman R, Toivio-Kinnucan M, Niemeyer GP, Vernau W, Hock T, Lothrop CD: Neutrophil elastase-processing defect in cyclic hematopoietic dogs. Exp Hematol. 2010, 38: 104-115. 10.1016/j.exphem.2009.09.010.
Itin PH, Pittelkow MR: Trichothiodystrophy with chronic neutropenia and mild mental retardation. J Am Acad Dermatol. 1991, 24: 356-358. 10.1016/0190-9622(91)70051-3.
Stoll C, Alembik Y, Lutz P: A syndrome of facial dysmorphia, birth defects, myelodysplasia and immunodeficiency in three sibs of consanguineous parents. Genetic Counseling. 1994, 5: 161-5.
Manicassamy S, Pulendran B: Modulation of adaptive immunity with Toll-like receptors. Semin Immunol. 2009, 21: 185-193. 10.1016/j.smim.2009.05.005.
Cham B, Bonilla MA, Winkelstein J: Neutropenia associated with primary immunodeficiency syndromes. Semin Hematol. 2002, 39: 107-112. 10.1053/shem.2002.31916.
Fiedler K, Sindrilaru A, Terszowski G, Kokai E, Feyerabend TB, Bullinger L, Rodewald HR, Brunner C: Neutrophil development and function critically depend on Bruton tyrosine kinase in a mouse model of X-linked agammaglobulinemia. Blood. 2011, 117: 1329-1339. 10.1182/blood-2010-04-281170.
Person RE, Li FQ, Duan Z, Benson KF, Wechsler J, Papadaki HA, Eliopoulos G, Kaufman C, Bertolone SJ, Nakamoto B: Mutations in proto-oncogene GFI1 cause human neutropenia and target ELA2. Nat Genet. 2003, 34: 308-312. 10.1038/ng1170.
Ancliff PJ, Blundell MP, Cory GO, Calle Y, Worth A, Kempski H, Burns S, Jones GE, Sinclair J, Kinnon C: Two novel activating mutations in the Wiskott-Aldrich syndrome protein result in congenital neutropenia. Blood. 2006, 108: 2182-2189. 10.1182/blood-2006-01-010249.
Beel K, Cotter MM, Blatny J, Bond J, Lucas G, Green F, Vanduppen V, Leung DW, Rooney S, Smith OP: A large kindred with X-linked neutropenia with an I294T mutation of the Wiskott-Aldrich syndrome gene 3. Br J Haematol. 2009, 144: 120-126. 10.1111/j.1365-2141.2008.07416.x.
Beel K, Vandenberghe P: G-CSF receptor (CSF3R) mutations in X-linked neutropenia evolving to acute myeloid leukemia or myelodysplasia. Haematologica. 2009, 94: 1449-1452. 10.3324/haematol.2009.009001.
Devriendt K, Kim AS, Mathijs G, Frints SG, Schwartz M, Van Den Oord JJ, Verhoef GE, Boogaerts MA, Fryns JP, You D: Constitutively activating mutation in WASP causes X-linked severe congenital neutropenia. Nat Genet. 2001, 27: 313-317. 10.1038/85886.
Moulding DA, Blundell MP, Spiller DG, White MR, Cory GO, Calle Y, Kempski H, Sinclair J, Ancliff PJ, Kinnon C: Unregulated actin polymerization by WASp causes defects of mitosis and cytokinesis in X-linked neutropenia. J Exp Med. 2007, 204: 2213-2224. 10.1084/jem.20062324.
Marmier-Savet C, Larosa F, Legrand F, Witz B, Michallet M, Ranta D, Louvat P, Puyraveau M, Raus N, Tavernier M: G-CSF-induced aneuploidy does not affect CD34+ cells and does not require cell division. Blood. 2010, 115: 910-911. 10.1182/blood-2009-10-250837.
Picard C, Puel A, Bonnet M, Ku CL, Bustamante J, Yang K, Soudais C, Dupuis S, Feinberg J, Fieschi C: Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science. 2003, 299: 2076-2079. 10.1126/science.1081902.
Bouma G, Doffinger R, Patel SY, Peskett E, Sinclair JC, Barcenas-Morales G, Cerron-Gutierrez L, Kumararatne DS, Davies EG, Thrasher AJ: Impaired neutrophil migration and phagocytosis in IRAK-4 deficiency. Br J Haematol. 2009, 147: 153-156. 10.1111/j.1365-2141.2009.07838.x.
Komiyama A, Kawai H, Yamada S, Aoyama K, Yamazaki M, Saitoh H, Miyagawa Y, Akabane T, Uehara Y: Impaired natural killer cell recycling in childhood chronic neutropenia with morphological abnormalities and defective chemotaxis of neutrophils. Blood. 1985, 66: 99-105.
Zuelwer WW: Myelokathexis - a new form of chronic granulocytopenia. Report of case. N Engl J Med. 1964, 270: 699-704. 10.1056/NEJM196404022701402.
Gorlin RJ, Gelb B, Diaz GA, Lofsness KG, Pittelkow MR, Fenyk JR: WHIM syndrome, an autosomal dominant disorder: clinical, hematological, and molecular studies. Am J Med Genet. 2000, 91: 368-376. 10.1002/(SICI)1096-8628(20000424)91:5<368::AID-AJMG10>3.0.CO;2-9.
Hernandez PA, Gorlin RJ, Lukens JN, Taniuchi S, Bohinjec J, Francois F, Klotman ME, Diaz GA: Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nat Genet. 2003, 34: 70-74. 10.1038/ng1149.
Arenzana-Seisdedos F, Virelizier JL, Rousset D, Clark-Lewis I, Loetscher P, Moser B, Baggiolini M: HIV blocked by chemokine antagonist. Nature. 1996, 383: 400. 10.1038/383400a0.
Aghamohammadi A, Cheraghi T, Rezaei N, Kanegane H, Abdollahzede S, Talaei-Khoei M, Heidari G, Zandieh F, Moin M, Miyawaki T: Neutropenia associated with X-linked Agammaglobulinemia in an Iranian referral center. Iran J Allergy Asthma Immunol. 2009, 8: 43-47.
