
Abstract
In recent years, a number of natural products isolated from Chinese herbs have been found to inhibit proliferation, induce apoptosis, suppress angiogenesis, retard metastasis and enhance chemotherapy, exhibiting anti-cancer potential both in vitro and in vivo. This article summarizes recent advances in in vitro and in vivo research on the anti-cancer effects and related mechanisms of some promising natural products. These natural products are also reviewed for their therapeutic potentials, including flavonoids (gambogic acid, curcumin, wogonin and silibinin), alkaloids (berberine), terpenes (artemisinin, β-elemene, oridonin, triptolide, and ursolic acid), quinones (shikonin and emodin) and saponins (ginsenoside Rg3), which are isolated from Chinese medicinal herbs. In particular, the discovery of the new use of artemisinin derivatives as excellent anti-cancer drugs is also reviewed.
Similar content being viewed by others


Background
Surgery, chemotherapy and radiotherapy are the main conventional cancer treatment often supplemented by other complementary and alternative therapies in China [1]. While chemotherapy is one of the most extensively studied methods in anti-cancer therapies, its efficacy and safety remain a primary concern as toxicity and other side effects of chemotherapy are severe. Moreover, multi-drug resistant cancer is even a bigger challenge. Medicinal herbs are main sources of new drugs. Newman et al. reported that more than half of the new chemicals approved between 1982 and 2002 were derived directly or indirectly from natural products [2]. Some active compounds have been isolated from Chinese medicinal herbs and tested for anti-cancer effects. For example, β-elemene, a compound isolated from Curcuma wenyujin Y. H. Chen et C. Ling (Wenyujin), is used as an anti-cancer drug in China. For this study, we searched three databases, namely PubMed, Scopus and Web of Science, using keywords "cancer", "tumor", "neoplastic" and "Chinese herbs" or "Chinese medicine". Publications including research and review papers covered in this review were dated between 1987 and 2011, the majority of which were published between 2007 and 2011. Chinese herb-derived ingredients, including flavonoids, alkaloids, terpenes, quinones and saponins, were found.
Gambogic acid (GA)
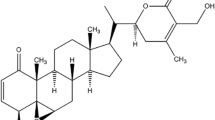
GA (Figure 1A) is the principal active ingredient of gamboges which is the resin from various Garcinia species including Garcinia hanburyi Hook.f. (Tenghuang) [3]. GA has various biological effects, such as anti-inflammatory, analgesic and anti-pyretic [3] as well as anti-cancer activities [4, 5]. In vitro and in vivo studies have demonstrated its potential as an excellent cytotoxicity against a variety of malignant tumors, including glioblastoma, as well as cancers of the breast, lung and liver. GA is currently investigated in clinical trials in China [6–8].

Chemical structures of the compounds. (A) gambogic acid; (B) curcumin; (C) wogonin; (D) silibinin; (E) berberine; (F) artemisinin; (G) artesunate; (H) β-elemene; (I) oridonin; (J) triptolide; (K) ursolic acid; (L) shikonin; (M) emodin; (N) ginsenoside Rg3.
GA induces apoptosis in various cancer cell types and the action mechanisms of GA remain unclear. Transferrin receptor (TfR) significantly over-expressed in a variety of cancers cells may be the primary target of GA [4]. The binding of GA to TfR in a manner independent of the transferrin binding site, leading to the rapid apoptosis of tumor cells [4]. Proteomics analysis suggests that stathmin may be another molecular target of GA [9]. The importance of the role of p53 in GA-induced apoptosis remains controversial [5, 10]. Furthermore, GA antagonizes the anti-apoptotic B-cell lymphoma 2 (Bcl-2) family of proteins and inhibits all six human Bcl-2 proteins to various extents, most potently inhibiting myeloid cell leukemia sequence 1 (Mcl-1) and Bcl-B, as evidenced by a half maximal inhibitory concentration (IC50) lower than 1 μM [11]. Moreover, GA also influences other anti-cancer targets, such as nuclear factor-kappa B (NF-κB) [12] and topoisomerase IIα [13].
GA causes a dose-dependent suppression of cell invasion and inhibits lung metastases of MDA-MB-435 cells in vivo through protein kinase C (PKC)-mediated matrix metalloproteinase-2 (MMP-2) and matrix metallopeptidase-9 (MMP-9) inhibition [8]. GA also exhibits significant anti-metastatic activities on B16-F10 melanoma cancer cells partially through the inhibition of the cell surface expression of integrin α4 in C57BL/6 mice [14].
Notably, the combination of GA with other compounds enhances their anti-cancer activities [15–17]. For example, He et al.[15] reports that proliferative inhibition and apoptosis induction are much more visibly increased when Tca8113 cells are treated with combined GA and celastrol, indicating that the combination of GA and celastrol can be a promising modality for treating oral squamous cell carcinoma. Another study showed that GA in combined use with 5-fluorouracil (5-FU) induced considerably higher apoptosis rates in BGC-823 human gastric cells and inhibited tumor growth in human xenografts [16]. Furthermore, low concentrations of GA were found to cause a dramatic increase in docetaxel-induced cytotoxicity in docetaxel-resistant BGC-823/Doc cells [17]. Magnetic nanoparticles of Fe3O4 (MNPs-Fe3O4) were reported to enhance GA-induced cytotoxicity and apoptosis in K562 human leukemia cells [18].
Curcumin
Curcumin (Figure 1B) is the main active flavonoid derived from the rhizome of Curcuma longa (Jianghuang), with its dry herb weight consisting of up to 3.08% curcumin [19]. Curcumin has been used to treat cardiovascular disease, inflammation and arthritis [20]. Epidemiological studies have found that incidence of several cancers is low in India where curcumin is widely consumed, suggesting that curcumin intake plays a role in cancer prevention [21]. Other studies have also indicated that curcumin inhibits cell proliferation and survival in breast cancer, colon cancer, prostate cancer, gastric cancer, leukemia, lymphoma and melanoma [20]. Curcumin induces cell apoptosis through complex intrinsic and extrinsic pathways. Curcumin binds to more than 30 different protein targets, including transcript factors (NF-κB and activator protein-1), growth factor receptors [epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2)], kinases [mitogen-activated protein kinase (MAPK), PKC and protein kinase A (PKA)], inflammatory cytokines [tumor necrosis factor (TNF) and interleukins], cell cycle-related proteins (p53 and p21), matrix metalloproteinases (MMPs) and urokinase plasminogen activators (u-PA) [20, 22, 23]. Daily oral administration of curcumin suppresses metastasis in breast, colon, lung and medulloblastoma cancers. The suppression involves the regulation of metastatic proteins, such as vascular endothelial growth factor (VEGF), MMP-2, MMP-9 and intercellular adhesion molecules [24, 25].
Curcumin induces non-apoptotic cell death, such as autophagic cell death, which involves the degradation of the cell's own components through lysosomal machinery [23]. In vitro and in vivo studies have demonstrated that curcumin induces autophagic cell death, as evidenced by the immunoreactivity of microtubule-associated protein light chain 3 (LC3) in myeloid leukemia cells. The action mechanism is attributed to the inhibition of the Akt/mammalian target of rapamycin/p70 ribosomal protein S6 kinase pathway and activation of extracellular signal-regulated kinase 1/2 by curcumin in malignant glioma cells [26, 27]. In addition, autophagic inhibitor bafilomycin A1 suppresses curcumin-induced cell death [28]. Another type of non-apoptotic cell death induced by curcumin is paraptosis which is observed in malignant breast cancer cells but not in normal breast cells. Curcumin induces paraptotic events (eg the promotion of vacuolation accompanied with mitochondrial and/or endoplasmic reticular swelling and fusion) and decreases the level of paraptotic inhibitor protein AIP-1/Alix [29]. These paraptotic events are attributed to superoxide anion and proteasomal dysfunction [29].
Curcumin reduces toxicity induced by anti-cancer agents [30], sensitizes chemo-resistant cancer cells and demonstrates synergic effects with different chemotherapeutic agents such as doxorubicin, 5-FU, paclitaxel, vincristine, melphalan, butyrate, cisplatin, celecoxib, vinorelbine, gemcitabine, oxaliplatin, etoposide, sulfinosine, thalidomide, suberoylanilide hydroxamic acid, dasatinib and bortezomib [30]. Prior administration of curcumin reduces the DNA damage and oxidative stress induced by cyclophosphamide (CXC) [31], improves uroprotective efficacy in the CXC hemorrhagic cystitis model [32] and suppresses early lung damage in CXC-treated rats [33]. Curcumin alleviates the side effects of mitomycin C, as evidenced by decreased lipid peroxidation and DNA damage [34]. Furthermore, curcumin reduces weight loss and improves kidney function and bone marrow suppression in animal studies [35]. When combined with oxaliplatin, curcumin decreases the proliferative capacity of oxaliplatin-resistant cell lines and enhances the cytotoxicity of oxaliplatin in an in vitro oxaliplatin-resistant model [36]. Additionally, curcumin protects healthy cells against radiation and sensitizes tumor cells to radiation therapy [37, 38].
Clinical trials have been or are currently being conducted to evaluate the tolerance, safety, pharmacokinetics and efficiency of curcumin as well as its combination therapy with current anti-cancer drugs [39]. A phase I clinical trial found no dose-limiting toxicity in patients treated with an oral-dose of up to 8g/day of curcumin. The recommendation is seven consecutive doses (6g/day) of curcumin every three weeks in combination with a standard dose of docetaxel [40]. Improvements in biological and clinical responses were observed in most treated patients [40]. A phase II trial of gemcitabine-resistant pancreatic cancer found chemotherapeutic drugs in combined use with curcumin to be sufficiently safe, feasible and efficient. While the bioavailability of curcumin is relatively poor, two out of 21 patients in the phase II trial showed clinical biological responses; one patient exhibited marked tumor regression coupled with a significant increase in serum cytokine levels [41, 42].
Wogonin
Wogonin (Figure 1C) is one of the flavonoids isolated from Scutellaria baicalensis Georgi (Huangqin), with its dry herb weight consisting of up to 0.39 mg/100 mg of wogonin [43]. Wogonin has been widely used in the treatment of various inflammatory diseases owing to its inhibition of nitric oxide (NO), prostaglandin E2 and pro-inflammatory cytokines production, as well as its reduction of cyclooxygenase-2 (COX-2). In vitro studies [44–48] have shown wogonin to possess cytostatic and cytotoxic activities against several human tumor cell lines.
Wogonin induces apoptosis through the mediation of Ca2+ and/or inhibition of NF-κB, shifting O2- to H2O2 to some extent; H2O2, in turn, serves as a signaling molecule that activates phospholipase Cγ. Ca2+ efflux from the endoplasmic reticulum is then regulated, leading to the activation of Bcl-2-associated agonist of cell death [44]. Wogonin may also directly activate the mitochondrial Ca2+ channel uniporter and enhance Ca2+ uptake, resulting in Ca2+ overload and mitochondrial damage [44]. Furthermore, wogonin induces cell type-dependent cell cycle inhibitions in cancer cells, such as those observed in human cervical carcinoma HeLa cells at the G1 phase [48] and in THP-1 cells at the G2/M phase [46] respectively. Unlike the inhibitory effect of baicalein and baicalin on normal human fetal lung diploid TIG-1 cells [46], wogonin imposes minor or almost no toxicity on normal peripheral T cells [44], TIG-1 cells [46] and human prostate epithelial cells [47]. This selective inhibition of wogonin is due to a high expression of L-type voltage dependent Ca2+ channels in cancer cells [44]. In addition, wogonin suppresses VEGF-stimulated migration and tube formation in HUVEC by inhibiting VEGF receptor 2 (VEGFR2) instead of VEGFR1 phosphorylation [49].
The synergistic effect of wogonin on chemotherapy drugs, such as etoposide, has also been investigated. Wogonin significantly improves etoposide-induced apoptosis in cancer cells in a similar capacity as the typical P-glycoprotein (P-gp) inhibitors verapamil and cyclosporine A [50–52]. However, other P-gp substrates, such as doxorubicin and vinblastine, do not show any synergistic effect [52]. Similar effect was also found when combination treatment with 5-FU in human gastric MGC-803 cells and in MGC-803 transplanted nude mice [53]. The underlying mechanisms might be due to its pro-apoptotic effect and inhibition of NF-κB nuclear translocation activity [53].
Anti-inflammatory and anti-viral activities of wogonin may also contribute to tumor prevention [54]. Wogonin is a good anti-cancer candidate due to its broad toxicities to various types of tumor cell lines and the low toxicities to normal tissues, as well as the synergistic effects.
Silibinin
Silibinin (Figure 1D), a mixture of flavonoids derived from Silybum marianum (Shuifeiji), is therapeutically used for the treatment of hepatic diseases in China, Germany and Japan. Silibinin has effects on many cancers, such as prostate, colon, bladder and lung cancers [55, 56], particularly the migration, invasion and metastasis of cancer cells [57]. In a transgenic adenocarcinoma of the mouse prostate (TRAMP) mouse model, silibinin inhibits tumor growth, progression, local invasion and distant metastasis [56]. Silibinin induces both death receptor-mediated and mitochondrial-mediated apoptosis in human breast cancer MCF-7 cells [58]. Silibinin also reduces hepatocellular carcinoma xenograft growth through the inhibition of cell proliferation, cell cycle progression, as well as phosphatase and tensin homolog/P-Akt and extracellular signal-regulated kinase (ERK) signaling. These effects induce apoptosis and increase histone acetylation and superoxide dismutase-1 (SOD-1) expression on human hepatocellular carcinoma xenografts [59]. Not only does silibinin inhibit primary prostatic tumor progress but also protects against angiogenesis and late-stage metastasis. Therefore, silibinin may have a potential for improving survival and reducing morbidity in prostate cancer patients [60].
Silibinin exerts anti-cancer activity mainly by blocking cell cycle progression and induces G1 cell cycle arrest in a dose- and time-dependent manner in large cell carcinoma H1299 and H460 cells and bronchioalveolar carcinoma H322 cells [61]. Silibinin modulates the protein levels of cyclin-dependent kinases (CDKs; 4, 6 and 2), cyclins (D1, D3 and E), and CDK inhibitors (p18/INK4C, p21/Waf1 and p27/Kip1) in a differential manner in the above-mentioned cell lines [61]. Silibinin also regulates multiple cellular proliferative pathways in cancer cells, including receptor tyrosine kinases (RTKs), androgen receptors, signal transducers and activators of transcription (STATs), NF-κB [62]. Moreover, silibinin inhibits the constitutive activation of STAT3 and causes caspase activation and apoptotic cell death in human prostate carcinoma DU145 cells [63].
