
Abstract
Background
Several forms of progressive retinal atrophy (PRA) segregate in more than 100 breeds of dog with each PRA segregating in one or a few breeds. This breed specificity may be accounted for by founder effects and genetic drift, which have reduced the genetic heterogeneity of each breed, thereby facilitating the identification of causal mutations. We report here a new form of PRA segregating in the Border Collie breed. The clinical signs, including the loss of night vision and a progressive loss of day vision, resulting in complete blindness, occur at the age of three to four years and may be detected earlier through systematic ocular fundus examination and electroretinography (ERG).
Results
Ophthalmic examinations performed on 487 dogs showed that affected dogs present a classical form of PRA. Of those, 274 have been sampled for DNA extraction and 87 could be connected through a large pedigree. Segregation analysis suggested an X-linked mode of transmission; therefore both XLPRA1 and XLPRA2 mutations were excluded through the genetic tests.
Conclusion
Having excluded these mutations, we suggest that this PRA segregating in Border Collie is a new XLPRA (XLPRA3) and propose it as a potential model for the homologous human disease, X-Linked Retinitis Pigmentosa.
Similar content being viewed by others

Background
Progressive Retinal Atrophy (PRA) has been described in more than 100 breeds of dog [1–3], providing a powerful resource for the identification of new retinopathy-causing genes and a unique model for treatments for homologous human retinal diseases [4, 5]. The strong founder effect and genetic drift occurring during the breeding of dogs may have significantly reduced the genetic heterogeneity of diseases in each breed, making it easier to identify causal mutations in dogs than in humans. Several genes responsible for canine retinopathies have yet been identified (Table 1). We focused on PRA, a clinically homogeneous group of diseases characterized by a loss of night vision in the first few years of life (2 to 5 years). This night blindness is followed by a progressive loss of the peripheral visual field and finally a total loss of vision, involving an initial loss of rods and then cone photoreceptors [2, 6].
Age-at-onset differs between breeds. PRA are also highly heterogeneous genetically, with several modes of transmission and a large number of genes and mutations involved. Each PRA generally occurs in only one or a few breeds, as demonstrated for PRA with a known genetic basis [3] (Table 1). PRA-prcd is a notable exception, affecting more than 20 breeds [7, 8]. Only two X-linked PRA have been described both involving the RPGR gene (Retinitis Pigmentosa GTPase Regulator) with a different mutation in exon 15 (ORF15) in each breed. XLPRA1 is caused by a deletion of five nucleotides, leading to a frameshift and immediate premature stop in the Siberian Husky and Samoyed. XLPRA2 is caused by a deletion of two nucleotides leading to a frameshift that has been shown to result in significant changes in the deduced peptide sequence in a mongrel dog [9].
PRA are naturally occurring retinal diseases in dogs, and have a phenotype similar to that of Retinitis Pigmentosa in humans. Retinitis Pigmentosa (RP) is the most prevalent group of inherited retinopathies in humans, affecting about 1 in 3600 individuals [10]. RP display considerable clinical and genetical heterogeneity, with wide variations in disease onset, progression and severity [11] and several transmission modes. Up to now, 54 loci for non-syndromic RP have been mapped, for which 39 genes have been identified [12]. Those genes account for an estimated 50% of dominant RP, 40% of recessive RP and 80% of X-linked RP cases [13].
Studies aiming to identify the genes responsible for X-linked RP have led to the identification of four loci (RP6, RP23, RP24, RP34) and only two genes (RPGR and RP2) [14–19]. Other genes for X-linked RP remain to be identified, indeed, known genes and loci involved in those diseases have been excluded in several families [20]. The RPGR gene, a GTPase regulator that is essential for the maintenance of photoreceptor viability, is involved in the X-linked RP3 disease. In this gene nearly 100 mutations have been already described in several families [21]. The RP2 gene responsible for X-Linked RP2 disease is thought to be involved in the beta-tubulin folding [17]. Up to 17 mutations have been identified as associated with RP2 [21]. In the last decade, the canine model has displayed considerable genetic potential, as individual breeds correspond to isolated populations, it has facilitated the identification of a number of dog genes and invaluable candidates for the homologous diseases in humans [22, 23]. This applied to retinal diseases and PRA in particular, with the example of the identification of a new canine gene (PRCD), responsible for PRA-prcd in different breeds. This gene, which was not annotated in the human genome, constituted a new candidate gene for human RP and indeed, a mutation in this gene has been identified in a patient from Bangladesh with RP [7].