Andrews FJ, Katz F, Jones A, Smith S, Finn A: CD40 ligand deficiency presenting as unresponsive neutropenia. Archives of Disease in Childhood. 1996, 74: 458-9. 10.1136/adc.74.5.458.
Jacobs ZD, Guajardo JR, Anderson KM: XLA-associated neutropenia treatment: a case report and review of the literature. J Pediatr Hematol Oncol. 2008, 30: 631-634. 10.1097/MPH.0b013e3181758947.
Kozlowski C, Evans DI: Neutropenia associated with X-linked agammaglobulinaemia. J Clin Pathol. 1991, 44: 388-390. 10.1136/jcp.44.5.388.
Agarwal BR, Currimbhoy Z: Resolution of cyclic neutropenia by intramuscular gamma globulin in a case of common variable immunodeficiency with predominantly antibody deficiency [see comments]. Indian Pediatrics. 1994, 31: 320-2.
Buckley RH: Molecular defects in human severe combined immunodeficiency and approaches to immune reconstitution. Annu Rev Immunol. 2004, 22: 625-655. 10.1146/annurev.immunol.22.012703.104614.
Dupuis-Girod S, Medioni J, Haddad E, Quartier P, Cavazzana-Calvo M, Le Deist F, de Saint BG, Delaunay J, Schwarz K, Casanova JL: Autoimmunity in Wiskott-Aldrich syndrome: risk factors, clinical features, and outcome in a single-center cohort of 55 patients. Pediatrics. 2003, 111: e622-e627. 10.1542/peds.111.5.e622.
Lagresle-Peyrou C, Six EM, Picard C, Rieux-Laucat F, Michel V, Ditadi A, merens-de CC, Morillon E, Valensi F, Simon-Stoos KL: Human adenylate kinase 2 deficiency causes a profound hematopoietic defect associated with sensorineural deafness. Nat Genet. 2009, 41: 106-111. 10.1038/ng.278.
Pannicke U, Honig M, Hess I, Friesen C, Holzmann K, Rump EM, Barth TF, Rojewski MT, Schulz A, Boehm T: Reticular dysgenesis (aleukocytosis) is caused by mutations in the gene encoding mitochondrial adenylate kinase 2. Nat Genet. 2009, 41: 101-105. 10.1038/ng.265.
Ozbek N, Derbent M, Olcay L, Yilmaz Z, Tokel K: Dysplastic changes in the peripheral blood of children with microdeletion 22q11.2. Am J Hematol. 2004, 77: 126-131. 10.1002/ajh.20139.
Latger-Cannard V, Bensoussan D, Gregoire MJ, Marcon F, Cloez JL, Leheup B, Jonveaux P, Lecompte T, Bordigoni P: Frequency of thrombocytopenia and large platelets correlates neither with conotruncal cardiac anomalies nor immunological features in the chromosome 22q11.2 deletion syndrome. Eur J Pediatr. 2004, 163: 327-328. 10.1007/s00431-004-1426-9.
Kratz CP, Niehues T, Lyding S, Heusch A, Janssen G, Gobel U: Evans syndrome in a patient with chromosome 22q11.2 deletion syndrome: a case report. Pediatr Hematol Oncol. 2003, 20: 167-172. 10.1080/0880010390158685.
de Saint BG, Fischer A: Defective cytotoxic granule-mediated cell death pathway impairs T lymphocyte homeostasis. Curr Opin Rheumatol. 2003, 15: 436-445. 10.1097/00002281-200307000-00011.
Ward DM, Shiflett SL, Kaplan J: Chediak-Higashi syndrome: a clinical and molecular view of a rare lysosomal storage disorder. Curr Mol Med. 2002, 2: 469-477. 10.2174/1566524023362339.
Feldmann J, Callebaut I, Raposo G, Certain S, Bacq D, Dumont C, Lambert N, Ouachee-Chardin M, Chedeville G, Tamary H: Munc13-4 is essential for cytolytic granules fusion and is mutated in a form of familial hemophagocytic lymphohistiocytosis (FHL3). Cell. 2003, 115: 461-473. 10.1016/S0092-8674(03)00855-9.
Menasche G, Ho CH, Sanal O, Feldmann J, Tezcan I, Ersoy F, Houdusse A, Fischer A, de Saint BG: Griscelli syndrome restricted to hypopigmentation results from a melanophilin defect (GS3) or a MYO5A F-exon deletion (GS1). J Clin Invest. 2003, 112: 450-456.
Kwong YL, Ng MH, Ma SK: Familial acute myeloid leukemia with monosomy 7: late onset and involvement of a multipotential progenitor cell. Cancer Genet Cytogenet. 2000, 116: 170-173. 10.1016/S0165-4608(99)00121-1.
Maserati E, Minelli A, Menna G, Cecchini MP, Bernardo ME, Rossi G, De FP, Lo CF, Danesino C, Locatelli F: Familial myelodysplastic syndromes, monosomy 7/trisomy 8, and mutator effects. Cancer Genet Cytogenet. 2004, 148: 155-158. 10.1016/S0165-4608(03)00271-1.
Minelli A, Maserati E, Giudici G, Tosi S, Olivieri C, Bonvini L, De FP, Biondi A, Lo CF, Pasquali F: Familial partial monosomy 7 and myelodysplasia: different parental origin of the monosomy 7 suggests action of a mutator gene. Cancer Genet Cytogenet. 2001, 124: 147-151. 10.1016/S0165-4608(00)00344-7.
Soriano JR, Taitz LS, Finberg L, Edelmann CM: Hyperglycinemia with ketoacidosis and leukopenia. Metabolic studies on the nature of the defect. Pediatrics. 1967, 39: 818-828.
Duval M, Fenneteau O, Doireau V, Faye A, Emilie D, Yotnda P, Drapier JC, Schlegel N, Sterkers G, de Baulny HO: Intermittent hemophagocytic lymphohistiocytosis is a regular feature of lysinuric protein intolerance. J Pediatr. 1999, 134: 236-239. 10.1016/S0022-3476(99)70423-3.