The combined use of silibinin with 1,25-dihydroxyvitamin D3 promotes the expression of both differentiation-promoting and -inhibiting genes in acute myelogenous leukemia cells and the latter can be neutralized by a highly specific pharmacological inhibitor, suggesting the therapeutic potential of silibinin [64].
Berberine
Berberine (Figure 1E) is an isoquinoline alkaloid isolated from Coptidis Rhizoma (Huanglian), which is a Chinese medicinal herb for heat dissipation and detoxification, with its dry herb weight consisting of up to 7.1 mg/100 mg of berberine [65]. Berberine has diverse pharmacological activities [66–70] and is especially used as an antibacterial and anti-inflammatory gastrointestinal remedy in China [71]. Berberine has anti-proliferative effects on cancer cells has been documented [72–78]. Multiple targets of berberine have been identified, including mitochondria, DNA or RNA, DNA topoisomerases, estrogen receptors, MMPs, p53 and NF-κB [74, 79–82]. Berberine exerts cytotoxicity and inhibits telomerase and topoisomerase in cancer cells by specifically binding to oligonucleotides or polymorphic nucleic acid and by stabilizing DNA triplexes or G-quadruplexes [81, 83, 84]; the electrostatic interactions may be quantified in terms of the Hill model of cooperative interactions [85].
Cell cycle regulation is a common target mechanism in anti-cancer therapies. A low-dose (12.5-50 μM) berberine treatment induces G1 phase arrest whereas doses higher than 50 μM induce G2 phase arrest in mouse melanoma K1735-M2 and human melanoma WM793 cells [86]. Moreover, 50 μM berberine decreases cyclin B1 levels and induces cycle arrest at the G1 phase in human lung cancer H1299 and A549 cell lines [75]. Even in anoikis-resistant human breast cancer MDA-MB-231 and MCF-7 cells, 10 or 20 μM doses of berberine is superior to 5 or 10 nM of doxorubicine respectively by inducing cell cycle arrest at the G0/G1 phase [87].
In human breast cancer MCF-7 cells, berberine induces apoptosis through a mitochondrial dependent pathway by increasing the Bcl-2-associated × protein (Bax)/Bcl-2 protein ratio, activating caspases and inducing poly (ADP-ribose) polymerase (PARP) cleavage [76]. These apoptotic processes also occur in human tongue squamous carcinoma cancer-4 and human glioblastoma T98G cells [73, 88]. Accumulation of berberine on mitochondrial membranes alters the binding between adenine nucleotide translocator and bongkrekic acid, thereby inducing depolarization and fragmentation which may contribute to mitochondrial respiration inhibition and mitochondrial dysfunction [89]. In the p53-expressing human neuroblastoma SK-N-SH and p53-deficient SK-N-MC cells, the role of p53 in berberine's anti-neoplastic function is highlighted by the cytotoxic effects and apoptotic gene expression accompanied by caspase-3 activation [72].
In addition to apoptotic alteration induced by berberine, recent findings are about anti-cancer mechanisms that have a higher propensity to cause autophagy. Berberine induces autophagic cell death in human hepatocellular liver carcinoma cell lines (HepG2) and MHCC97-L cells, which may be diminished by cell death inhibitor 3-methyladenine through beclin-1 activation and mammalian target of rapamycin (mTOR) signaling pathway inhibition [90]. In addition, berberine also modifies LC3, an autophagic marker, in human lung cancer A549 cells, indicating that autophagy may play a crucial role in berberine-induced cancer cell death [91].
Berberine also inhibits tumor metastasis and invasion. For example, berberine inhibits 12-O-Tetradecanoylphorbol 13-acetate (TPA)-induced cell migration and blocks prostaglandin E (EP) receptor 4 agonist-induced migration by reducing EP receptors 2 and 4 in A375 and Hs294 cells [92]. Even at low doses, berberine suppresses Rho GTPase activation and induces migration and motility inhibition in HONE1 cells [93]. Berberine also inhibits Rho kinase-mediated Ezrin phosphorylation at Thr (567) in 5-8F cells, leading to a 51.1% inhibition of tumor metastasis to the lymph nodes in vivo[94]. A combination of As2O3 (5 μM) and berberine (10 μM) inhibit the formation of a cell confluent layer by blocking PKCα and ξ, consistent with reduced levels of myelocytomatosis oncogene (Myc), Jun proto-oncogene, metallothionein 1-MMP and MMP-2 [95].
Berberine enhances chemo- and radio-sensitivity, implying its potential as an adjuvant in cancer therapy. Combined with chemotherapy drugs such as cisplatin or As2O3, berberine exhibits significant cytotoxicity in HeLa and SH-SY5Y cells compared with monotherapy [96, 97]. When combined with γ radiation, the apoptotic effect is significantly enhanced in HepG2 cells [98]. Berberine also alleviates chemo-resistance by down-regulating overexpressed transformed mouse 3T3 cell double minute-2 and activating p53 in acute lymphoblastic leukemia cells [99]. Berberine's poor bioavailability makes it less likely to be an independent anti-tumor agent [100–102]. Berberine is nevertheless a potential natural compound for alternative cancer therapy.
Artemisinin and its derivatives (ARTs)
Artemisinin (Figure 1F) is an active terpene of the Chinese medicinal herb Artemisia annua L. (Huanghuahao) used in China to treat malaria and fever. ARTs, such as dihydroartemisinin (DHA) and artesunate (Figure 1G), exhibit anti-cancer activities in vitro and in vivo[103–106]. DHA is one of the main metabolites of ARTs and artesunate is a semi-synthesized derivative of ARTs; both compounds exhibit anti-cancer potentials.
The anti-cancer potential of ARTs has been demonstrated in various cancer cells including those of leukemia and other cancer cells of breast, ovary, liver, lung, pancreas and colon [104, 105]. The selective anti-cancer potential of ARTs was related with the expression of different molecules such as c-MYC, cdc25A, EGFR, γ-glutamycysteine synthetase (GLCLR) [105, 106]. ARTs also exert anti-cancer effects in vivo in multiple cancer types [103, 107, 108]. For example, either DHA or artesunate has anti-cancer activity against pancreatic cancer xenografts [107, 109].
The anti-cancer mechanism of ARTs is likely to be related to the cleavage of the iron- or heme-mediated peroxide bridge, followed by the generation of reactive oxygen species (ROS) [110–112]. According to Efferth et al.[113], CCRF-CEM and U373 cells are sensitive to a combined treatment of ARTs and iron (II)-glycine sulfate or holotransferring. Pretreatment with deferoxamine mesylate salt (an iron chelator) visibly reduces DHA-induced apoptosis in HL-60 leukemia cells [104]. The anti-cancer potential of ARTs is possibly connected to the expression of TfR. The synergism of artesunate and iron (II)-glycine sulfate co-treatment is unsuitable for all types of tumor cells [114]. Endoplasmic reticulum stress is partially involved in some cases of ARTs-mediated anti-proliferation [115, 116].
ARTs induce cell cycle arrest in various cell types [103, 115, 117]. For example, DHA and artesunate effectively mediate G1 phase arrest in HepG2 and Hep3B cells [103]. DHA reduces cell number in the S phase in HCT116 colon cancer cells [115]. Interestingly, DHA also arrests the G2 phase in OVCA-420 ovarian cancer cells [117]. Thus, ART-mediated cell cycle arrest is possibly cell type dependent. ARTs also induce apoptotic cell death in a number of cell types, in which the mitochondrial-mediated apoptotic pathway plays a decisive role [104, 106]. For instance, DHA enhances Bax and reduces Bcl-2 expression in cancer cells [103, 107]. DHA-induced apoptosis is abrogated by the loss of Bak and is largely reduced in cells with siRNA-mediated downregulation of Bak or NOXA [118]. However, DHA activates caspase-8, which is related to the death receptor-mediated apoptotic pathway in HL-60 cells [104]. DHA enhances Fas expression and activates caspase-8 in ovarian cancer cells [119]. DHA also enhances death receptor 5 and activates both mitochondrial- and death receptor-mediated apoptotic pathways in prostate cancer cells [120]. ARTs-induced apoptosis in cancer cells may involve p38 MAPK rather than p53 [103, 104].
ARTs inhibit angiogenesis which is a vital process in metastasis [121–124]. DHA inhibits chorioallantoic membrane angiogenesis at low concentrations and decreases the levels of two major VEGF receptors on HUVEC [122]. Conditioned media from K562 cells pre-treated with DHA inhibits VEGF expression and secretion in chronic myeloid leukemia K562 cells, leading to angiogenetic activity decrease [121, 124]. Artemisinin inhibits cell migration and concomitantly decreases the expression of MMP2 and the αvβ3 integrins in human melanoma cells [125]. ARTs also regulate the levels of u-PA, MMP2, MMP7 and MMP9 all of which are related to metastasis [126].
ARTs exert synergistic effects with other compounds. Combination of DHA and caboplatin significantly reduces the development of ovarian cancer as compared with DHA only [119]. Combined use of DHA or artesunate with gencitabine inhibits the growth of HepG2 and Hep3B transplanted tumors [103]. Supra-additive inhibition of cell growth in some glioblastoma multiforme cells is observable when artesunate is in combined use with EGFR inhibitor OSI-774 [127]. DHA not only up-regulates death receptor 5 expression but also cooperates with TNF-related apoptosis-inducing ligand (TRAIL) to induce apoptosis in human prostate cancer cells [120]. Therefore, either used alone or in combination with other compounds, ARTs are promising compounds for chemotherapy.
β-elemene
Elemene (Figure 1H) is a sesquiterpene mixture isolated from more than 50 Chinese herbs and plants, such as Curcuma wenyujin Y. H. Chen et C. Ling (Wenyujin) [128]. Elemene is mainly composed of β- and δ- and γ-elemene, with β-elemene accounting for 60%-72% of all three isoforms. β-elemene exerts anti-cancer potential in brain, laryngeal, lung, breast, prostate, cervical, colon and ovarian carcinomas [128–130]. Elemene shows synergistic effects in combination with other chemotherapeutic drugs [131], leading to the blockade of cell cycle progression by modulating the G2 cell cycle checkpoint and inducing G2/M arrest in human non-small cell lung cancer (NSCLC) and ovarian carcinoma cells while inducing G0/G1 phase arrest in glioblastoma cell lines through phosphorylation of p38 MAPK [129, 130, 132]. In NSCLC cells, β-elemene induces cell arrest at the G2/M phase by increasing phospho-Cdc2 (Tyr15) and p27/Kip1, and by decreasing phospho-Cdc2 (Thr161) and cyclin B1. Moreover, elemene reduces the expression of Cdc25C, activates Cdc2 and increases Chk2 [129]. β-elemene combined with cisplatin also mediate G2/M cell cycle arrest in chemo-resistant ovarian carcinoma cells through down-regulation of cyclin B1 and Cdc2 by elevating the levels of phosphorylation of Cdc2, Cdc25C, p53, p21/Waf1, p27/Kip1 and GADD45 [130]. β-elemene also induces mitochondrial-mediated apoptosis in prostate cancer and NSCLC cells [128, 129]. Combining β-elemene with cisplatin, docetaxel and taxanes significantly increases its inhibitory effect in androgen-independent prostate carcinoma DU145 and PC-3 cells, as well as in NSCLC H460 and A549 cells [131]. β-elemene enhances cellular uptake of taxanes due to the alteration of cell membrane permeability may partly account for its synergistic effects with taxanes [131]. Elemene inhibits the growth of human epidermoid and thyroid cancer cells in vivo[133], and passes through the blood-brain barrier [134], suggesting its potential for treating cerebral malignancy.
β-elemene has been approved by China's State Food and Drug Administration as a second class innovative drug and is prescribed as an adjuvant drug for some tumor therapies in China.
Oridonin
Oridonin (Figure 1I) is a diterpenoid isolated from Rabdosia rubescens (Hemsl.) Hara (Donglingcao), with its dry raw herb consisting of up to 0.35% of oridonin [135]. Rabdosia rubescens (Hemsl.) Hara has long been used to treat sore throat, tonsillitis, and esophageal cancer by native residents of Henan Province. Oridonin was included in the Chinese Pharmacopoeia in 1977. Main chemical constituents of Rabdosia rubescens (Hemsl.) Hara are ent-Kaurene diterpenoids, which have multiple biological activities, such as anti-inflammatory, anti-bacterial and anti-tumor effects.
Oridonin significantly inhibits tumor cell proliferation, induces cell cycle arrest and promotes cell death. In anti-proliferation tests, different cell lines exhibited similar sensitivity to oridonin with an IC50 of about 40-80 μM after 24 hours of treatment [136–141]. Oridonin induces G2/M cell cycle arrest by up-regulation of heat shock 70 kDa protein 1, serine-threonine kinase receptor-associated protein, translationally controlled tumor protein, stress-induced phosphoprotein 1, trifunctional purine biosynthetic protein adenosine-3 and inorganic pyrophosphatase as well as down-regulation of poly(rC)-binding protein 1 [142] in a p53-independent and p21/Waf1-dependent manner [143]. Induction of apoptosis contributes to oridonin-induced cell death, mainly through mitochondrial-mediated pathways. The up-regulation of Fas, Fas ligand (FasL) and Fas (TNFRSF6)-associated via death domain (FADD) expression, as well as the down-regulation of pro-caspase-8 expression suggests that the activation of the Fas/FasL pathway may also be partially involved in oridonin-induced apoptosis [144]. Possible downstream responses include the induction of loss of mitochondrial transmembrane potential [145], the activation of several caspases [136, 146], the down-regulation of Bcl-2, the up-regulation of Bax and Bid [136, 147] as well as the promotion of cytochrome c release [147] and PARP cleavage [148]. However, the regulation of Bcl-xL and participation of caspase-3/9 remain controversial [136, 143, 146, 148–150]. Oridonin-induced intracellular ROS formation may be an initiator of this process [143, 151]. Other proteins may also be involved in oridonin-induced cell cycle arrest and apoptosis; these proteins include ERK [144, 152], p38MAPK [149], insulin-like growth factor 1 receptor [153], EGFR [154], NF-κB [155], as well as p16, p21/Waf1, p27/Kip1 and c-MYC [156]. Oridonin induce cell death by affecting the balance of apoptosis and necrosis. In A375-S2 cells, low concentrations (34.3 μM) of oridonin induce p53 and ERK-dependent apoptosis whereas high concentrations (137.4 μM) induce necrosis [146]. In L929 cells, oridonin induces a caspase-independent and mitochondria- or MAPK-dependent cell death through both apoptosis and necrosis [139, 149]. Similar results are also observed in A431 cells [154]. Oridonin also induces simultaneous autophagy and apoptosis in MCF-7 [157] and HeLa cells [138]. This autophagy may be attributed to the inactivation of Ras, changes in mitochondrial membrane potential [158], activation of PKC, Raf-1 or c-jun N-terminal kinase (JNK) signaling [141] and even NF-κB signaling pathways [159]. Inhibition of autophagy is attributed to apoptotic up-regulation because oridonin-induced apoptosis augmentation is accompanied by reduced autophagy [138] whereas oridonin-induced autophagy inhibits ROS-mediated apoptosis by activating the p38 MAPK-NF-κB survival pathways in L929 cells [160]. Oridonin inhibits DNA, RNA, and protein syntheses [161], decrease telomerase, as well as down-regulate human telomerase reverse transcriptase mRNA expression [162]. The in vivo anti-tumor activities of oridonin have been demonstrated in different tumors such as Ehrlich ascites carcinoma, sarcoma-180 solid tumors and in leukemic mice models [163, 164].