We searched for potential candidate genes for human RP, by investigating a PRA segregating with a high frequency in the Border Collie breed. Three retinopathies have been described in Border Collie: the Colley Eye Anomaly (CEA) [24], the Central Progressive Retinal Atrophy (CPRA) [25, 26] and the (non central) Progressive Retinal Atrophy (PRA) [27]. Border Collie belongs to the Collie lineage in which CEA is frequently diagnosed in Colley but less frequently in Border Collie. The primary clinical sign in CEA is choroidal hypoplasia corresponding to the under-development of the choroid. Moderately affected dogs present a normal vision throughout life but in severely affected individuals, colobomas at the optic nerve can lead to retinal detachments and blindness [24]. CPRA has an incidence of 12% in the breed in the United Kingdom, its occurrence is between 1 and 2 years of age, with primary brown spots in the tapetal area along the vessels, the later stages looking like PRA. The both eyes were generally identically affected and secondary cataract is rarer than in PRA [25, 26]. The PRA that occurs in the Border Collie has been clinically described previously [27]. In the present study, the clinical examination of 487 Border Collie dogs from France demonstrated the occurrence of PRA at a high frequency in this breed. We present a precise clinical description of this canine PRA and the first steps of the genetic analysis. The genetic study consisted in collecting cases and controls as well as related individuals to investigate the exact mode of transmission and to test known canine mutations. Over 250 Border Collie blood samples were collected, most of them could be connected through a 375 dog pedigree. Segregation analysis suggested an X-linked mode of transmission. We investigated the possibility of the molecular cause of this PRA being a known mutation, by carrying out the genetic tests for XLPRA1 and XLPRA2. The exclusion of these mutations as possible causes of the disease suggested that this PRA in Border Collie might correspond to a new form of X-linked PRA. We are currently collecting a larger sample of dogs for linkage analysis.
Results
Clinical features of PRA in the Border Collie
In total, 487 dogs were examined by the same veterinary ophthalmologist. All of them are working Border Collies and were examined all over France, for purpose of clinical prediction of inherited eye diseases. In total, the 241 males and 246 females were aged of 2 months to 13 years, 293 of which were at least 2 years old at the time of the first examination (mean age at the first examination: 3.25 years); 357 dogs were examined once, 86 twice, 23 three times, 17 four times, 4 five times, on average in the space of a one year period.
Clinical examination consisted in the evaluation of menace responses and pupillary light reflexes and indirect ophthalmoscopic examination (30δ & 20δ lenses) after pupillary dilatation. A total of 60 dogs (54 males, 6 females) aged between six months and nine years (mean age of diagnostic: 3.85 year old) presented lesions of the fundus, mostly in the tapetum: 31 dogs were bilaterally affected, 26 dogs were unilaterally affected at the first examination and bilaterally at the second examination and three dogs were unilaterally affected at the second examination and bilaterally at the third examination.
Lesions of the fundus were variable: at early stages, focal or multifocal hyperreflective lesions of the tapetum resulting in thinning of the retina, with an inconstant symmetry were frequently observed (Figure 1a), but the hyperreflective band surrounding the tapetal-nontapetal junction was rare; at advanced stages, generalized hyperreflexion coexisted either with hyperreflective coalescent foci in the tapetum (Figure 1b), or with diffuse tapetal hyperreflexion (Figure 1c) and depigmentation/hyperpigmentation in the nontapetum (Figure 1c and 1d), whereas the vessels became thinner before disappearing and the optic disc became grayish (atrophic myelin loss) (Figure 1b and 1c); secondary cortical equatorial cataracts appeared after the age of seven years, but this finding was rare in the affected dogs, some of which were totally blind [28].