Lacbawan F, Tifft CJ, Luban NL, Schmandt SM, Guerrera M, Weinstein S, Pennybacker M, Wong LJ: Clinical heterogeneity in mitochondrial DNA deletion disorders: a diagnostic challenge of Pearson syndrome. Am J Med Genet. 2000, 95: 266-268. 10.1002/1096-8628(20001127)95:3<266::AID-AJMG13>3.0.CO;2-0.
Makitie O, Pukkala E, Kaitila I: Increased mortality in cartilage-hair hypoplasia. Arch Dis Child. 2001, 84: 65-67. 10.1136/adc.84.1.65.
Ridanpaa M, Sistonen P, Rockas S, Rimoin DL, Makitie O, Kaitila I: Worldwide mutation spectrum in cartilage-hair hypoplasia: ancient founder origin of the major70A-->G mutation of the untranslated RMRP. Eur J Hum Genet. 2002, 10: 439-447. 10.1038/sj.ejhg.5200824.
van der Hilst JC, Simon A, Drenth JP: Hereditary periodic fever and reactive amyloidosis. Clin Exp Med. 2005, 5: 87-98. 10.1007/s10238-005-0071-6.
Samuels J, Aksentijevich I, Torosyan Y, Centola M, Deng Z, Sood R, Kastner DL: Familial Mediterranean fever at the millennium. Clinical spectrum, ancient mutations, and a survey of 100 American referrals to the National Institutes of Health. Medicine (Baltimore). 1998, 77: 268-297. 10.1097/00005792-199807000-00005.
Ganiou TK, Ailal F, Najib J, Bellanne-Chantelot C, Donadieu J, Bousfiha AA: Intermittent chronic neutropenia in a patient with familial Mediterranean fever. Pediatr Blood Cancer. 2008, 51: 701-703. 10.1002/pbc.21685.
Jennings GH, Levi AJ, Reeve J: A case of chronic granulocytopenia associated with vasculitis and amyloidosis. J Clin Pathol. 1973, 26: 592-595. 10.1136/jcp.26.8.592.
Metin A, Ersoy F, Tinaztepe K, Besbas N, Tezcan I, Sanal O: Cyclic neutropenia complicated by renal AA amyloidosis. Turk J Pediatr. 2000, 42: 61-64.
Shiomura T, Ishida Y, Matsumoto N, Sasaki K, Ishihara T, Miwa S: A case of generalized amyloidosis associated with cyclic neutropenia. Blood. 1979, 54: 628-635.
Demiroglu H, Dundar S: Behcet's disease and chronic neutropenia. Scandinavian Journal of Rheumatology. 1997, 26: 130-2. 10.3109/03009749709115832.
Becton DL, Schultz WH, Kinney TR: Severe neutropenia caused by copper deficiency in a child receiving continuous ambulatory peritoneal dialysis. J Pediatr. 1986, 108: 735-737. 10.1016/S0022-3476(86)81056-3.
Halfdanarson TR, Kumar N, Li CY, Phyliky RL, Hogan WJ: Hematological manifestations of copper deficiency: a retrospective review. Eur J Haematol. 2008, 80: 523-531. 10.1111/j.1600-0609.2008.01050.x.
Karimbakas J, Langkamp-Henken B, Percival SS: Arrested maturation of granulocytes in copper deficient mice. J Nutr. 1998, 128: 1855-1860.
Ulinski T, Aoun B, Toubiana J, Vitkevic R, Bensman A, Donadieu J: Neutropenia in congenital nephrotic syndrome of the Finnish type: role of urinary ceruloplasmin loss. Blood. 2009, 113: 4820-4821. 10.1182/blood-2009-02-204099.
Bitoun M, Stojkovic T, Prudhon B, Maurage CA, Latour P, Vermersch P, Guicheney P: A novel mutation in the dynamin 2 gene in a Charcot-Marie-Tooth type 2 patient: clinical and pathological findings. Neuromuscul Disord. 2008, 18: 334-338. 10.1016/j.nmd.2008.01.005.
Claeys KG, Zuchner S, Kennerson M, Berciano J, Garcia A, Verhoeven K, Storey E, Merory JR, Bienfait HM, Lammens M: Phenotypic spectrum of dynamin 2 mutations in Charcot-Marie-Tooth neuropathy. Brain. 2009, 132: 1741-1752. 10.1093/brain/awp115.
Gbadoe AD, Fenneteau O, Duval M, Rohrlich P, Cartron J, Vilmer E: Phagocytose élective des polynucléaires neutrophiles par les macrophages médullaires et neutropénie auto immune de l'enfant. Arch Pediatr. 1997, 4: 398-405. 10.1016/S0929-693X(97)86659-6.
Dresch C, Flandrin G, Breton-Gorius J: Phagocytosis of neutrophil polymorphonuclears by macrophages in human bone marrow: importance in granulopoiesis. J Clin Pathol. 1980, 33: 1110-1113. 10.1136/jcp.33.11.1110.
Parmley RT, Crist WM, Ragab AH, Boxer LA, Malluh A, Findley H: Phagocytosis of neutrophils by marrow macrophages in childhood chronic benign neutropenia. Journal of Pediatrics. 1981, 98: 207-12. 10.1016/S0022-3476(81)80636-1.
Stahlie TD: Chronic benign neutropenia in infancy and early childhood; report of a case with a review of the literature. J Pediatr. 1956, 48: 710-721. 10.1016/S0022-3476(56)80201-1.
Bux J, Behrens G, Jaeger G, Welte K: Diagnosis and clinical course of autoimmune neutropenia in infancy: analysis of 240 cases. Blood. 1998, 91: 181-186.
Lalezari P, Khorshidi M, Petrosova M: Autoimmune neutropenia of infancy. Journal of Pediatrics. 1986, 109: 764-9. 10.1016/S0022-3476(86)80690-4.