Triptolide
Triptolide (Figure 1J) is a diterpenoid triepoxide and the principal active ingredient of Tripterygium wilfordii Hook. f. (Leigongteng) used in Chinese medicine to treat inflammation and autoimmune diseases [165]. Triptolide exhibits potent anti-inflammation, immunomodulation and anti-tumor activities [166–170]. Triptolide exerts multiple effects on apoptosis, angiogenesis, metastasis and drug-resistance [166–170].
Triptolide is active in pro-apoptosis in diverse tumor cell types including ovarian cancer [166], myeloma [167], myeloid leukemia [168], thyroid carcinoma [169] and pancreatic tumor cells [170]. Many in vitro and in vivo studies have tried to elucidate the potential mechanism of triptolide; however, conclusions have been inconsistent. Triptolide seems to induce apoptosis via different pathways in various cell lines. For example, triptolide induces apoptosis by the overexpression of cytomembrane death receptor in a caspase-8-dependent manner in pancreatic tumor [170] and cholangiocarcinoma cells [171]. Triptolide also promotes apoptosis in leukemic and hepatocarcinoma cells by the mitochondrial-mediated pathway [172, 173].
Triptolide is a potent inhibitor of tumor angiogenesis in a zebrafish embryo model and demonstrates potent activities against vessel formation by nearly 50% at 1.2 μM [165]. In a xenograft model, triptolide (0.75 mg/kg/day) blocks tumor angiogenesis and progression in a murine tumorigenesis assay possibly correlated with the down-regulation of proangiogenic Tie2 and VEGFR-2 expression [174]. In vitro studies have shown that triptolide inhibits the proliferation of HUVEC. A chick embryo chorioallantoic membrane test shows that triptolide inhibits angiogenesis as well. Triptolide impairs VEGF expression in thyroid carcinoma TA-K cells and down-regulates NF-κB pathway activity; the target genes of triptolide are associated with endothelial cell mobilization in HUVEC [165]. The down-regulation of NF-κB signaling [175], in combination with the inhibition of VEGF expression [176], may be the anti-angiogenesis action of triptolide.
Furthermore, triptolide inhibits tumor metastasis, reducing basal and stimulated colon cancer cell migration through collagen by 65% to 80% and decreasing the expression of VEGF and COX-2 [174]. Triptolide inhibits the expression of multiple cytokine receptors potentially involved in cell migration and cancer metastasis, including the thrombin receptor, CXCR4, TNF receptors and TGF-β receptors [174]. Triptolide also inhibits interferon-γ-induced programmed death-1-ligand 1 surface expression whose up-regulation is an important mechanism of tumor immune evasion in human breast cancer cells [177]. Triptolide inhibits the experimental metastasis of melanoma cells to the lungs and spleens of mice [178]. Moreover, triptolide inhibits the migration of lymphoma cells via lymph nodes, a result which may be related to its anti-proliferative effects and blockage of the SDF-1/CXCR4 axis [179].
Triptolide enhances the anti-neoplastic activity of chemotherapy [180, 181]. The combination index-isobologram indicates that the effect of triptolide on 5-FU is synergistic on colon carcinoma [180]. In a tumor xenograft model, the combined effects of triptolide (0.25 mg/kg/day) and 5-FU (12 mg/kg/day) on the growth of colon carcinoma are superior to those of individual agents [180]. Triptolide is synergistic with other anti-cancer agents or therapies including hydroxycamptothecin [181], idarubicin, AraC [182], TRAIL [183] and ionizing radiation [184]. These results indicate the therapeutic potential of triptolide in treating cancer.
Ursolic acid (UA)
UA (Figure 1K) is a ubiquitous pentacyclic triterpenoid compound from many plants such as Ligustrum lucidum Ait. (Nuzhen). UA exerts proliferation inhibition in human ovarian cancer CAOV3 cells and doxorubicin-resistant human hepatoma R-HepG2 cells [185, 186]. UA disrupts cell cycle progression and induces necrosis in a clonal MMTV-Wnt-1 mammary tumor cell line [187]. Eight novel UA derivatives with substitutions at positions C-3, C-11, and C-28 of UA show cytotoxicity to some degree in HeLa, SKOV3 and BGC-823 in vitro; only one derivative exhibits more potent cytotoxicity than UA [188].
UA induces apoptosis via both extrinsic and intrinsic signaling pathways in cancer cells [189]. In PC-3 cells, UA inhibits proliferation by activating caspase-9 and JNK as well as FasL activation and Akt inhibition [190]. A significant proliferation inhibition and invasion suppression in both a dose- and time-dependent manner is observed in highly metastatic breast cancer MDA-MB-231 cells; this inhibition is related to the down-regulation of MMP2 and u-PA expression [191]. Moreover, UA reduces IL-1β- or TNF-α-induced rat C6 glioma cell invasion and inhibits the interaction of ZIP/p62 and PKC-ζ [192]. Nontoxic UA concentrations inhibit vessel growth in rat aortic ring and down-regulate matrix MMPs such as MMP2 and MMP9 [193]. In other cancer cell lines, such as Hep3B, Huh7 and HA22T cells, UA exerts a potential anti-angiogenic effect by decreasing HIF-1α, VEGF and IL-8 gene expression [194].
Shikonin
Shikonin (Figure 1L) is a natural anthraquinone derivative isolated from the roots of Lithospermum erythrorhizon (Zicao) and exerts anti-tumor effects mainly by inhibiting cell growth and inducing apoptosis. The underlying molecular mechanisms vary with cell types and treatment methods. Shikonin induces apoptosis in a classic caspase-dependent pathway in cervical, bladder and melanoma cancer cells [195–198]. Shikonin induces necroptosis regardless of the drug concentration in caspase-3-negative MCF-7 cells [199]. Different concentrations of shikonin induce either apoptosis or necroptosis, and necroptosis converts to apoptosis in the presence of Nec-1 in HL-60 and K562 cells [200]. The growth inhibition and apoptosis induced by shikonin in some cancer cells may be attributed to the inactivation of NF-κB activity or increasing Annexin V signal and CD95 (Fas/APO) expression [201, 202]. Shikonin also induces apoptosis via ROS production in osteosarcoma and Bcr/Abl-positive CML cells [203, 204].
Several different mechanisms contribute to the anti-cancer activities of shikonin. For example, shikonin suppresses proteasomal activities thereby inhibiting tumor growth in both H22 allografts and PC-3 xenografts [205]. Shikonin also inhibits topoisomerase II [206] and down-regulates ER2 and activates NFE2-related factor 2 as an anti-estrogen agent in human breast cancer [207, 208]. Shikonin modulates an estrogen enzyme by down-regulating the expression of steroid sulfatase which is important for estrogen biosynthesis [205]. Shikonin inhibits tumor invasion via the NF-κB signaling pathway in human high-metastatic adenoid cystic carcinoma cells [209]. Therefore, shikonin may directly or indirectly inhibit or modulate disease-related cellular targets in cancer.
Emodin
Emodin (Figure 1M) is a natural anthraquinone derivative isolated from Rheum palmatum L. (Zhangyedahuang), with its dry raw herb consisting of up to 0.20 mg/100 mg of emodin [210]. Emodin exerts anti-tumor activity against various human cancers [211]. Emodin induces cell cycle arrest and apoptosis in cancer cells [212–214] and the oxidative injury acts upstream of anti-proliferation. Emodin inhibits IL-6-induced Janus-activated kinase 2/STAT3 pathways and induces apoptosis in myeloma cells via the down-regulation of Mcl-1 [213]. Emodin down-regulates androgen receptors and inhibits prostate cancer cell growth [215]. Moreover, emodin stabilizes topoisomerase II-DNA cleavage complexes, thereby inducing DNA double-strand breaks [216]. The suppression of excision repair cross complementation 1 (ERCC1) and Rad51 expression through ERK1/2 inactivation is vital in emodin-induced cytotoxicity in human NSCLC cells [217].
Emodin inhibits basic fibroblast growth factor (bFGF)-induced proliferation and migration in HUVEC and VEGF-A-induced tube formation [218]. Emodin inhibits tumor cell migration through suppression of the phosphatidylinositol 3-kinase-Cdc42/Rac1 pathway [219]. The disruption of the membrane lipid raft-associated integrin signaling pathway by emodin may inhibit cell adhesion and spreading [220].
Emodin sensitizes chemotherapy associated with ROS production [221, 222]. In combined use with cisplatin, emodin elevates ROS generation and enhances chemosensitivity in DU-145 cells, accompanied by the down-regulation of MDR1 expression and suppression of HIF-1α transactivation [223]. Emodin enhances the sensitivity of gallbladder cancer SGC996 cells to platinum drugs via glutathione depletion and multidrug resistance-related protein 1 down-regulation [224]. The mechanisms of the synergistic effects of emodin with cisplatin or gencitabin may be attributed to the emodin-induced down-regulation of ERCC1 and Rad51 expression, respectively [225, 226]. These results suggest that emodin may be used as an adjuvant to enhance the anti-cancer effects of chemotherapeutic agents.
Ginsenoside Rg3
Extracted from Panax ginseng C.A. Mey. (Renshen) and Panax quinquefolius L., Araliaceae (Xiyangshen), ginsenoside Rg3 (Figure 1N) is a biologically active component with both in vitro and in vivo anti-cancer activities [227, 228]. The anti-proliferative mechanism of ginsenoside Rg3 is associated with the inactivation of NF-κB [229, 230], modulation of MAPKs [231] and the down-regulation of Wnt/β-catenin signaling [232]. Ginsenoside Rg3 affects the ephrin receptor pathway in HCT-116 human colorectal cancer cells [233]. The anti-proliferative mechanism of ginsenoside Rg3 is also associated with the molecules of mitotic inhibition, DNA replication, repair, and growth factor signaling [234].
Ginsenoside Rg3 inhibits the proliferation of HUVEC and suppresses the capillary tube formation of HUVEC on a matrigel at nanomole scales in the presence or absence of VEGF. Ginsenoside Rg3 attenuates VEGF-induced chemo-invasion of HUVEC and ex vivo microvascular sprouting in rat aortic ring. bFGF-induced angiogenesis may be abolished by ginsenoside Rg3[227]. In lung metastasis models of ovarian cancer, ginsenoside Rg3 decreases the number of tumor colonies in the lung and vessels oriented toward the tumor mass [235]. This effect may be partially due to the inhibition of angiogenesis and the decrease in MMP9 expression [235].
Ginsenoside Rg3 increases the efficacy of cancer chemotherapy. Combined treatments with ginsenoside Rg3 enhance the susceptibility of colon cancer cells to docetaxel, paclitaxel, cisplatin and doxorubicin; the mechanism of such an enhancement is related to the inhibition of the constitutively activated NF-κB [229]. A similar phenomenon has been observed in prostate cancer cells, in which the combination of ginsenoside Rg3 and docetaxel more effectively induces apoptosis and G1 cell cycle arrest, accompanied by the inhibition of NF-κB activity [230]. Low-dose administration of cyclophosphamide (CTX) with ginsenoside Rg3 increases the efficacy of targeting the tumor microvasculature and the two-drug combination treatment results demonstrate the longest patient survival rates [236]. Ginsenoside Rg3 combined with gemcitabine not only enhances the efficacy of tumor growth suppression and survival prolongation, but also decreases VEGF expression and microvessel density in tumors [228].
Conclusion
Natural products such as GA, curcumin, β-elemene et al. derived from Chinese medicinal herbs are potential candidates for anti-cancer therapeutic drugs.
Abbreviations
- 5-FU:
-
5-fluorouracil
- AIF:
-
apoptosis inducing factor
- AP-1:
-
activator protein-1
- ARTs:
-
artemisinin and its derivatives
- ATRA:
-
all-trans retinoic acid
- bFGF:
-
basic fibroblast growth factor
- CDKs:
-
cyclin-dependent kinases
- CTX:
-
cyclophosphamide DHA: dihydroartemisinin
- DPD:
-
dihydropyrimidine dehydrogenase
- EGFR:
-
epidermal growth factor receptor
- ERK1/2:
-
extracellular signal-regulated kinase 1/2
- FasL:
-
Fas ligand
- GA:
-
gambogic acid
- HDMEC:
-
human dermal microvascular endothelial cells
- HUVEC:
-
human umbilical vascular endothelial cells
- ICAM-1:
-
intercellular cell adhesion molecule-1
- IL-1β:
-
interleukin-1β
- LC3:
-
light chain 3
- MMP2:
-
matrix metalloproteinase-2
- MMP9:
-
matrix metalloproteinase9
- MRP1:
-
multidrug resistance-associated protein 1
- NF-κB:
-
nuclear factor-kappa B
- P-gp:
-
P-glycoprotein
- PI3K:
-
phosphoinositide 3-kinase
- ROS:
-
reactive oxygen species
- STAT:
-
signal transducer and activator of transcription
- STS:
-
steroid sulfatase
- TfR:
-
transferrin receptor
- TPA:
-
12-O-tetradeca noylphorbol-13-acetate
- TRAMP:
-
transgenic adenocarcinoma of the mouse prostate
- UA:
-
ursolic acid
- u-PA:
-
urokinase plasminogen activators
- VCAM-1:
-
vascular cell adhesion molecule-1
- VEGF:
-
vascular endothelial growth factor
- VEGFR1:
-
vascular endothelial growth factor receptor 1
- VEGFR2:
-
vascular endothelial growth factor receptor 2.
References
Bell RM: A review of complementary and alternative medicine practices among cancer survivors. Clin J Oncol Nurs. 2010, 14: 365-370. 10.1188/10.CJON.365-370.
Newman DJ, Cragg GM, Snader KM: Natural products as sources of new drugs over the period 1981-2002. J Nat Prod. 2003, 66: 1022-1037. 10.1021/np030096l.