Bareau B, Rey J, Hamidou M, Donadieu J, Morcet J, Reman O, Schleinitz N, Tournilhac O, Roussel M, Fest T: Analysis of a French cohort of patients with large granular lymphocyte leukemia: a report on 229 cases. Haematologica. 2010, 95: 1534-1541. 10.3324/haematol.2009.018481.
Palmblad JE, dem Borne AE: Idiopathic, immune, infectious, and idiosyncratic neutropenias. Semin Hematol. 2002, 39: 113-120. 10.1053/shem.2002.31919.
Starkebaum G: Chronic neutropenia associated with autoimmune disease. Semin Hematol. 2002, 39: 121-127. 10.1053/shem.2002.31918.
Mathew P, Chen G, Wang W: Evans syndrome: results of a national survey. J Pediatr Hematol Oncol. 1997, 19: 433-437. 10.1097/00043426-199709000-00005.
Yip D, Rasko JE, Lee C, Kronenberg H, O'Neill B: Thymoma and agranulocytosis: two case reports and literature review. Br J Haematol. 1996, 95: 52-56. 10.1046/j.1365-2141.1996.d01-1880.x.
Lamy T, Hamidou M, Loughran TP: Spectre des proliférations LGL et nouveaux concepts physiopathogéniques. Hematologie. 1999, 300-308.
Le Deist F, de Saint BG, Coulombel L, Breton-Gorius J, Maier-Redelsperger M, Beljorde K, Bremard C, Griscelli C: A familial occurrence of natural killer cell--T-lymphocyte proliferation disease in two children. Cancer. 1991, 67: 2610-2617. 10.1002/1097-0142(19910515)67:10<2610::AID-CNCR2820671035>3.0.CO;2-W.
Forbes WH, Johnson RECF: Leukopenia in negro workmen. Am J Med Sci. 1941, 201: 407-412. 10.1097/00000441-194103000-00013.
Hsieh MM, Everhart JE, Byrd-Holt DD, Tisdale JF, Rodgers GP: Prevalence of neutropenia in the U.S. population: age, sex, smoking status, and ethnic differences. Ann Intern Med. 2007, 146: 486-492.
Denic S, Showqi S, Klein C, Takala M, Nagelkerke N, Agarwal MM: Prevalence, phenotype and inheritance of benign neutropenia in Arabs. BMC Blood Disord. 2009, 9: 3. 10.1186/1471-2326-9-3.
Papadaki HA, Xylouri I, Coulocheri S, Kalmanti M, Kafatos A, Eliopoulos GD: Prevalence of chronic idiopathic neutropenia of adults among an apparently healthy population living on the island of Crete. Annals of Hematology. 1999, 78: 293-7. 10.1007/s002770050518.
Bain BJ, Phillips D, Thomson K, Richardson D, Gabriel I: Investigation of the effect of marathon running on leucocyte counts of subjects of different ethnic origins: relevance to the aetiology of ethnic neutropenia. Br J Haematol. 2000, 108: 483-487. 10.1046/j.1365-2141.2000.01922.x.
Grann VR, Ziv E, Joseph CK, Neugut AI, Wei Y, Jacobson JS, Horwitz MS, Bowman N, Beckmann K, Hershman DL: Duffy (Fy), DARC, and neutropenia among women from the United States, Europe and the Caribbean. Br J Haematol. 2008, 143: 288-293. 10.1111/j.1365-2141.2008.07335.x.
Malcov M, Reches A, Ben-Yosef D, Cohen T, Amit A, Dgany O, Tamary H, Yaron Y: Resolving a genetic paradox throughout preimplantation genetic diagnosis for autosomal dominant severe congenital neutropenia. Prenat Diagn. 2010, 30: 207-211.
Benson KF, Horwitz M: Possibility of somatic mosaicism of ELA2 mutation overlooked in an asymptomatic father transmitting severe congenital neutropenia to two offspring. Br J Haematol. 2002, 118: 923-924.
Rappeport JM, Parkman R, Newburger P, Camitta BM, Chusid MJ: Correction of infantile agranulocytosis (Kostmann's syndrome) by allogeneic bone marrow transplantation. Am J Med. 1980, 68: 605-609. 10.1016/0002-9343(80)90312-5.
Bonilla MA, Gillio AP, Ruggeiro M, Kernan NA, Brochstein JA, Abboud M, Fumagalli L, Vincent M, Gabrilove JL, Welte K: Effects of recombinant human granulocyte colony-stimulating factor on neutropenia in patients with congenital agranulocytosis. N Engl J Med. 1989, 320: 1574-1580. 10.1056/NEJM198906153202402.
Lucas KG, Brown AE, Armstrong D, Chapman D, Heller G: The identification of febrile, neutropenic children with neoplastic disease at low risk for bacteremia and complications of sepsis. Cancer. 1996, 77: 791-798. 10.1002/(SICI)1097-0142(19960215)77:4<791::AID-CNCR27>3.0.CO;2-V.
Rackoff WR, Gonin R, Robinson C, Kreissman SG, Breitfeld PB: Predicting the risk of bacteremia in childen with fever and neutropenia. Journal of Clinical Oncology. 1996, 14: 919-24.
Cordonnier C, Leverger G, Schlemmer B, Andremont A, Boasson M, Herbrecht R, Kazmierczak A, Marie JP, Marit G, Miclea JM: Stratégie antibiotique dans les épisodes fébriles au cours des neutropénies profondes (inférieures à 500 PNN) et prolongées (supérieures ou égales à 7 jours). Recommandations du College Francais des Hematologistes. Nouv Rev Fr Hematol. 1994, 36: 289-291.
Gurwith MJ, Brunton JL, Lank BA, Harding GK, Ronald AR: A prospective controlled investigation of prophylactic trimethoprim/sulfamethoxazole in hospitalized granulocytopenic patients. Am J Med. 1979, 66: 248-256. 10.1016/0002-9343(79)90539-4.
Margolis DM, Melnick DA, Alling DW, Gallin JI: Trimethoprim-sulfamethoxazole prophylaxis in the management of chronic granulomatous disease. J Infect Dis. 1990, 162: 723-726. 10.1093/infdis/162.3.723.
Keisu M, Wiholm BE, Palmblad J: Trimethoprim-sulphamethoxazole-associated blood dyscrasias. Ten years' experience of the Swedish spontaneous reporting system. J Intern Med. 1990, 228: 353-360. 10.1111/j.1365-2796.1990.tb00245.x.
Freund MR, Luft S, Schober C, Heussner P, Schrezenmaier H, Porzsolt F, Welte K: Differential effect of GM-CSF and G-CSF in cyclic neutropenia [letter]. Lancet. 1990, 336: 313. 10.1016/0140-6736(90)91849-6.
Schroten H, Roesler J, Breidenbach T, Wendel U, Elsner J, Schweitzer S, Zeidler C, Burdach S, Lohmann-Matthes ML, Wahn V: Granulocyte and granulocyte-macrophage colony-stimulating factors for treatment of neutropenia in glycogen storage disease type Ib. Journal of Pediatrics. 1991, 119: 748-54. 10.1016/S0022-3476(05)80290-2.
Welte K, Zeidler C, Reiter A, Muller W, Odenwald E, Souza L, Riehm H: Differential effects of granulocyte-macrophage colony-stimulating factor and granulocyte colony-stimulating factor in children with severe congenital neutropenia. Blood. 1990, 75: 1056-63.
Carlsson G, Ahlin A, Dahllof G, Elinder G, Henter JI, Palmblad J: Efficacy and safety of two different rG-CSF preparations in the treatment of patients with severe congenital neutropenia. Br J Haematol. 2004, 126: 127-132. 10.1111/j.1365-2141.2004.05008.x.
Fioredda F, Calvillo M, Lanciotti M, Lanza T, Giunti L, Castagnola E, Lorenzi I, Tonelli R, Ghezzi P, Dufour C: Pegfilgrastim in children with severe congenital neutropenia. Pediatr Blood Cancer. 2010, 54: 465-467. 10.1002/pbc.22350.
Donadieu J, Beaupain B, Rety-Jacob F, Nove-Josserand R: Respiratory distress and sudden death of a patient with GSDIb chronic neutropenia: possible role of pegfilgrastim. Haematologica. 2009, 94: 1175-1177. 10.3324/haematol.2008.005330.
Beaupain B, Leblanc T, Reman O, Hermine O, Vannier JP, Suarez F, Lutz P, Bordigoni P, Jourdain A, Schoenvald M: Is pegfilgrastim safe and effective in congenital neutropenia? An analysis of the French Severe Chronic Neutropenia registry. Pediatr Blood Cancer. 2009, 53: 1068-1073. 10.1002/pbc.22147.
Donadieu J, Boutard P, Bernatowska E, Tchernia G, Couillaud G, Philippe N, Le Gall E: A European phase II study of recombinant human granulocyte colony-stimulating factor (lenograstim) in the treatment of severe chronic neutropenia in children. Lenograstim Study Group. Eur J Pediatr. 1997, 156: 693-700. 10.1007/s004310050692.
Dale DC, Bonilla MA, Davis MW, Nakanishi AM, Hammond WP, Kurtzberg J, Wang W, Jakubowski A, Winton E, Lalezari P: A randomized controlled phase III trial of recombinant human granulocyte colony-stimulating factor (filgrastim) for treatment of severe chronic neutropenia. Blood. 1993, 81: 2496-502.
Cottle TE, Fier CJ, Donadieu J, Kinsey SE: Risk and benefit of treatment of severe chronic neutropenia with granulocyte colony-stimulating factor. Semin Hematol. 2002, 39: 134-140. 10.1053/shem.2002.31914.
Sandor V, Hassan R, Kohn E: Exacerbation of pseudogout by granulocyte colony-stimulating factor. Ann Intern Med. 1996, 125: 781.
Park JW, Mehrotra B, Barnett BO, Baron AD, Venook AP: The Sweet syndrome during therapy with granulocyte colony-stimulating factor. Ann Intern Med. 1992, 116: 996-998.
Yakisan E, Schirg E, Zeidler C, Bishop NJ, Reiter A, Hirt A, Riehm H, Welte K: High incidence of significant bone loss in patients with severe congenital neutropenia (Kostmann's syndrome). Journal of Pediatrics. 1997, 131: 592-7. 10.1016/S0022-3476(97)70068-4.
Kannourakis G: Glycogen storage disease. Semin Hematol. 2002, 39: 103-106. 10.1053/shem.2002.31920.
Ferry C, Ouachee M, Leblanc T, Michel G, Notz-Carrere A, Tabrizi R, Flood T, Lutz P, Fischer A, Gluckman E: Hematopoietic stem cell transplantation in severe congenital neutropenia: experience of the French SCN register. Bone Marrow Transplant. 2005, 35: 45-50. 10.1038/sj.bmt.1704718.
Oshima K, Hanada R, Kobayashi R, Kato K, Nagatoshi Y, Tabuchi K, Kato S: Hematopoietic stem cell transplantation in patients with severe congenital neutropenia: An analysis of 18 Japanese cases. Pediatr Transplant. 2010, 14 (5): 657-63. 10.1111/j.1399-3046.2010.01319.x.
Zeidler C, Welte K, Barak Y, Barriga F, Bolyard AA, Boxer L, Cornu G, Cowan MJ, Dale DC, Flood T: Stem cell transplantation in patients with severe congenital neutropenia without evidence of leukemic transformation. Blood. 2000, 95: 1195-1198.
Donadieu J, Michel G, Merlin E, Bordigoni P, Monteux B, Beaupain B, Leverger G, Laporte JP, Hermine O, Buzyn A: Hematopoietic stem cell transplantation for Shwachman-Diamond syndrome: experience of the French neutropenia registry. Bone Marrow Transplant. 2005, 36: 787-792. 10.1038/sj.bmt.1705141.
Bhatla D, Davies SM, Shenoy S, Harris RE, Crockett M, Shoultz L, Smolarek T, Bleesing J, Hansen M, Jodele S: Reduced-intensity conditioning is effective and safe for transplantation of patients with Shwachman-Diamond syndrome. Bone Marrow Transplant. 2008, 42: 159-165. 10.1038/bmt.2008.151.