Panthong A, Norkaew P, Kanjanapothi D, Taesotikul T, Anantachoke N, Reutrakul V: Anti-inflammatory, analgesic and antipyretic activities of the extract of gamboge from Garcinia hanburyi Hook f. J Ethnopharmacol. 2007, 111: 335-340. 10.1016/j.jep.2006.11.038.
Kasibhatla S, Jessen KA, Maliartchouk S, Wang JY, English NM, Drewe J, Qiu L, Archer SP, Ponce AE, Sirisoma N, Jiang S, Zhang HZ, Gehlsen KR, Cai SX, Green DR, Tseng B: A role for transferrin receptor in triggering apoptosis when targeted with gambogic acid. Proc Natl Acad Sci USA. 2005, 102: 12095-12100. 10.1073/pnas.0406731102.
Gu H, Wang X, Rao S, Wang J, Zhao J, Ren FL, Mu R, Yang Y, Qi Q, Liu W, Lu N, Ling H, You Q, Guo Q: Gambogic acid mediates apoptosis as a p53 inducer through down-regulation of mdm2 in wild-type p53-expressing cancer cells. Mol Cancer Ther. 2008, 7: 3298-3305. 10.1158/1535-7163.MCT-08-0212.
Wu ZQ, Guo QL, You QD, Zhao L, Gu HY: Gambogic acid inhibits proliferation of human lung carcinoma SPC-A1 cells in vivo and in vitro and represses telomerase activity and telomerase reverse transcriptase mRNA expression in the cells. Biol Pharm Bull. 2004, 27: 1769-1774. 10.1248/bpb.27.1769.
Qiang L, Yang Y, You QD, Ma YJ, Yang L, Nie FF, Gu HY, Zhao L, Lu N, Qi Q, Liu W, Wang XT, Guo QL: Inhibition of glioblastoma growth and angiogenesis by gambogic acid: an in vitro and in vivo study. Biochem Pharmacol. 2008, 75: 1083-1092. 10.1016/j.bcp.2007.10.033.
Qi Q, Gu H, Yang Y, Lu N, Zhao J, Liu W, Ling H, You QD, Wang X, Guo Q: Involvement of matrix metalloproteinase 2 and 9 in gambogic acid induced suppression of MDA-MB-435 human breast carcinoma cell lung metastasis. J Mol Med. 2008, 86: 1367-1377. 10.1007/s00109-008-0398-z.
Wang X, Chen Y, Han QB, Chan CY, Wang H, Liu Z, Cheng CH, Yew DT, Lin MC, He ML, Xu HX, Sung JJ, Kung HF: Proteomic identification of molecular targets of gambogic acid: role of stathmin in hepatocellular carcinoma. Proteomics. 2009, 9: 242-253. 10.1002/pmic.200800155.
Rong JJ, Hu R, Qi Q, Gu HY, Zhao Q, Wang J, Mu R, You QD, Guo QL: Gambogic acid down-regulates MDM2 oncogene and induces p21(Waf1/CIP1) expression independent of p53. Cancer Lett. 2009, 284: 102-112. 10.1016/j.canlet.2009.04.011.
Zhai D, Jin C, Shiau CW, Kitada S, Satterthwait AC, Reed JC: Gambogic acid is an antagonist of antiapoptotic Bcl-2 family proteins. Mol Cancer Ther. 2008, 7: 1639-1646. 10.1158/1535-7163.MCT-07-2373.
Pandey MK, Sung B, Ahn KS, Kunnumakkara AB, Chaturvedi MM, Aggarwal BB: Gambogic acid, a novel ligand for transferrin receptor, potentiates TNF-induced apoptosis through modulation of the nuclear factor-kappaB signaling pathway. Blood. 2007, 110: 3517-3525. 10.1182/blood-2007-03-079616.
Qin Y, Meng L, Hu C, Duan W, Zuo Z, Lin L, Zhang X, Ding J: Gambogic acid inhibits the catalytic activity of human topoisomerase IIalpha by binding to its ATPase domain. Mol Cancer Ther. 2007, 6: 2429-2440. 10.1158/1535-7163.MCT-07-0147.
Zhao J, Qi Q, Yang Y, Gu HY, Lu N, Liu W, Wang W, Qiang L, Zhang LB, You QD, Guo QL: Inhibition of alpha(4) integrin mediated adhesion was involved in the reduction of B16-F10 melanoma cells lung colonization in C57BL/6 mice treated with gambogic acid. Eur J Pharmacol. 2008, 589: 127-131. 10.1016/j.ejphar.2008.04.063.
He D, Xu Q, Yan M, Zhang P, Zhou X, Zhang Z, Duan W, Zhong L, Ye D, Chen W: The NF-kappa B inhibitor, celastrol, could enhance the anti-cancer effect of gambogic acid on oral squamous cell carcinoma. BMC Cancer. 2009, 9: 343-10.1186/1471-2407-9-343.
Wang J, Liu W, Zhao Q, Qi Q, Lu N, Yang Y, Nei FF, Rong JJ, You QD, Guo QL: Synergistic effect of 5-fluorouracil with gambogic acid on BGC-823 human gastric carcinoma. Toxicology. 2009, 256: 135-140. 10.1016/j.tox.2008.11.014.
Wang T, Wei J, Qian X, Ding Y, Yu L, Liu B: Gambogic acid, a potent inhibitor of survivin, reverses docetaxel resistance in gastric cancer cells. Cancer Lett. 2008, 262: 214-222. 10.1016/j.canlet.2007.12.004.
Chen B, Liang Y, Wu W, Cheng J, Xia G, Gao F, Ding J, Gao C, Shao Z, Li G, Chen W, Xu W, Sun X, Liu L, Li X, Wang X: Synergistic effect of magnetic nanoparticles of Fe(3)O(4) with gambogic acid on apoptosis of K562 leukemia cells. Int J Nanomedicine. 2009, 4: 251-259.
Minami M, Nishio K, Ajioka Y, Kyushima H, Shigeki K, Kinjo K, Yamada K, Nagai M, Satoh K, Sakurai Y: Identification of Curcuma plants and curcumin content level by DNA polymorphisms in the trnS-trnfM intergenic spacer in chloroplast DNA. J Nat Med. 2009, 63: 75-79. 10.1007/s11418-008-0283-7.
Goel A, Kunnumakkara AB, Aggarwal BB: Curcumin as "Curecumin": from kitchen to clinic. Biochem Pharmacol. 2008, 75: 787-809. 10.1016/j.bcp.2007.08.016.
Lopez-Lazaro M: Anticancer and carcinogenic properties of curcumin: considerations for its clinical development as a cancer chemopreventive and chemotherapeutic agent. Mol Nutr Food Res. 2008, 52 (Suppl 1): S103-127.
Bhandarkar SS, Arbiser JL: Curcumin as an inhibitor of angiogenesis. Adv Exp Med Biol. 2007, 595: 185-195. 10.1007/978-0-387-46401-5_7.
Ravindran J, Prasad S, Aggarwal BB: Curcumin and cancer cells: how many ways can curry kill tumor cells selectively?. AAPS J. 2009, 11: 495-510. 10.1208/s12248-009-9128-x.
Aggarwal BB, Shishodia S, Takada Y, Banerjee S, Newman RA, Bueso-Ramos CE, Price JE: Curcumin suppresses the paclitaxel-induced nuclear factor-kappaB pathway in breast cancer cells and inhibits lung metastasis of human breast cancer in nude mice. Clin Cancer Res. 2005, 11: 7490-7498. 10.1158/1078-0432.CCR-05-1192.
Binion DG, Otterson MF, Rafiee P: Curcumin inhibits VEGF-mediated angiogenesis in human intestinal microvascular endothelial cells through COX-2 and MAPK inhibition. Gut. 2008, 57: 1509-1517. 10.1136/gut.2008.152496.
Shinojima N, Yokoyama T, Kondo Y, Kondo S: Roles of the Akt/mTOR/p70S6K and ERK1/2 signaling pathways in curcumin- induced autophagy. Autophagy. 2007, 3: 635-637.
Aoki H, Takada Y, Kondo S, Sawaya R, Aggarwal BB, Kondo Y: Evidence that curcumin suppresses the growth of malignant gliomas in vitro and in vivo through induction of autophagy: role of Akt and extracellular signal-regulated kinase signaling pathways. Mol Pharmacol. 2007, 72: 29-39. 10.1124/mol.106.033167.
Jia YL, Li J, Qin ZH, Liang ZQ: Autophagic and apoptotic mechanisms of curcumin-induced death in K562 cells. J Asian Nat Prod Res. 2009, 11: 918-928. 10.1080/10286020903264077.
Yoon MJ, Kim EH, Lim JH, Kwon TK, Choi KS: Superoxide anion and proteasomal dysfunction contribute to curcumin-induced paraptosis of malignant breast cancer cells. Free Radic Biol Med. 2009, 48: 713-726.
Nautiyal J, Banerjee S, Kanwar SS, Yu Y, Patel BB, Sarkar FH, Majumdar AP: Curcumin enhances dasatinib-induced inhibition of growth and transformation of colon cancer cells. Int J Cancer. 2011, 128: 951-961. 10.1002/ijc.25410.
Ibrahim MA, Elbehairy AM, Ghoneim MA, Amer HA: Protective effect of curcumin and chlorophyllin against DNA mutation induced by cyclophosphamide or benzo[a]pyrene. Z Naturforsch C. 2007, 62: 215-222.
Arafa HM: Uroprotective effects of curcumin in cyclophosphamide -induced haemorrhagic cystitis paradigm. Basic Clin Pharmacol Toxicol. 2009, 104: 393-399. 10.1111/j.1742-7843.2009.00379.x.
Venkatesan N, Chandrakasan G: Modulation of cyclophosphamide-induced early lung injury by curcumin, an anti-inflammatory antioxidant. Mol Cell Biochem. 1995, 142: 79-87. 10.1007/BF00928916.
Siddique YH, Ara G, Beg T, Gupta J, Afzal M: Assessment of cell viability, lipid peroxidation and quantification of DNA fragmentation after the treatment of anticancerous drug mitomycin C and curcumin in cultured human blood lymphocytes. Exp Toxicol Pathol. 2010, 62: 503-508. 10.1016/j.etp.2009.06.010.
Zhou QM, Zhang H, Lu YY, Wang XF, Su SB: Curcumin reduced the side effects of mitomycin C by inhibiting GRP58-mediated DNA cross-linking in MCF-7 breast cancer xenografts. Cancer Sci. 2009, 100: 2040-2045. 10.1111/j.1349-7006.2009.01297.x.
Howells LM, Sale S, Sriramareddy SN, Irving GR, Jones DJ, Ottley CJ, Pearson DG, Mann CD, Manson MM, Berry DP, Gescher A, Steward WP, Brown K: Curcumin ameliorates oxaliplatin-induced chemoresistance in HCT116 colorectal cancer cells in vitro and in vivo. Int J Cancer. 2010,
Goel A, Aggarwal BB: Curcumin, the golden spice from Indian saffron, is a chemosensitizer and radiosensitizer for tumors and chemoprotector and radioprotector for normal organs. Nutr Cancer. 2010, 62: 919-930. 10.1080/01635581.2010.509835.
Yallapu MM, Maher DM, Sundram V, Bell MC, Jaggi M, Chauhan SC: Curcumin induces chemo/radio-sensitization in ovarian cancer cells and curcumin nanoparticles inhibit ovarian cancer cell growth. J Ovarian Res. 2010, 3: 11-10.1186/1757-2215-3-11.
Hatcher H, Planalp R, Cho J, Torti FM, Torti SV: Curcumin: from ancient medicine to current clinical trials. Cell Mol Life Sci. 2008, 65: 1631-1652. 10.1007/s00018-008-7452-4.
Bayet-Robert M, Kwiatkowski F, Leheurteur M, Gachon F, Planchat E, Abrial C, Mouret-Reynier MA, Durando X, Barthomeuf C, Chollet P: Phase I dose escalation trial of docetaxel plus curcumin in patients with advanced and metastatic breast cancer. Cancer Biol Ther. 2009, 9: 8-14.
Kanai M, Yoshimura K, Asada M, Imaizumi A, Suzuki C, Matsumoto S, Nishimura T, Mori Y, Masui T, Kawaguchi Y, Yanagihara K, Yazumi S, Chiba T, Guha S, Aggarwal BB: A phase I/II study of gemcitabine-based chemotherapy plus curcumin for patients with gemcitabine-resistant pancreatic cancer. Cancer Chemother Pharmacol. 2011, 68: 157-164. 10.1007/s00280-010-1470-2.
Dhillon N, Aggarwal BB, Newman RA, Wolff RA, Kunnumakkara AB, Abbruzzese JL, Ng CS, Badmaev V, Kurzrock R: Phase II trial of curcumin in patients with advanced pancreatic cancer. Clin Cancer Res. 2008, 14: 4491-4499. 10.1158/1078-0432.CCR-08-0024.
Li C, Zhou L, Lin G, Zuo Z: Contents of major bioactive flavones in proprietary traditional Chinese medicine products and reference herb of radix Scutellariae. J Pharm Biomed Anal. 2009, 50: 298-306. 10.1016/j.jpba.2009.04.028.
Baumann S, Fas SC, Giaisi M, Müller WW, Merling A, Gülow K, Edler L, Krammer PH, Li-Weber M: Wogonin preferentially kills malignant lymphocytes and suppresses T-cell tumor growth by inducing PLCγ1- and Ca2+ -dependent apoptosis. Blood. 2008, 111: 2354-2363. 10.1182/blood-2007-06-096198.
Yu JQ, Liu HB, Tian DZ, Liu YW, Lei JC, Zou GL: Changes in mitochondrial membrane potential and reactive oxygen species during wogonin-induced cell death in human hepatoma cells. Hepatol Res. 2007, 37: 68-76. 10.1111/j.1872-034X.2007.00003.x.
Himeji M, Ohtsuki T, Fukazawa H, Tanaka M, Yazaki S-i, Ui S, Nishio K, Yamamoto H, Tasaka K, Mimura A: Difference of growth-inhibitory effect of Scutellaria baicalensis-producing flavonoid wogonin among human cancer cells and normal diploid cell. Cancer Lett. 2007, 245: 269-274. 10.1016/j.canlet.2006.01.011.
Lee D-H, Kim C, Zhang L, Lee YJ: Role of p53, PUMA, and Bax in wogonin-induced apoptosis in human cancer cells. Biochem Pharmacol. 2008, 75: 2020-2033. 10.1016/j.bcp.2008.02.023.
Yang L, Zhang HW, Hu R, Yang Y, Qi Q, Lu N, Liu W, Chu YY, You QD, Guo QL: Wogonin induces G1 phase arrest through inhibiting Cdk4 and cyclin D1 concomitant with an elevation in p21Cip1 in human cervical carcinoma HeLa cells. Biochem Cell Biol. 2009, 87: 933-942. 10.1139/O09-060.