Sauer M, Zeidler C, Meissner B, Rehe K, Hanke A, Welte K, Lohse P, Sykora KW: Substitution of cyclophosphamide and busulfan by fludarabine, treosulfan and melphalan in a preparative regimen for children and adolescents with Shwachman-Diamond syndrome. Bone Marrow Transplant. 2007, 39: 143-147. 10.1038/sj.bmt.1705553.
Reimann HA, DeBERARDINIS CT: Periodic (cyclic) neutropenia, an entity; a collection of 16 cases. Blood. 1949, 4: 1109-1116.
Rosenberg PS, Alter BP, Bolyard AA, Bonilla MA, Boxer LA, Cham B, Fier C, Freedman M, Kannourakis G, Kinsey S: The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood. 2006, 107: 4628-4635. 10.1182/blood-2005-11-4370.
Carlsson G, Wahlin YB, Johansson A, Olsson A, Eriksson T, Claesson R, Hanstrom L, Henter JI: Periodontal disease in patients from the original Kostmann family with severe congenital neutropenia. J Periodontol. 2006, 77: 744-751. 10.1902/jop.2006.050191.
Tassone L, Notarangelo LD, Bonomi V, Savoldi G, Sensi A, Soresina A, Smith CI, Porta F, Plebani A, Notarangelo LD: Clinical and genetic diagnosis of warts, hypogammaglobulinemia, infections, and myelokathexis syndrome in 10 patients. J Allergy Clin Immunol. 2009, 123 (5): 1170-3. 10.1016/j.jaci.2008.12.1133. 1173.e1-3.
De Vries A, Peketh L, Joshua H: Leukaemia and agranulocytosis in a member of a family with hereditary leukopenia. Acta Med Orient. 1958, 17: 26-32.
Gilman PA, Jackson DP, Guild HG: Congenital agranulocytosis: prolonged survival and terminal acute leukemia. Blood. 1970, 36: 576-585.
Rosen R, Kang S: Congenital agranulocytosis terminating in acute myelomonocytic leukemia. J Pediatr. 1979, 94: 406-408. 10.1016/S0022-3476(79)80581-8.
Rosenberg PS, Alter BP, Link DC, Stein S, Rodger E, Bolyard AA, Aprikyan AA, Bonilla MA, Dror Y, Kannourakis G: Neutrophil elastase mutations and risk of leukaemia in severe congenital neutropenia. Br J Haematol. 2008, 140: 210-213.
Yetgin S, Olcay L, Koc A, Germeshausen M: Transformation of severe congenital neutropenia to early acute lymphoblastic leukemia in a patient with HAX1 mutation and without G-CSF administration or receptor mutation. Leukemia. 2008, 22: 1797. 0.1038/leu.2008.64.
Dror Y: Shwachman-Diamond syndrome. Pediatr Blood Cancer. 2005, 45: 892-901. 10.1002/pbc.20478.
Schroeder T, Hildebrandt B, Mayatepek E, Germing U, Haas R: A patient with glycogen storage disease type Ib presenting with acute myeloid leukemia (AML) bearing monosomy 7 and translocation t(3;8)(q26;q24) after 14 years of treatment with granulocyte colony-stimulating factor (G-CSF): a case report. J Med Case Reports. 2008, 2: 319. 10.1186/1752-1947-2-319.
Pinsk M, Burzynski J, Yhap M, Fraser RB, Cummings B, Ste-Marie M: Acute myelogenous leukemia and glycogen storage disease 1b. J Pediatr Hematol Oncol. 2002, 24: 756-758. 10.1097/00043426-200212000-00015.
Germeshausen M, Ballmaier M, Welte K: Incidence of CSF3R mutations in severe congenital neutropenia and relevance for leukemogenesis: Results of a long-term survey. Blood. 2007, 109: 93-99. 10.1182/blood-2006-02-004275.
Link DC, Kunter G, Kasai Y, Zhao Y, Miner T, McLellan MD, Ries RE, Kapur D, Nagarajan R, Dale DC: Distinct patterns of mutations occurring in de novo AML versus AML arising in the setting of severe congenital neutropenia. Blood. 2007, 110: 1648-1655. 10.1182/blood-2007-03-081216.
Touw IP, Bontenbal M: Granulocyte colony-stimulating factor: key (f)actor or innocent bystander in the development of secondary myeloid malignancy?. J Natl Cancer Inst. 2007, 99: 183-186. 10.1093/jnci/djk057.
Ward AC, Gits J, Majeed F, Aprikyan AA, Lewis RS, O'Sullivan LA, Freedman M, Shigdar S, Touw IP, Dale DC: Functional interaction between mutations in the granulocyte colony-stimulating factor receptor in severe congenital neutropenia. Br J Haematol. 2008, 142: 653-656. 10.1111/j.1365-2141.2008.07224.x.
Sloand EM, Yong AS, Ramkissoon S, Solomou E, Bruno TC, Kim S, Fuhrer M, Kajigaya S, Barrett AJ, Young NS: Granulocyte colony-stimulating factor preferentially stimulates proliferation of monosomy 7 cells bearing the isoform IV receptor. Proc Natl Acad Sci USA. 2006, 103: 14483-14488. 10.1073/pnas.0605245103.
Germeshausen M, Ballmaier M, Schulze H, Welte K, Flohr T, Beiske K, Storm-Mathisen I, Abrahamsen TG: Granulocyte colony-stimulating factor receptor mutations in a patient with acute lymphoblastic leukemia secondary to severe congenital neutropenia. Blood. 2001, 97: 829-830. 10.1182/blood.V97.3.829.
Jeha S, Chan KW, Aprikyan AG, Hoots WK, Culbert S, Zietz H, Dale DC, Albitar M: Spontaneous remission of granulocyte colony-stimulating factor-associated leukemia in a child with severe congenital neutropenia. Blood. 2000, 96: 3647-3649.
Nibu K, Yanai F, Hirota O, Hatazoe M, Yamaguchi S, Akamatsu M, Kikuchi M, Morimoto Y, Kuwano A: Acute monocytic leukemia in a patient with severe congenital neutropenia after treatment with recombinant human granulocyte colony-stimulating factor. J Pediatr Hematol Oncol. 1996, 18: 422-424.
Smith OP, Reeves BR, Kempski HM, Evans JP: Kostmann's disease, recombinant HuG-CSF, monosomy 7 and MDS/AML. Br J Haematol. 1995, 91: 150-153. 10.1111/j.1365-2141.1995.tb05260.x.
Weinblatt ME, Scimeca P, James-Herry A, Sahdev I, Kochen J: Transformation of congenital neutropenia into monosomy 7 and acute nonlymphoblastic leukemia in a child treated with granulocyte colony-stimulating factor. Journal of Pediatrics. 1995, 126: 263-5. 10.1016/S0022-3476(95)70557-0.
Wong WY, Williams D, Slovak ML, Charak B, Mazumder A, Snyder D, Powars DR, Brynes RK: Terminal acute myelogenous leukemia in a patient with congenital agranulocytosis. Am J Hematol. 1993, 43: 133-138. 10.1002/ajh.2830430212.
Dror Y, Durie P, Ginzberg H, Herman R, Banerjee A, Champagne M, Shannon K, Malkin D, Freedman MH: Clonal evolution in marrows of patients with Shwachman-Diamond syndrome: a prospective 5-year follow-up study. Exp Hematol. 2002, 30: 659-669. 10.1016/S0301-472X(02)00815-9.
Huijgens PC, van der Veen EA, Meijer S, Muntinghe OG: Syndrome of Shwachman and leukaemia. Scandinavian Journal of Haematology. 1977, 18: 20-4.
Raj AB, Bertolone SJ, Barch MJ, Hersh JH: Chromosome 20q deletion and progression to monosomy 7 in a patient with Shwachman-Diamond syndrome without MDS/AML. J Pediatr Hematol Oncol. 2003, 25: 508-509. 10.1097/00043426-200306000-00018.
Rosenberg PS, Zeidler C, Bolyard AA, Alter BP, Bonilla MA, Boxer LA, Dror Y, Kinsey S, Link DC, Newburger PE: Stable long-term risk of leukaemia in patients with severe congenital neutropenia maintained on G-CSF therapy. Br J Haematol. 2010, 150 (2): 196-9.
Kojima S, Ohara A, Tsuchida M, Kudoh T, Hanada R, Okimoto Y, Kaneko T, Takano T, Ikuta K, Tsukimoto I: Risk factors for evolution of acquired aplastic anemia into myelodysplastic syndrome and acute myeloid leukemia after immunosuppressive therapy in children. Blood. 2002, 100: 786-790. 10.1182/blood.V100.3.786.
Socie G, Mary JY, Schrezenmeier H, Marsh J, Bacigalupo A, Locasciulli A, Fuehrer M, Bekassy A, Tichelli A, Passweg J: Granulocyte-stimulating factor and severe aplastic anemia: a survey by the European Group for Blood and Marrow Transplantation (EBMT). Blood. 2007, 109: 2794-2796.
Hershman D, Neugut AI, Jacobson JS, Wang J, Tsai WY, McBride R, Bennett CL, Grann VR: Acute myeloid leukemia or myelodysplastic syndrome following use of granulocyte colony-stimulating factors during breast cancer adjuvant chemotherapy. J Natl Cancer Inst. 2007, 99: 196-205. 10.1093/jnci/djk028.
Le Deley MC, Leblanc T, Shamsaldin A, Raquin MA, Lacour B, Sommelet D, Chompret A, Cayuela JM, Bayle C, Bernheim A: Risk of secondary leukemia after a solid tumor in childhood according to the dose of epipodophyllotoxins and anthracyclines: a case-control study by the Societe Francaise d'Oncologie Pediatrique. J Clin Oncol. 2003, 21: 1074-1081. 10.1200/JCO.2003.04.100.
Smith RE, Bryant J, DeCillis A, Anderson S: Acute myeloid leukemia and myelodysplastic syndrome after doxorubicin-cyclophosphamide adjuvant therapy for operable breast cancer: the National Surgical Adjuvant Breast and Bowel Project Experience. J Clin Oncol. 2003, 21: 1195-1204. 10.1200/JCO.2003.03.114.
Morley AA: A neutrophil cycle in healthy individuals. Lancet. 1966, 2: 1220-1222.
Morley A, Stohlman F: Cyclophosphamide-induced cyclical neutropenia. An animal model of a human periodic disease. N Engl J Med. 1970, 282: 643-646. 10.1056/NEJM197003192821202.
May RM: Simple mathematical models with very complicated dynamics. Nature. 1976, 261: 459-467. 10.1038/261459a0.
Duan Z, Person RE, Lee HH, Huang S, Donadieu J, Badolato R, Grimes HL, Papayannopoulou T, Horwitz MS: Epigenetic Regulation of Protein-Coding and MicroRNA Genes by the Gfi1-Interacting, Tumor Suppressor PRDM5. Mol Cell Biol. 2007, 27 (19): 6889-902. 10.1128/MCB.00762-07.
Mackey MC, Aprikyan AA, Dale DC: The rate of apoptosis in post mitotic neutrophil precursors of normal and neutropenic humans. Cell Prolif. 2003, 36: 27-34. 10.1046/j.1365-2184.2003.00251.x.
Ganapathi KA, Austin KM, Lee CS, Dias A, Malsch MM, Reed R, Shimamura A: The human Shwachman-Diamond syndrome protein, SBDS, associates with ribosomal RNA. Blood. 2007, 110: 1458-1465. 10.1182/blood-2007-02-075184.
Salipante SJ, Rojas ME, Korkmaz B, Duan Z, Wechsler J, Benson KF, Person RE, Grimes HL, Horwitz MS: Contributions to neutropenia from PFAAP5 (N4BP2L2), a novel protein mediating transcriptional repressor cooperation between Gfi1 and neutrophil elastase. Mol Cell Biol. 2009, 29: 4394-4405. 10.1128/MCB.00596-09.