Lu N, Gao Y, Ling Y, Chen Y, Yang Y, Gu H-Y, Qi Q, Liu W, Wang X-T, You Q-D, Guo Q-L: Wogonin suppresses tumor growth in vivo and VEGF-induced angiogenesis through inhibiting tyrosine phosphorylation of VEGFR2. Life Sci. 2008, 82: 956-963. 10.1016/j.lfs.2008.02.013.
Lee E, Enomoto R, Koshiba C, Hirano H: Inhibition of P-glycoprotein by wogonin is involved with the potentiation of etoposide-induced apoptosis in cancer cells. Ann N Y Acad Sci. 2009, 1171: 132-136. 10.1111/j.1749-6632.2009.04722.x.
Lee E, Enomoto R, Suzuki C, Ohno M, Ohashi T, Miyauchi A, Tanimoto E, Maeda K, Hirano H, Yokoi T, Sugahara C: Wogonin, a plant flavone, potentiates etoposide-induced apoptosis in cancer cells. Ann N Y Acad Sci. 2007, 1095: 521-526. 10.1196/annals.1397.056.
Enomoto R, Koshiba C, Suzuki C, Lee E: Wogonin potentiates the antitumor action of etoposide and ameliorates its adverse effects. Cancer Chemother Pharmacol. 2011, 67: 1063-1072. 10.1007/s00280-010-1396-8.
Zhao Q, Wang J, Zou MJ, Hu R, Zhao L, Qiang L, Rong JJ, You QD, Guo QL: Wogonin potentiates the antitumor effects of low dose 5-fluorouracil against gastric cancer through induction of apoptosis by down-regulation of NF-kappaB and regulation of its metabolism. Toxicol Lett. 2010, 197: 201-210. 10.1016/j.toxlet.2010.05.019.
Li-Weber M: New therapeutic aspects of flavones: The anticancer properties of Scutellaria and its main active constituents Wogonin, Baicalein and Baicalin. Cancer Treatment Reviews. 2009, 35: 57-68. 10.1016/j.ctrv.2008.09.005.
Rajamanickam S, Velmurugan B, Kaur M, Singh RP, Agarwal R: Chemoprevention of intestinal tumorigenesis in APCmin/+ mice by silibinin. Cancer Res. 2010, 70: 2368-2378. 10.1158/0008-5472.CAN-09-3249.
Singh RP, Raina K, Deep G, Chan D, Agarwal R: Silibinin suppresses growth of human prostate carcinoma PC-3 orthotopic xenograft via activation of extracellular signal-regulated kinase 1/2 and inhibition of signal transducers and activators of transcription signaling. Clin Cancer Res. 2009, 15: 613-621. 10.1158/1078-0432.CCR-08-1846.
Singh RP, Raina K, Sharma G, Agarwal R: Silibinin inhibits established prostate tumor growth, progression, invasion, and metastasis and suppresses tumor angiogenesis and epithelial-mesenchymal transition in transgenic adenocarcinoma of the mouse prostate model mice. Clin Cancer Res. 2008, 14: 7773-7780. 10.1158/1078-0432.CCR-08-1309.
Tiwari P, Kumar A, Balakrishnan S, Kushwaha HS, Mishra KP: Silibinin-induced apoptosis in MCF7 and T47D human breast carcinoma cells involves caspase-8 activation and mitochondrial pathway. Cancer Invest. 2011, 29: 12-20. 10.3109/07357907.2010.535053.
Cui W, Gu F, Hu KQ: Effects and mechanisms of silibinin on human hepatocellular carcinoma xenografts in nude mice. World J Gastroenterol. 2009, 15: 1943-1950. 10.3748/wjg.15.1943.
Raina K, Rajamanickam S, Singh RP, Deep G, Chittezhath M, Agarwal R: Stage-specific inhibitory effects and associated mechanisms of silibinin on tumor progression and metastasis in transgenic adenocarcinoma of the mouse prostate model. Cancer Res. 2008, 68: 6822-6830. 10.1158/0008-5472.CAN-08-1332.
Mateen S, Tyagi A, Agarwal C, Singh RP, Agarwal R: Silibinin inhibits human nonsmall cell lung cancer cell growth through cell-cycle arrest by modulating expression and function of key cell-cycle regulators. Mol Carcinog. 2010, 49: 247-258.
Li L, Zeng J, Gao Y, He D: Targeting silibinin in the antiproliferative pathway. Expert Opin Investig Drugs. 2010, 19: 243-255. 10.1517/13543780903533631.
Agarwal C, Tyagi A, Kaur M, Agarwal R: Silibinin inhibits constitutive activation of Stat3, and causes caspase activation and apoptotic death of human prostate carcinoma DU145 cells. Carcinogenesis. 2007, 28: 1463-1470. 10.1093/carcin/bgm042.
Wang X, Gocek E, Novik V, Harrison JS, Danilenko M, Studzinski GP: Inhibition of Cot1/Tlp2 oncogene in AML cells reduces ERK5 activation and up-regulates p27Kip1 concomitant with enhancement of differentiation and cell cycle arrest induced by silibinin and 1,25-dihydroxyvitamin D(3). Cell Cycle. 2010, 9: 4542-4551.
Ong ES, Woo SO, Yong YL: Pressurized liquid extraction of berberine and aristolochic acids in medicinal plants. J Chromatogr A. 2000, 904: 57-64. 10.1016/S0021-9673(00)00914-6.
Lee IA, Hyun YJ, Kim DH: Berberine ameliorates TNBS-induced colitis by inhibiting lipid peroxidation, enterobacterial growth and NF-kappaB activation. Eur J Pharmacol. 2010, 648: 162-170. 10.1016/j.ejphar.2010.08.046.
Liu X, Li G, Zhu H, Huang L, Liu Y, Ma C, Qin C: Beneficial effect of berberine on hepatic insulin resistance in diabetic hamsters possibly involves in SREBPs, LXRalpha and PPARalpha transcriptional programs. Endocr J. 2010, 57: 881-893. 10.1507/endocrj.K10E-043.
Zhou J, Zhou S: Berberine regulates peroxisome proliferator-activated receptors and positive transcription elongation factor b expression in diabetic adipocytes. Eur J Pharmacol. 2010, 649: 390-397. 10.1016/j.ejphar.2010.09.030.
Wu M, Wang J, Liu LT: Advance of studies on anti-atherosclerosis mechanism of berberine. Chin J Integr Med. 2010, 16: 188-192. 10.1007/s11655-010-0188-7.
Zhao L, Li W, Han F, Hou L, Baillargeon JP, Kuang H, Wang Y, Wu X: Berberine reduces insulin resistance induced by dexamethasone in theca cells in vitro. Fertil Steril. 2011, 95: 461-463. 10.1016/j.fertnstert.2010.07.1090.
Remppis A, Bea F, Greten HJ, Buttler A, Wang H, Zhou Q, Preusch MR, Enk R, Ehehalt R, Katus H, Blessing E: Rhizoma Coptidis inhibits LPS-induced MCP-1/CCL2 production in murine macrophages via an AP-1 and NFkappaB-dependent pathway. Mediators Inflamm. 2010, 2010: 194896-
Choi MS, Yuk DY, Oh JH, Jung HY, Han SB, Moon DC, Hong JT: Berberine inhibits human neuroblastoma cell growth through induction of p53-dependent apoptosis. Anticancer Res. 2008, 28: 3777-3784.
Ho YT, Lu CC, Yang JS, Chiang JH, Li TC, Ip SW, Hsia TC, Liao CL, Lin JG, Wood WG, Chung JG: Berberine induced apoptosis via promoting the expression of caspase-8, -9 and -3, apoptosis-inducing factor and endonuclease G in SCC-4 human tongue squamous carcinoma cancer cells. Anticancer Res. 2009, 29: 4063-4070.
Hsu WH, Hsieh YS, Kuo HC, Teng CY, Huang HI, Wang CJ, Yang SF, Liou YS, Kuo WH: Berberine induces apoptosis in SW620 human colonic carcinoma cells through generation of reactive oxygen species and activation of JNK/p38 MAPK and FasL. Arch Toxicol. 2007, 81: 719-728. 10.1007/s00204-006-0169-y.
James MA, Fu H, Liu Y, Chen DR, You M: Dietary administration of berberine or Phellodendron amurense extract inhibits cell cycle progression and lung tumorigenesis. Mol Carcinog. 2011, 50: 1-7. 10.1002/mc.20690.
Patil JB, Kim J, Jayaprakasha GK: Berberine induces apoptosis in breast cancer cells (MCF-7) through mitochondrial-dependent pathway. Eur J Pharmacol. 2010, 645: 70-78. 10.1016/j.ejphar.2010.07.037.
Auyeung KK, Ko JK: Coptis chinensis inhibits hepatocellular carcinoma cell growth through nonsteroidal anti-inflammatory drug-activated gene activation. Int J Mol Med. 2009, 24: 571-577.
Yu FS, Yang JS, Lin HJ, Yu CS, Tan TW, Lin YT, Lin CC, Lu HF, Chung JG: Berberine inhibits WEHI-3 leukemia cells in vivo. In Vivo. 2007, 21: 407-412.
Meeran SM, Katiyar S, Katiyar SK: Berberine-induced apoptosis in human prostate cancer cells is initiated by reactive oxygen species generation. Toxicol Appl Pharmacol. 2008, 229: 33-43. 10.1016/j.taap.2007.12.027.
Pandey MK, Sung B, Kunnumakkara AB, Sethi G, Chaturvedi MM, Aggarwal BB: Berberine modifies cysteine 179 of IkappaBalpha kinase, suppresses nuclear factor-kappaB-regulated antiapoptotic gene products, and potentiates apoptosis. Cancer Res. 2008, 68: 5370-5379. 10.1158/0008-5472.CAN-08-0511.
Qin Y, Pang JY, Chen WH, Zhao ZZ, Liu L, Jiang ZH: Inhibition of DNA topoisomerase I by natural and synthetic mono- and dimeric protoberberine alkaloids. Chem Biodivers. 2007, 4: 481-487. 10.1002/cbdv.200790040.
Katiyar SK, Meeran SM, Katiyar N, Akhtar S: p53 Cooperates berberine-induced growth inhibition and apoptosis of non-small cell human lung cancer cells in vitro and tumor xenograft growth in vivo. Mol Carcinog. 2009, 48: 24-37. 10.1002/mc.20453.
Maiti M, Kumar GS: Polymorphic nucleic Acid binding of bioactive isoquinoline alkaloids and their role in cancer. J Nucleic Acids. 2010, 2010:
Bhadra K, Kumar GS: Therapeutic potential of nucleic acid-binding isoquinoline alkaloids: Binding aspects and implications for drug design. Med Res Rev. 2010,
Tian X, Song Y, Dong H, Ye B: Interaction of anticancer herbal drug berberine with DNA immobilized on the glassy carbon electrode. Bioelectrochemistry. 2008, 73: 18-22. 10.1016/j.bioelechem.2008.02.005.
Serafim TL, Oliveira PJ, Sardao VA, Perkins E, Parke D, Holy J: Different concentrations of berberine result in distinct cellular localization patterns and cell cycle effects in a melanoma cell line. Cancer Chemother Pharmacol. 2008, 61: 1007-1018. 10.1007/s00280-007-0558-9.
Kim JB, Yu JH, Ko E, Lee KW, Song AK, Park SY, Shin I, Han W, Noh DY: The alkaloid Berberine inhibits the growth of Anoikis-resistant MCF-7 and MDA-MB-231 breast cancer cell lines by inducing cell cycle arrest. Phytomedicine. 2010, 17: 436-440. 10.1016/j.phymed.2009.08.012.
Eom KS, Hong JM, Youn MJ, So HS, Park R, Kim JM, Kim TY: Berberine induces G1 arrest and apoptosis in human glioblastoma T98G cells through mitochondrial/caspases pathway. Biol Pharm Bull. 2008, 31: 558-562. 10.1248/bpb.31.558.
Pereira CV, Machado NG, Oliveira PJ: Mechanisms of berberine (natural yellow 18)-induced mitochondrial dysfunction: interaction with the adenine nucleotide translocator. Toxicol Sci. 2008, 105: 408-417. 10.1093/toxsci/kfn131.
Wang N, Feng Y, Zhu M, Tsang CM, Man K, Tong Y, Tsao SW: Berberine induces autophagic cell death and mitochondrial apoptosis in liver cancer cells: the cellular mechanism. J Cell Biochem. 2010, 111: 1426-1436. 10.1002/jcb.22869.
Peng PL, Kuo WH, Tseng HC, Chou FP: Synergistic tumor-killing effect of radiation and berberine combined treatment in lung cancer: the contribution of autophagic cell death. Int J Radiat Oncol Biol Phys. 2008, 70: 529-542. 10.1016/j.ijrobp.2007.08.034.
Singh T, Vaid M, Katiyar N, Sharma S, Katiyar SK: Berberine, an isoquinoline alkaloid, inhibits melanoma cancer cell migration by reducing the expressions of cyclooxygenase-2, prostaglandin E and prostaglandin E receptors. Carcinogenesis. 2011, 32: 86-92. 10.1093/carcin/bgq215.
Tsang CM, Lau EP, Di K, Cheung PY, Hau PM, Ching YP, Wong YC, Cheung AL, Wan TS, Tong Y, Tsao SW, Feng Y: Berberine inhibits Rho GTPases and cell migration at low doses but induces G2 arrest and apoptosis at high doses in human cancer cells. Int J Mol Med. 2009, 24: 131-138.
Tang F, Wang D, Duan C, Huang D, Wu Y, Chen Y, Wang W, Xie C, Meng J, Wang L, Wu B, Liu S, Tian D, Zhu F, He Z, Deng F, Cao Y: Berberine inhibits metastasis of nasopharyngeal carcinoma 5-8F cells by targeting Rho kinase-mediated Ezrin phosphorylation at threonine 567. J Biol Chem. 2009, 284: 27456-27466. 10.1074/jbc.M109.033795.
Lin TH, Kuo HC, Chou FP, Lu FJ: Berberine enhances inhibition of glioma tumor cell migration and invasiveness mediated by arsenic trioxide. BMC Cancer. 2008, 8: 58-10.1186/1471-2407-8-58.
Kim DW, Ahan SH, Kim TY: Enhancement of Arsenic Trioxide (As(2)O(3))- Mediated Apoptosis Using Berberine in Human Neuroblastoma SH-SY5Y Cells. J Korean Neurosurg Soc. 2007, 42: 392-399. 10.3340/jkns.2007.42.5.392.
Youn MJ, So HS, Cho HJ, Kim HJ, Kim Y, Lee JH, Sohn JS, Kim YK, Chung SY, Park R: Berberine, a natural product, combined with cisplatin enhanced apoptosis through a mitochondria/caspase-mediated pathway in HeLa cells. Biol Pharm Bull. 2008, 31: 789-795. 10.1248/bpb.31.789.
Hur JM, Hyun MS, Lim SY, Lee WY, Kim D: The combination of berberine and irradiation enhances anti-cancer effects via activation of p38 MAPK pathway and ROS generation in human hepatoma cells. J Cell Biochem. 2009, 107: 955-964. 10.1002/jcb.22198.
Zhang X, Gu L, Li J, Shah N, He J, Yang L, Hu Q, Zhou M: Degradation of MDM2 by the interaction between berberine and DAXX leads to potent apoptosis in MDM2-overexpressing cancer cells. Cancer Res. 2010, 70: 9895-9904. 10.1158/0008-5472.CAN-10-1546.
Gui SY, Wu L, Peng DY, Liu QY, Yin BP, Shen JZ: Preparation and evaluation of a microemulsion for oral delivery of berberine. Pharmazie. 2008, 63: 516-519.
Cheng Z, Chen AF, Wu F, Sheng L, Zhang HK, Gu M, Li YY, Zhang LN, Hu LH, Li JY, Li J: 8,8-Dimethyldihydroberberine with improved bioavailability and oral efficacy on obese and diabetic mouse models. Bioorg Med Chem. 2010, 18: 5915-5924. 10.1016/j.bmc.2010.06.085.
Pan GY, Wang GJ, Liu XD, Fawcett JP, Xie YY: The involvement of P-glycoprotein in berberine absorption. Pharmacol Toxicol. 2002, 91: 193-197. 10.1034/j.1600-0773.2002.t01-1-910403.x.
Hou J, Wang D, Zhang R, Wang H: Experimental therapy of hepatoma with artemisinin and its derivatives: in vitro and in vivo activity, chemosensitization, and mechanisms of action. Clin Cancer Res. 2008, 14: 5519-5530. 10.1158/1078-0432.CCR-08-0197.
Lu JJ, Meng LH, Cai YJ, Chen Q, Tong LJ, Lin LP, Ding J: Dihydroartemisinin induces apoptosis in HL-60 leukemia cells dependent of iron and p38 mitogen-activated protein kinase activation but independent of reactive oxygen species. Cancer Biol Ther. 2008, 7: 1017-1023. 10.4161/cbt.7.7.6035.
Efferth T, Sauerbrey A, Olbrich A, Gebhart E, Rauch P, Weber HO, Hengstler JG, Halatsch ME, Volm M, Tew KD, Ross DD, Funk JO: Molecular modes of action of artesunate in tumor cell lines. Mol Pharmacol. 2003, 64: 382-394. 10.1124/mol.64.2.382.
Lu JJ, Meng LH, Shankavaram UT, Zhu CH, Tong LJ, Chen G, Lin LP, Weinstein JN, Ding J: Dihydroartemisinin accelerates c-MYC oncoprotein degradation and induces apoptosis in c-MYC-overexpressing tumor cells. Biochem Pharmacol. 2010, 80: 22-30. 10.1016/j.bcp.2010.02.016.
Chen H, Sun B, Pan S, Jiang H, Sun X: Dihydroartemisinin inhibits growth of pancreatic cancer cells in vitro and in vivo. Anticancer Drugs. 2009, 20: 131-140. 10.1097/CAD.0b013e3283212ade.
Li LN, Zhang HD, Yuan SJ, Tian ZY, Wang L, Sun ZX: Artesunate attenuates the growth of human colorectal carcinoma and inhibits hyperactive Wnt/beta-catenin pathway. Int J Cancer. 2007, 121: 1360-1365. 10.1002/ijc.22804.
Du JH, Zhang HD, Ma ZJ, Ji KM: Artesunate induces oncosis-like cell death in vitro and has antitumor activity against pancreatic cancer xenografts in vivo. Cancer Chemother Pharmacol. 2010, 65: 895-902. 10.1007/s00280-009-1095-5.
Mercer AE, Copple IM, Maggs JL, O'Neill PM, Park BK: The role of heme and the mitochondrion in the chemical and molecular mechanisms of mammalian cell death induced by the artemisinin antimalarials. J Biol Chem. 2011, 286: 987-996. 10.1074/jbc.M110.144188.
Zhang S, Chen H, Gerhard GS: Heme synthesis increases artemisinin -induced radical formation and cytotoxicity that can be suppressed by superoxide scavengers. Chem Biol Interact. 2010, 186: 30-35. 10.1016/j.cbi.2010.03.021.
Zhang S, Gerhard GS: Heme mediates cytotoxicity from artemisinin and serves as a general anti-proliferation target. PLoS ONE. 2009, 4: e7472-10.1371/journal.pone.0007472.
Efferth T, Benakis A, Romero MR, Tomicic M, Rauh R, Steinbach D, Hafer R, Stamminger T, Oesch F, Kaina B, Marschall M: Enhancement of cytotoxicity of artemisinins toward cancer cells by ferrous iron. Free Radic Biol Med. 2004, 37: 998-1009. 10.1016/j.freeradbiomed.2004.06.023.
Kelter G, Steinbach D, Konkimalla VB, Tahara T, Taketani S, Fiebig HH, Efferth T: Role of transferrin receptor and the ABC transporters ABCB6 and ABCB7 for resistance and differentiation of tumor cells towards artesunate. PLoS ONE. 2007, 2: e798-10.1371/journal.pone.0000798.
Lu JJ, Chen SM, Zhang XW, Ding J, Meng LH: The anti-cancer activity of dihydroartemisinin is associated with induction of iron-dependent endoplasmic reticulum stress in colorectal carcinoma HCT116 cells. Invest New Drugs. 2010,
Stockwin LH, Han B, Yu SX, Hollingshead MG, ElSohly MA, Gul W, Slade D, Galal AM, Newton DL, Bumke MA: Artemisinin dimer anticancer activity correlates with heme-catalyzed reactive oxygen species generation and endoplasmic reticulum stress induction. Int J Cancer. 2009, 125: 1266-1275. 10.1002/ijc.24496.
Jiao Y, Ge CM, Meng QH, Cao JP, Tong J, Fan SJ: Dihydroartemisinin is an inhibitor of ovarian cancer cell growth. Acta pharmacol Sin. 2007, 28: 1045-1056. 10.1111/j.1745-7254.2007.00612.x.
Handrick R, Ontikatze T, Bauer KD, Freier F, Rubel A, Durig J, Belka C, Jendrossek V: Dihydroartemisinin induces apoptosis by a Bak-dependent intrinsic pathway. Mol Cancer Ther. 2010, 9: 2497-2510. 10.1158/1535-7163.MCT-10-0051.
Chen T, Li M, Zhang R, Wang H: Dihydroartemisinin induces apoptosis and sensitizes human ovarian cancer cells to carboplatin therapy. J Cell Mol Med. 2009, 13: 1358-1370. 10.1111/j.1582-4934.2008.00360.x.
He Q, Shi J, Shen XL, An J, Sun H, Wang L, Hu YJ, Sun Q, Fu LC, Sheikh MS, Huang Y: Dihydroartemisinin upregulates death receptor 5 expression and cooperates with TRAIL to induce apoptosis in human prostate cancer cells. Cancer Biol Ther. 2010, 9: 819-824. 10.4161/cbt.9.10.11552.
Zhou HJ, Wang WQ, Wu GD, Lee J, Li A: Artesunate inhibits angiogenesis and downregulates vascular endothelial growth factor expression in chronic myeloid leukemia K562 cells. Vascul Pharmacol. 2007, 47: 131-138. 10.1016/j.vph.2007.05.002.
Chen HH, Zhou HJ, Wang WQ, Wu GD: Antimalarial dihydroartemisinin also inhibits angiogenesis. Cancer Chemother Pharmacol. 2004, 53: 423-432. 10.1007/s00280-003-0751-4.
Anfosso L, Efferth T, Albini A, Pfeffer U: Microarray expression profiles of angiogenesis-related genes predict tumor cell response to artemisinins. Pharmacogenomics J. 2006, 6: 269-278.
Lee J, Zhou HJ, Wu XH: Dihydroartemisinin downregulates vascular endothelial growth factor expression and induces apoptosis in chronic myeloid leukemia K562 cells. Cancer Chemother Pharmacol. 2006, 57: 213-220. 10.1007/s00280-005-0002-y.
Buommino E, Baroni A, Canozo N, Petrazzuolo M, Nicoletti R, Vozza A, Tufano MA: Artemisinin reduces human melanoma cell migration by down-regulating alpha V beta 3 integrin and reducing metalloproteinase 2 production. Invest New Drugs. 2009, 27: 412-418. 10.1007/s10637-008-9188-2.
Rasheed SA, Efferth T, Asangani IA, Allgayer H: First evidence that the antimalarial drug artesunate inhibits invasion and in vivo metastasis in lung cancer by targeting essential extracellular proteases. Int J Cancer. 2010, 127: 1475-1485. 10.1002/ijc.25315.
Efferth T, Ramirez T, Gebhart E, Halatsch ME: Combination treatment of glioblastoma multiforme cell lines with the anti-malarial artesunate and the epidermal growth factor receptor tyrosine kinase inhibitor OSI-774. Biochem Pharmacol. 2004, 67: 1689-1700. 10.1016/j.bcp.2003.12.035.
Li QQ, Wang GD, Huang FR, Banda M, Reed E: Antineoplastic effect of beta-elemene on prostate cancer cells and other types of solid tumour cells. J Pharm Pharmacol. 2010, 62: 1018-1027.
Wang G, Li X, Huang F, Zhao J, Ding H, Cunningham C, Coad JE, Flynn DC, Reed E, Li QQ: Antitumor effect of beta-elemene in non-small-cell lung cancer cells is mediated via induction of cell cycle arrest and apoptotic cell death. Cell Mol Life Sci. 2005, 62: 881-893. 10.1007/s00018-005-5017-3.
Li X, Wang G, Zhao J, Ding H, Cunningham C, Chen F, Flynn DC, Reed E, Li QQ: Antiproliferative effect of beta-elemene in chemoresistant ovarian carcinoma cells is mediated through arrest of the cell cycle at the G2-M phase. Cell Mol Life Sci. 2005, 62: 894-904. 10.1007/s00018-005-5027-1.
Zhao J, Li QQ, Zou B, Wang G, Li X, Kim JE, Cuff CF, Huang L, Reed E, Gardner K: In vitro combination characterization of the new anticancer plant drug beta-elemene with taxanes against human lung carcinoma. Inter J Oncol. 2007, 31: 241-252.
Yao YQ, Ding X, Jia YC, Huang CX, Wang YZ, Xu YH: Anti-tumor effect of beta-elemene in glioblastoma cells depends on p38 MAPK activation. Cancer Lett. 2008, 264: 127-134. 10.1016/j.canlet.2008.01.049.
Tao L, Zhou L, Zheng L, Yao M: Elemene displays anti-cancer ability on laryngeal cancer cells in vitro and in vivo. Cancer Chemother Pharmacol. 2006, 58: 24-34. 10.1007/s00280-005-0137-x.
Wu XS, Xie T, Lin J, Fan HZ, Huang-Fu HJ, Ni LF, Yan HF: An investigation of the ability of elemene to pass through the blood-brain barrier and its effect on brain carcinomas. J Pharm Pharmacol. 2009, 61: 1653-1656.
Liao WL, Du Z, Zhang T, Guo XL, Wen YT, Yu Q, Luo AQ, Li WM: Qualitative and quantitative evaluation of Rabdosia Rubescens (Hemls.) Hara of different areas. Central South Pharmacy. 2010, 12: 912-915.
Liu JJ, Huang RW, Lin DJ, Wu XY, Peng J, Pan XL, Lin Q, Hou M, Zhang MH, Chen F: Antiproliferation effects of oridonin on HPB-ALL cells and its mechanisms of action. Am J Hematol. 2006, 81: 86-94. 10.1002/ajh.20524.
Liu JJ, Huang RW, Lin DJ, Peng J, Wu XY, Pan XL, Li MQ, Lin Q: Anti-proliferative effects of oridonin on SPC-A-1 cells and its mechanism of action. J Int Med Res. 2004, 32: 617-625.
Cui Q, Tashiro S, Onodera S, Ikejima T: Augmentation of oridonin-induced apoptosis observed with reduced autophagy. J Pharmacol Sci. 2006, 101: 230-239. 10.1254/jphs.FPJ06003X.
Wu JN, Huang J, Yang J, Tashiro S, Onodera S, Ikejima T: Caspase inhibition augmented oridonin-induced cell death in murine fibrosarcoma l929 by enhancing reactive oxygen species generation. J Pharmacol Sci. 2008, 108: 32-39. 10.1254/jphs.FP0072079.
Kang N, Zhang JH, Qiu F, Tashiro S, Onodera S, Ikejima T: Inhibition of EGFR signaling augments oridonin-induced apoptosis in human laryngeal cancer cells via enhancing oxidative stress coincident with activation of both the intrinsic and extrinsic apoptotic pathways. Cancer Lett. 2010, 294: 147-158. 10.1016/j.canlet.2010.01.032.
Zhang Y, Wu Y, Tashiro S, Onodera S, Ikejima T: Involvement of PKC signal pathways in oridonin-induced autophagy in HeLa cells: a protective mechanism against apoptosis. Biochem Biophys Res Commun. 2009, 378: 273-278. 10.1016/j.bbrc.2008.11.038.
Wang H, Ye Y, Pan SY, Zhu GY, Li YW, Fong DW, Yu ZL: Proteomic identification of proteins involved in the anticancer activities of oridonin in HepG2 cells. Phytomedicine. 2011, 18: 163-169. 10.1016/j.phymed.2010.06.011.
Kang N, Zhang JH, Qiu F, Chen S, Tashiro S, Onodera S, Ikejima T: Induction of G(2)/M phase arrest and apoptosis by oridonin in human laryngeal carcinoma cells. J Nat Prod. 2010, 73: 1058-1063. 10.1021/np9008199.
Liu YQ, Mu ZQ, You S, Tashiro S, Onodera S, Ikejima T: Fas/FasL signaling allows extracelluar-signal regulated kinase to regulate cytochrome c release in oridonin-induced apoptotic U937 cells. Biol Pharm Bull. 2006, 29: 1873-1879. 10.1248/bpb.29.1873.
Li D, Wu LJ, Tashiro S, Onodera S, Ikejima T: Oridonin induces human epidermoid carcinoma A431 cell apoptosis through tyrosine kinase and mitochondrial pathway. J Asian Nat Prod Res. 2008, 10: 77-87. 10.1080/10286020701273866.