Skokowa J, Cario G, Uenalan M, Schambach A, Germeshausen M, Battmer K, Zeidler C, Lehmann U, Eder M, Baum C: LEF-1 is crucial for neutrophil granulocytopoiesis and its expression is severely reduced in congenital neutropenia. Nat Med. 2006, 12: 1191-1197. 10.1038/nm1474.
Li FQ, Person RE, Takemaru K, Williams K, Meade-White K, Ozsahin AH, Gungor T, Moon RT, Horwitz M: Lymphoid enhancer factor-1 links two hereditary leukemia syndromes through core-binding factor alpha regulation of ELA2. J Biol Chem. 2004, 279: 2873-2884.
Karlsson J, Carlsson G, Ramme KG, Hagglund H, Fadeel B, Nordenskjold M, Henter JI, Palmblad J, Putsep K, Andersson M: Low plasma levels of the protein pro-LL-37 as an early indication of severe disease in patients with chronic neutropenia. Br J Haematol. 2007, 137: 166-169. 10.1111/j.1365-2141.2007.06530.x.
Karlsson J, Carlsson G, Larne O, Andersson M, Putsep K: Vitamin D3 induces pro-LL-37 expression in myeloid precursors from patients with severe congenital neutropenia. J Leukoc Biol. 2008, 84: 1279-1286. 10.1189/jlb.0607437.
Skokowa J, Lan D, Thakur BK, Wang F, Gupta K, Cario G, Brechlin AM, Schambach A, Hinrichsen L, Meyer G: NAMPT is essential for the G-CSF-induced myeloid differentiation via a NAD(+)-sirtuin-1-dependent pathway. Nat Med. 2009, 15: 151-158. 10.1038/nm.1913.
Faiyaz-Ul-Haque M, Al-Jefri A, Al-Dayel F, Bhuiyan JA, Abalkhail HA, Al-Nounou R, Al-Abdullatif A, Pulicat MS, Gaafar A, Alaiya AA: A novel HAX1 gene mutation in severe congenital neutropenia (SCN) associated with neurological manifestations. Eur J Pediatr. 2010, 169 (6): 661-6. 10.1007/s00431-010-1150-6.
Ishikawa N, Okada S, Miki M, Shirao K, Kihara H, Tsumura M, Nakamura K, Kawaguchi H, Ohtsubo M, Yasunaga S: Neurodevelopmental abnormalities associated with severe congenital neutropenia due to the R86X mutation in the HAX1 gene. J Med Genet. 2008, 45: 802-807. 10.1136/jmg.2008.058297.
Veiga-da-Cunha M, Gerin I, Chen YT, Lee PJ, Leonard JV, Maire I, Wendel U, Vikkula M, Van Schaftingen E: The putative glucose 6-phosphate translocase gene is mutated in essentially all cases of glycogen storage disease type I non-a. Eur J Hum Genet. 1999, 7: 717-723. 10.1038/sj.ejhg.5200366.
Acknowledgements
We are indebted to David Young for his editorial assistance. Our work was supported by a grant from Inserm Invs (registre des neutropénies) and by the e Rare funds (NEUTRO NET) Agence Nationale de la Recherche 09-RARE-010-02
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JD conceived the design and the first draft of the manuscript. OF collect the cytological iconography, all authors critically review the initial draft and participate to the final version of the manuscript.
Electronic supplementary material
13023_2010_298_MOESM1_ESM.PPT
Additional file 1: Plate #1: Large aphthae on inner lip of a patient with severe congenital neutropenia. Plate #2: Inflammatory gum lesion in a 12-y-old body with severe congenital neutropenia. Note the enamel damage and loss of parodontal tissue. Plate #3: Aspects of maturation arrest at the promyelocytic stage associated with hypereosinophilia and monocytosis in a patient with ELANE severe congenital neutropenia. Plate #4: Marrow smear in a patients with Shwachman-Diamond syndrome complicated by bone marrow aplasia: Poor cellularity, fat cells and mast cells. Plate #5: Marrow smear in a patient with Shwachman-Diamond syndrome complicated by acute erytrhoid leukemia. Plate #6: Marrow smear in a patient with Shwachman-Diamond syndrome complicated by cytopenia and monosomy 7. Left: monolobated micromegacaryocyte (arrow) Right: double nucleus of the granulocyte lineage (arrow). Plate #7: Marrow smear in a patient with glycogen storage disease 1b: Hyperplasia of the granulocyte lineage with no maturation arrest. Plate #8: Marrow smear of a patient with WHIM syndrome: The PN nuclear lobes are separated by long, thin filaments; the cytoplasm is occasionally vacuolated. Plate #9: Blood smear of a patient with Chediak-Higashi Syndrome: Left: lymphocyte with a voluminous bright red inclusion (MGG staining) Middle: PN with large sparse granulation, Right Marrow smear of a patient with Chediak Higashi Syndrome: Voluminous inclusions in the cytoplasm of myeloid precursors. Plate #10: Marrow smear of a patient with Griscelli Syndrome: Numerous histiocytes reflecting a histiocyte activation syndrome. Plate #11: Marrow smear of a patient with dibasic protein intolerance: Left: PN with picnotic nuclei phagocyted, by immatures myeloid cells (centre) and by histiocytes (right). Plate #12: Marrow smear of a patient with Pearson's syndrome: Vacuolization of precursors (left) associated with dyserythropoiesis with acidophilic cells with laminated cytoplasm (centre) and ring sideroblasts (Perls stain) (right). Plate #13: Marrow smear in a young patient with autoimmune neutropenia Left: Phagocytosis of a neutrophil by a histiocyte. Right: Two histiocytes having engulfed several neutrophils that are at various stages of breakdown (PPT 20 MB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Donadieu, J., Fenneteau, O., Beaupain, B. et al. Congenital neutropenia: diagnosis, molecular bases and patient management. Orphanet J Rare Dis 6, 26 (2011). https://doi.org/10.1186/1750-1172-6-26
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1750-1172-6-26