Zhang CL, Wu LJ, Tashiro S, Onodera S, Ikejima T: Oridonin induced A375-S2 cell apoptosis via bax-regulated caspase pathway activation, dependent on the cytochrome c/caspase-9 apoptosome. J Asian Nat Prod Res. 2004, 6: 127-138. 10.1080/1028602031000147375.
Zhang CL, Wu LJ, Zuo HJ, Tashiro S, Onodera S, Ikejima T: Cytochrome c release from oridonin-treated apoptotic A375-S2 cells is dependent on p53 and extracellular signal-regulated kinase activation. J Pharmacol Sci. 2004, 96: 155-163. 10.1254/jphs.FPJ04008X.
Liu J, Huang R, Lin D, Wu X, Peng J, Lin Q, Pan X, Zhang M, Hou M, Chen F: Apoptotic effect of oridonin on NB4 cells and its mechanism. Leuk Lymphoma. 2005, 46: 593-597. 10.1080/10428190400019800.
Zhang CL, Wu LJ, Tashiro S, Onodera S, Ikejima T: Oridonin induces a caspase-independent but mitochondria- and MAPK-dependent cell death in the murine fibrosarcoma cell line L929. Biol Pharm Bull. 2004, 27: 1527-1531. 10.1248/bpb.27.1527.
Cui Q, Yu JH, Wu JN, Tashiro S, Onodera S, Minami M, Ikejima T: P53-mediated cell cycle arrest and apoptosis through a caspase-3- independent, but caspase-9-dependent pathway in oridonin-treated MCF-7 human breast cancer cells. Acta Pharmacol Sin. 2007, 28: 1057-1066. 10.1111/j.1745-7254.2007.00588.x.
Cheng Y, Qiu F, Ikejima T: Molecular mechanisms of oridonin-induced apoptosis and autophagy in murine fibrosarcoma L929 cells. Autophagy. 2009, 5: 430-431. 10.4161/auto.5.3.7896.
Cheng Y, Qiu F, Ye YC, Tashiro S, Onodera S, Ikejima T: Oridonin induces G2/M arrest and apoptosis via activating ERK-p53 apoptotic pathway and inhibiting PTK-Ras-Raf-JNK survival pathway in murine fibrosarcoma L929 cells. Arch Biochem Biophys. 2009, 490: 70-75. 10.1016/j.abb.2009.08.011.
Wang HJ, Li D, Yang FY, Tashiro S, Onodera S, Ikejima T: Oridonin induces human melanoma A375-S2 cell death partially through inhibiting insulin-like growth factor 1 receptor signaling. J Asian Nat Prod Res. 2008, 10: 787-798.
Li D, Wu LJ, Tashiro S, Onodera S, Ikejima T: Oridonin inhibited the tyrosine kinase activity and induced apoptosis in human epidermoid carcinoma A431 cells. Biol Pharm Bull. 2007, 30: 254-260. 10.1248/bpb.30.254.
Ikezoe T, Yang Y, Bandobashi K, Saito T, Takemoto S, Machida H, Togitani K, Koeffler HP, Taguchi H: Oridonin, a diterpenoid purified from Rabdosia rubescens, inhibits the proliferation of cells from lymphoid malignancies in association with blockade of the NF-kappa B signal pathways. Mol Cancer Ther. 2005, 4: 578-586. 10.1158/1535-7163.MCT-04-0277.
Gao FH, Hu XH, Li W, Liu H, Zhang YJ, Guo ZY, Xu MH, Wang ST, Jiang B, Liu F, Zhao YZ, Fang Y, Chen FY, Wu YL: Oridonin induces apoptosis and senescence in colorectal cancer cells by increasing histone hyperacetylation and regulation of p16, p21, p27 and c-myc. BMC Cancer. 2010, 10: 610-10.1186/1471-2407-10-610.
Cui Q, Tashiro S, Onodera S, Minami M, Ikejima T: Autophagy preceded apoptosis in oridonin-treated human breast cancer MCF-7 cells. Biol Pharm Bull. 2007, 30: 859-864. 10.1248/bpb.30.859.
Li D, Cui Q, Chen SG, Wu LJ, Tashiro S, Onodera S, Ikejima T: Inactivation of ras and changes of mitochondrial membrane potential contribute to oridonin-induced autophagy in a431 cells. J Pharmacol Sci. 2007, 105: 22-33. 10.1254/jphs.FPJ06022X.
Zhang Y, Wu Y, Wu D, Tashiro S, Onodera S, Ikejima T: NF-kappab facilitates oridonin-induced apoptosis and autophagy in HT1080 cells through a p53-mediated pathway. Arch Biochem Biophys. 2009, 489: 25-33. 10.1016/j.abb.2009.07.017.
Cheng Y, Qiu F, Ye YC, Guo ZM, Tashiro S, Onodera S, Ikejima T: Autophagy inhibits reactive oxygen species-mediated apoptosis via activating p38-nuclear factor-kappa B survival pathways in oridonin-treated murine fibrosarcoma L929 cells. FEBS J. 2009, 276: 1291-1306. 10.1111/j.1742-4658.2008.06864.x.
Wang MY, Lin C, Zhang TM: Effects of oridonin on DNA, RNA and protein syntheses of leukemia L 1210 cells studied by autoradiography. Zhongguo Yao Li Xue Bao. 1987, 8: 164-165.
Liu JJ, Wu XY, Peng J, Pan XL, Lu HL: Antiproliferation effects of oridonin on HL-60 cells. Ann Hematol. 2004, 83: 691-695. 10.1007/s00277-004-0919-y.
Lou H, Zhang X, Gao L, Feng F, Wang J, Wei X, Yu Z, Zhang D, Zhang Q: In vitro and in vivo antitumor activity of oridonin nanosuspension. Int J Pharm. 2009, 379: 181-186. 10.1016/j.ijpharm.2009.06.022.
Zhou GB, Kang H, Wang L, Gao L, Liu P, Xie J, Zhang FX, Weng XQ, Shen ZX, Chen J, Gu LJ, Yan M, Zhang DE, Chen SJ, Wang ZY, Chen Z: Oridonin, a diterpenoid extracted from medicinal herbs, targets AML1-ETO fusion protein and shows potent antitumor activity with low adverse effects on t(8;21) leukemia in vitro and in vivo. Blood. 2007, 109: 3441-3450. 10.1182/blood-2006-06-032250.
He MF, Liu L, Ge W, Shaw PC, Jiang R, Wu LW, But PP: Antiangiogenic activity of Tripterygium wilfordii and its terpenoids. J Ethnopharmacol. 2009, 121: 61-68. 10.1016/j.jep.2008.09.033.
Li H, Takai N, Yuge A, Furukawa Y, Tsuno A, Tsukamoto Y, Kong S, Moriyama M, Narahara H: Novel target genes responsive to the anti-growth activity of triptolide in endometrial and ovarian cancer cells. Cancer Lett. 2010, 297: 198-206. 10.1016/j.canlet.2010.05.012.
Zhao F, Chen Y, Zeng L, Li R, Zeng R, Wen L, Liu Y, Zhang C: Role of triptolide in cell proliferation, cell cycle arrest, apoptosis and histone methylation in multiple myeloma U266 cells. Eur J Pharmacol. 2010, 646: 1-11. 10.1016/j.ejphar.2010.05.034.
Zhou GS, Hu Z, Fang HT, Zhang FX, Pan XF, Chen XQ, Hu AM, Xu L, Zhou GB: Biologic activity of triptolide in t(8;21) acute myeloid leukemia cells. Leuk Res. 2011, 35: 214-218. 10.1016/j.leukres.2010.07.013.
Zhu W, Hu H, Qiu P, Yan G: Triptolide induces apoptosis in human anaplastic thyroid carcinoma cells by a p53-independent but NF-kappaB- related mechanism. Oncol Rep. 2009, 22: 1397-1401.
Zhou GX, Ding XL, Huang JF, Zhang H, Wu SB: Suppression of 5-lipoxygenase gene is involved in triptolide-induced apoptosis in pancreatic tumor cell lines. Biochim Biophys Acta. 2007, 1770: 1021-1027.
Clawson KA, Borja-Cacho D, Antonoff MB, Saluja AK, Vickers SM: Triptolide and TRAIL combination enhances apoptosis in cholangiocarcinoma. J Surg Res. 2010, 163: 244-249. 10.1016/j.jss.2010.03.067.
Carter BZ, Mak DH, Schober WD, McQueen T, Harris D, Estrov Z, Evans RL, Andreeff M: Triptolide induces caspase-dependent cell death mediated via the mitochondrial pathway in leukemic cells. Blood. 2006, 108: 630-637. 10.1182/blood-2005-09-3898.
Wan CK, Wang C, Cheung HY, Yang M, Fong WF: Triptolide induces Bcl-2 cleavage and mitochondria dependent apoptosis in p53-deficient HL-60 cells. Cancer Lett. 2006, 241: 31-41. 10.1016/j.canlet.2005.10.001.
Johnson SM, Wang X, Mark Evers B: Triptolide Inhibits Proliferation and Migration of Colon Cancer Cells by Inhibition of Cell Cycle Regulators and Cytokine Receptors. J Surg Res. 2011, 168: 197-205. 10.1016/j.jss.2009.07.002.
Chang HJ, Kim MH, Baek MK, Park JS, Chung IJ, Shin BA, Ahn BW, Jung YD: Triptolide inhibits tumor promoter-induced uPAR expression via blocking NF-kappaB signaling in human gastric AGS cells. Anticancer Res. 2007, 27: 3411-3417.
Zhu W, He S, Li Y, Qiu P, Shu M, Ou Y, Zhou Y, Leng T, Xie J, Zheng X, Xu D, Su X, Yan G: Anti-angiogenic activity of triptolide in anaplastic thyroid carcinoma is mediated by targeting vascular endothelial and tumor cells. Vascul Pharmacol. 2010, 52: 46-54. 10.1016/j.vph.2009.10.006.
Liang M, Fu J: Triptolide inhibits interferon-gamma-induced programmed death-1-ligand 1 surface expression in breast cancer cells. Cancer Lett. 2008, 270: 337-341. 10.1016/j.canlet.2008.05.025.
Yang S, Chen J, Guo Z, Xu XM, Wang L, Pei XF, Yang J, Underhill CB, Zhang L: Triptolide inhibits the growth and metastasis of solid tumors. Mol Cancer Ther. 2003, 2: 65-72.
Zhang C, Cui GH, Liu F, Wu QL, Chen Y: Inhibitory effect of triptolide on lymph node metastasis in patients with non-Hodgkin lymphoma by regulating SDF-1/CXCR4 axis in vitro. Acta pharmacologica Sinica. 2006, 27: 1438-1446. 10.1111/j.1745-7254.2006.00415.x.
Tang XY, Zhu YQ, Tao WH, Wei B, Lin XL: Synergistic effect of triptolide combined with 5-fluorouracil on colon carcinoma. Postgrad Med J. 2007, 83: 338-343. 10.1136/pgmj.2006.055426.
Yang SW, Wang W, Xie XY, Zhu WP, Li FQ: In vitro synergistic cytotoxic effect of triptolide combined with hydroxycamptothecin on pancreatic cancer cells. Am J Chin Med. 2011, 39: 121-134. 10.1142/S0192415X11008695.
Pigneux A, Mahon FX, Uhalde M, Jeanneteau M, Lacombe F, Milpied N, Reiffers J, Belloc F: Triptolide cooperates with chemotherapy to induce apoptosis in acute myeloid leukemia cells. Exp Hematol. 2008, 36: 1648-1659. 10.1016/j.exphem.2008.08.002.
Carter BZ, Mak DH, Schober WD, Dietrich MF, Pinilla C, Vassilev LT, Reed JC, Andreeff M: Triptolide sensitizes AML cells to TRAIL-induced apoptosis via decrease of XIAP and p53-mediated increase of DR5. Blood. 2008, 111: 3742-3750. 10.1182/blood-2007-05-091504.
Wang W, Yang S, Su Y, Xiao Z, Wang C, Li X, Lin L, Fenton BM, Paoni SF, Ding I, Keng P, Okunieff P, Zhang L: Enhanced antitumor effect of combined triptolide and ionizing radiation. Clin Cancer Res. 2007, 13: 4891-4899. 10.1158/1078-0432.CCR-07-0416.
Wang X, Li L, Wang B, Xiang J: Effects of ursolic acid on the proliferation and apoptosis of human ovarian cancer cells. J Huazhong Uni Sci Technol Med Sci. 2009, 29: 761-764. 10.1007/s11596-009-0618-y.
Yang L, Liu X, Lu Z, Yuet-Wa Chan J, Zhou L, Fung KP, Wu P, Wu S: Ursolic acid induces doxorubicin-resistant HepG2 cell death via the release of apoptosis-inducing factor. Cancer Lett. 2010, 298: 128-138. 10.1016/j.canlet.2010.06.010.
De Angel RE, Smith SM, Glickman RD, Perkins SN, Hursting SD: Antitumor effects of ursolic acid in a mouse model of postmenopausal breast cancer. Nutr Cancer. 2010, 62: 1074-1086. 10.1080/01635581.2010.492092.
Meng Y, Song Y, Yan Z, Xia Y: Synthesis and in vitro cytotoxicity of novel ursolic acid derivatives. Molecules (Basel, Switzerland). 2010, 15: 4033-4040.
Kwon SH, Park HY, Kim JY, Jeong IY, Lee MK, Seo KI: Apoptotic action of ursolic acid isolated from Corni fructus in RC-58T/h/SA#4 primary human prostate cancer cells. Bioorg Med Chem Lett. 2010, 20: 6435-6438. 10.1016/j.bmcl.2010.09.073.
Zhang Y, Kong C, Zeng Y, Wang L, Li Z, Wang H, Xu C, Sun Y: Ursolic acid induces PC-3 cell apoptosis via activation of JNK and inhibition of Akt pathways in vitro. Mol Carcinog. 2010, 49: 374-385.
Yeh CT, Wu CH, Yen GC: Ursolic acid, a naturally occurring triterpenoid, suppresses migration and invasion of human breast cancer cells by modulating c-Jun N-terminal kinase, Akt and mammalian target of rapamycin signaling. Mol Nutr Food Res. 2010, 54: 1285-1295. 10.1002/mnfr.200900414.
Huang HC, Huang CY, Lin-Shiau SY, Lin JK: Ursolic acid inhibits IL-1beta or TNF-alpha-induced C6 glioma invasion through suppressing the association ZIP/p62 with PKC-zeta and downregulating the MMP-9 expression. Mol Carcinog. 2009, 48: 517-531. 10.1002/mc.20490.
Kanjoormana M, Kuttan G: Antiangiogenic activity of ursolic acid. Integr Cancer Ther. 2010, 9: 224-235. 10.1177/1534735410367647.
Lin CC, Huang CY, Mong MC, Chan CY, Yin MC: Antiangiogenic Potential of Three Triterpenic Acids in Human Liver Cancer Cells. J Agric Food Chem. 2010, 755-762.
Wu Z, Wu L, Li L, Tashiro S, Onodera S, Ikejima T: p53-mediated cell cycle arrest and apoptosis induced by shikonin via a caspase-9-dependent mechanism in human malignant melanoma A375-S2 cells. J Pharmacol Sci. 2004, 94: 166-176. 10.1254/jphs.94.166.
Wu Z, Wu LJ, Li LH, Tashiro S, Onodera S, Ikejima T: Shikonin regulates HeLa cell death via caspase-3 activation and blockage of DNA synthesis. J Asian Nat Prod Res. 2004, 6: 155-166. 10.1080/1028602032000169622.
Yeh CC, Kuo HM, Li TM, Lin JP, Yu FS, Lu HF, Chung JG, Yang JS: Shikonin-induced apoptosis involves caspase-3 activity in a human bladder cancer cell line (T24). In Vivo. 2007, 21: 1011-1019.
Yoon Y, Kim YO, Lim NY, Jeon WK, Sung HJ: Shikonin, an ingredient of Lithospermum erythrorhizon induced apoptosis in HL60 human premyelocytic leukemia cell line. Planta Med. 1999, 65: 532-535. 10.1055/s-1999-14010.
Han W, Li L, Qiu S, Lu Q, Pan Q, Gu Y, Luo J, Hu X: Shikonin circumvents cancer drug resistance by induction of a necroptotic death. Mol Cancer Ther. 2007, 6: 1641-1649. 10.1158/1535-7163.MCT-06-0511.
Han W, Xie J, Li L, Liu Z, Hu X: Necrostatin-1 reverts shikonin-induced necroptosis to apoptosis. Apoptosis. 2009, 14: 674-686. 10.1007/s10495-009-0334-x.
Min R, Tong J, Wenjun Y, Wenhu D, Xiaojian Z, Jiacai H, Jian Z, Wantao C, Chenping Z: Growth inhibition and induction of apoptosis in human oral squamous cell carcinoma Tca-8113 cell lines by Shikonin was partly through the inactivation of NF-kappaB pathway. Phytother Res. 2008, 22: 407-415. 10.1002/ptr.2340.
Nie YK, Zhu LS, Yu HM: Shikonin inhibits the proliferation and induces the apoptosis of human HepG2 cells. Can J Physiol Pharm. 2010, 88: 1138-1146. 10.1139/Y10-085.
Chang IC, Huang YJ, Chiang TI, Yeh CW, Hsu LS: Shikonin Induces Apoptosis through Reactive Oxygen Species/Extracellular Signal- Regulated Kinase Pathway in Osteosarcoma Cells. Biol Pharm Bull. 2010, 33: 816-824. 10.1248/bpb.33.816.
Mao X, Yu CR, Li WH, Li WX: Induction of apoptosis by shikonin through a ROS/JNK-mediated process in Bcr/Abl-positive chronic myelogenous leukemia (CML) cells. Cell Res. 2008, 18: 879-888. 10.1038/cr.2008.86.
Yang H, Zhou P, Huang H, Chen D, Ma N, Cui QC, Shen S, Dong W, Zhang X, Lian W, Wang X, Dou QP, Liu J: Shikonin exerts antitumor activity via proteasome inhibition and cell death induction in vitro and in vivo. Int J Cancer. 2009, 124: 2450-2459. 10.1002/ijc.24195.
Yang F, Chen Y, Duan W, Zhang C, Zhu H, Ding J: SH-7, a new synthesized shikonin derivative, exerting its potent antitumor activities as a topoisomerase inhibitor. Int J Cancer. 2006, 119: 1184-1193. 10.1002/ijc.21943.
Yao Y, Zhou Q: A novel antiestrogen agent Shikonin inhibits estrogen- dependent gene transcription in human breast cancer cells. Breast Cancer Res Tr. 2010, 121: 233-240. 10.1007/s10549-009-0547-2.
Yao YA, Brodie AMH, Davidson NE, Kensler TW, Zhou Q: Inhibition of estrogen signaling activates the NRF2 pathway in breast cancer. Breast Cancer Res Tr. 2010, 124: 585-591. 10.1007/s10549-010-1023-8.
Min R, Zun Z, Min Y, Wenhu D, Wenjun Y, Chenping Z: Shikonin inhibits tumor invasion via down-regulation of NF-kappaB-mediated MMP-9 expression in human ACC-M cells. Oral Dis. 2011, 17: 362-369. 10.1111/j.1601-0825.2010.01758.x.
Wang L, Li D, Bao C, You J, Wang Z, Shi Y, Zhang H: Ultrasonic extraction and separation of anthraquinones from Rheum palmatum L. Ultrason Sonochem. 2008, 15: 738-746. 10.1016/j.ultsonch.2007.12.008.
Srinivas G, Babykutty S, Sathiadevan PP, Srinivas P: Molecular mechanism of emodin action: transition from laxative ingredient to an antitumor agent. Med Res Rev. 2007, 27: 591-608. 10.1002/med.20095.
Chun-Guang W, Jun-Qing Y, Bei-Zhong L, Dan-Ting J, Chong W, Liang Z, Dan Z, Yan W: Anti-tumor activity of emodin against human chronic myelocytic leukemia K562 cell lines in vitro and in vivo. Eur J Pharmacol. 2010, 627: 33-41. 10.1016/j.ejphar.2009.10.035.
Muto A, Hori M, Sasaki Y, Saitoh A, Yasuda I, Maekawa T, Uchida T, Asakura K, Nakazato T, Kaneda T, Kizaki M, Ikeda Y, Yoshida T: Emodin has a cytotoxic activity against human multiple myeloma as a Janus-activated kinase 2 inhibitor. Mol Cancer Ther. 2007, 6: 987-994.
Su YT, Chang HL, Shyue SK, Hsu SL: Emodin induces apoptosis in human lung adenocarcinoma cells through a reactive oxygen species-dependent mitochondrial signaling pathway. Biochem Pharmacol. 2005, 70: 229-241. 10.1016/j.bcp.2005.04.026.
Cha TL, Qiu L, Chen CT, Wen Y, Hung MC: Emodin down-regulates androgen receptor and inhibits prostate cancer cell growth. Cancer Res. 2005, 65: 2287-2295. 10.1158/0008-5472.CAN-04-3250.
Li Y, Luan Y, Qi X, Li M, Gong L, Xue X, Wu X, Wu Y, Chen M, Xing G, Yao J, Ren Jl: Emodin triggers DNA double-strand breaks by stabilizing topoisomerase II-DNA cleavage complexes and by inhibiting ATP hydrolysis of topoisomerase II. Toxicol Sci. 2010, 118: 435-443. 10.1093/toxsci/kfq282.
Ko JC, Su YJ, Lin ST, Jhan JY, Ciou SC, Cheng CM, Lin YW: Suppression of ERCC1 and Rad51 expression through ERK1/2 inactivation is essential in emodin-mediated cytotoxicity in human non-small cell lung cancer cells. Biochem Pharmacol. 2010, 79: 655-664. 10.1016/j.bcp.2009.09.024.
Kwak HJ, Park MJ, Park CM, Moon SI, Yoo DH, Lee HC, Lee SH, Kim MS, Lee HW, Shin WS, Park IC, Rhee CH, Hong SI: Emodin inhibits vascular endothelial growth factor-A-induced angiogenesis by blocking receptor-2 (KDR/Flk-1) phosphorylation. Int J Cancer. 2006, 118: 2711-2720. 10.1002/ijc.21641.
Huang Q, Shen HM, Ong CN: Emodin inhibits tumor cell migration through suppression of the phosphatidylinositol 3-kinase-Cdc42/Rac1 pathway. Cell Mol Life Sci. 2005, 62: 1167-1175. 10.1007/s00018-005-5050-2.
Huang Q, Shen HM, Shui G, Wenk MR, Ong CN: Emodin inhibits tumor cell adhesion through disruption of the membrane lipid Raft-associated integrin signaling pathway. Cancer Res. 2006, 66: 5807-5815. 10.1158/0008-5472.CAN-06-0077.
Wang XJ, Yang J, Cang H, Zou YQ, Yi J: Gene expression alteration during redox-dependent enhancement of arsenic cytotoxicity by emodin in HeLa cells. Cell Res. 2005, 15: 511-522. 10.1038/sj.cr.7290321.
Cai J, Niu X, Chen Y, Hu Q, Shi G, Wu H, Wang J, Yi J: Emodin-induced generation of reactive oxygen species inhibits RhoA activation to sensitize gastric carcinoma cells to anoikis. Neoplasia. 2008, 10: 41-51. 10.1593/neo.07754.
Huang XZ, Wang J, Huang C, Chen YY, Shi GY, Hu QS, Yi J: Emodin enhances cytotoxicity of chemotherapeutic drugs in prostate cancer cells: the mechanisms involve ROS-mediated suppression of multidrug resistance and hypoxia inducible factor-1. Cancer Biol Ther. 2008, 7: 468-475. 10.4161/cbt.7.3.5457.
Wang W, Sun YP, Huang XZ, He M, Chen YY, Shi GY, Li H, Yi J, Wang J: Emodin enhances sensitivity of gallbladder cancer cells to platinum drugs via glutathion depletion and MRP1 downregulation. Biochem Pharmacol. 2010, 79: 1134-1140. 10.1016/j.bcp.2009.12.006.
Chen RS, Jhan JY, Su YJ, Lee WT, Cheng CM, Ciou SC, Lin ST, Chuang SM, Ko JC, Lin YW: Emodin enhances gefitinib-induced cytotoxicity via Rad51 downregulation and ERK1/2 inactivation. Exp Cell Res. 2009, 315: 2658-2672. 10.1016/j.yexcr.2009.06.002.
Ko JC, Su YJ, Lin ST, Jhan JY, Ciou SC, Cheng CM, Chiu YF, Kuo YH, Tsai MS, Lin YW: Emodin enhances cisplatin-induced cytotoxicity via down- regulation of ERCC1 and inactivation of ERK1/2. Lung Cancer. 2010, 69: 155-164. 10.1016/j.lungcan.2009.10.013.
Yue PY, Wong DY, Wu PK, Leung PY, Mak NK, Yeung HW, Liu L, Cai Z, Jiang ZH, Fan TP, Wong RN: The angiosuppressive effects of 20(R)- ginsenoside Rg3. Biochem Pharmacol. 2006, 72: 437-445. 10.1016/j.bcp.2006.04.034.
Liu TG, Huang Y, Cui DD, Huang XB, Mao SH, Ji LL, Song HB, Yi C: Inhibitory effect of ginsenoside Rg3 combined with gemcitabine on angiogenesis and growth of lung cancer in mice. BMC Cancer. 2009, 9: 250-10.1186/1471-2407-9-250.
Kim SM, Lee SY, Yuk DY, Moon DC, Choi SS, Kim Y, Han SB, Oh KW, Hong JT: Inhibition of NF-kappaB by ginsenoside Rg3 enhances the susceptibility of colon cancer cells to docetaxel. Arch Pharm Res. 2009, 32: 755-765. 10.1007/s12272-009-1515-4.
Kim SM, Lee SY, Cho JS, Son SM, Choi SS, Yun YP, Yoo HS, Yoon do Y, Oh KW, Han SB, Hong JT: Combination of ginsenoside Rg3 with docetaxel enhances the susceptibility of prostate cancer cells via inhibition of NF-kappaB. Eur J Pharmacol. 2010, 631: 1-9. 10.1016/j.ejphar.2009.12.018.
Kim HS, Lee EH, Ko SR, Choi KJ, Park JH, Im DS: Effects of ginsenosides Rg3 and Rh2 on the proliferation of prostate cancer cells. Arch Pharm Res. 2004, 27: 429-435. 10.1007/BF02980085.
He BC, Gao JL, Luo X, Luo J, Shen J, Wang L, Zhou Q, Wang YT, Luu HH, Haydon RC, Wang CZ, Du W, Yuan CS, He TC, Zhang BQ: Ginsenoside Rg3 inhibits colorectal tumor growth through the down-regulation of Wnt/ß-catenin signaling. Int J Oncol. 2011, 38: 437-445.
Luo X, Wang CZ, Chen J, Song WX, Luo J, Tang N, He BC, Kang Q, Wang Y, Du W, He TC, Yuan CS: Characterization of gene expression regulated by American ginseng and ginsenoside Rg3 in human colorectal cancer cells. Int J Oncol. 2008, 32: 975-983.
Lee SY, Kim GT, Roh SH, Song JS, Kim HJ, Hong SS, Kwon SW, Park JH: Proteomic analysis of the anti-cancer effect of 20S-ginsenoside Rg3 in human colon cancer cell lines. Biosci Biotechnol Biochem. 2009, 73: 811-816. 10.1271/bbb.80637.
Xu TM, Cui MH, Xin Y, Gu LP, Jiang X, Su MM, Wang DD, Wang WJ: Inhibitory effect of ginsenoside Rg3 on ovarian cancer metastasis. Chin Med J (Engl). 2008, 121: 1394-1397.
Zhang Q, Kang X, Zhao W: Antiangiogenic effect of low-dose cyclophosphamide combined with ginsenoside Rg3 on Lewis lung carcinoma. Biochem Biophys Res Commun. 2006, 342: 824-828. 10.1016/j.bbrc.2006.02.044.
Acknowledgements
The work was supported by the grant (029/2007/A2) from the Science and Technology Development Fund of Macau Special Administrative Region, China and supported in part by the National Natural Science of China (No. 81001450) awarded to JJL.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
WT, JJL, MQH, YBL, MWC, GSW, JG, ZFZ, ZTX, YYD and XPC wrote the manuscript (WT wrote berberine; JJL wrote GA and ARTs; MQH wrote emodin and ginsenoside Rg3; YBL wrote cucurmin; MWC wrote silibinin; GSW wrote shikonin; JG wrote wogonin; ZFZ wrote β-elemene; ZTX wrote triptolide; YYD wrote UA; XPC wrote oridonin). JJG drew the chemical structures in Figure 1. WT, JJL and XPC revised the manuscript. YTW designed and supervised this work. All authors read and approved the final version of the manuscript.
Wen Tan, Jinjian Lu contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Tan, W., Lu, J., Huang, M. et al. Anti-cancer natural products isolated from chinese medicinal herbs. Chin Med 6, 27 (2011). https://doi.org/10.1186/1749-8546-6-27
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1749-8546-6-27