Ophthalmoscopy; 1a: hyperreflective nasal tapetal focal lesion with a pigmented dot in a 3 years old affected male, OD: the optic disc, the vessels and the nontapetal area are still remaining normal. 1b: Multifocal coalescent hyperreflective lesions in a 3,5 years old affected male: a pigmented dot is visible at the center of a suprapapillar lesion; the tapetum appears hyperreflective due to the coalescence of multiple focal lesions; the optic disc looks grayish and the arterial vasculature has disappeared. 1c: Generalized hyperreflexion of the tapetum in a 3 years old affected male, OD: the tapetum appears hyperreflective, with the exception of a subnormal horizontal band close to the nontapetal area; the optic disc is grayish and the arteries have disappeared. 1d: OS, dog of the figure 1c: attenuation of the arterial diameter, temporal triangular suprapapillar hyperreflective area with a pigmented line at its center.
The correspondence between ophthalmoscopic examinations and evaluation of menace responses and pupillary light reflexes was the following:
Observations for unilateral lesions
Isolated peripheral or central hyperreflective or grayish focal tapetal lesions, with or without a pigmented dot at their centre: pupillary light reflexes and menace response were present in all dogs.
Multiple hyperreflective focal lesions with diffuse increased in tapetal reflexion, thinning of the vessels and multifocal depigmentation of the non tapetal zone: the menace response was present but pupillary light reflexes were delayed and incomplete.
Observations for bilateral lesions
Isolated symmetric focal hyperreflective tapetal lesions, with a pigmented center and focal discolored non tapetal lesions: the menace response was present but pupillary light reflexes were delayed and incomplete;
Symmetric horizontal hyperreflective tapetal band parallel to the tapetum/non tapetum juction: the menace response and pupillary light reflexes were present and unmodified;
Multiple symmetric hypereflective tapetal lesions and multifocal discolored non tapetal lesions: at reevaluation one year later, these dogs displayed either additional similar lesions or generalized atrophy of the fundus (coalescent lesions) with hyporeflective midriasis and thinning of the vessels in all cases and palor of the papilla in some cases; all these dogs had lost the menace response.
Electroretinography results
Electroretinography (ERG) was carried out as a complementary examination in 15 dogs (14 affected, 1 clinically unaffected but at risk). Based on recordings in the same conditions for a two-year-old male dog used as a reference, we found: normal traces in cases of isolated unilateral peripheral or central hyperreflective or grayish lesions; significant to severe decrease in a and b-wave amplitude and in some cases, non recordable scotopic blue ERGs when symmetric multifocal hyperreflective tapetal lesions or diffuse hyperreflective tapetum were observed (Figure 2). A severe decrease in b-wave amplitude in scotopic blue and achromatic white stimulations; an increase in culmination phases when multiple symmetric hyperreflective focal lesions or diffuse hyperreflective tapetal zones were present.

Electroretinograms; 2a, 2c and 2e: dark adaptation, blue light scotopic stimuli. 2b, 2d and 2f: dark adaptation, white light stimuli. 2a and 2b: 2 years old unaffected male. 2c and 2d: 3 years old affected male with isolated lesions. 2e and 2f: 9 years old affected dog with generalized lesions.
Fluorescein Angiography results
Fluorescein angiography (FA) was performed in 10 affected dogs. Dogs were declared unaffected if they remained asymptomatic after the age of five years. Younger dogs without symptoms were considered to have an unknown phenotype.
We observed either hypofluorescent or hyperfluorescent defects as follows:
early focal hyperfluorescence in the tapetum by window defect, which remained without any modification or became wider through peripheral epithelial fluorescein leakage throughout the angiogram (Figure 3a and 3b);

Fluorescein angiograms: 3a: fundus of the figure 1a, choroidal phase: combined window effect and masking effect (arrows). 3b: fundus of the figure 1a, late venous phase: epithelial fluorescein leakage at the level of lesions. 3c: fundus of the figure 1d, laminar venous phase: masking effect by retinal pigment at the level of the lesion (arrow); linear leakage of fluorescein in the pigment epithelium (points of arrows). 3d: fundus of the figure 1d, late venous phase: most important diffusion of fluorescein at the periphery of the lesion; the hyperfluorescent lines (linear leakage) are corresponding to localized ruptures of the Bruch's membrane.
constant focal hypofluorescence linked to masking effect by retinal pigment at the level of the lesion and progressive diffusion of the fluorescein by leakage in the pigment epithelium (Figure 3c);
linear ruptures of Bruch's membrane in the area centralis (Figures 3c and 3d);
epithelial fluorescein leakage starting from neovessels either in the tapetum or nontapetum and increasing during late phases of the angiogram in advanced atrophic retinal lesions (Figure 4).

Fluorescein angiogram; 7 years old affected male, late venous phase: epithelial leakage of fluorescein in the tapetal area (arrows) and in the nontapetal area (hollowed arrow), visible choroidal vasculature (bold arrows).
All these lesions corresponded to primary retinal involvement, but were not suggestive of CPRA, neither of XLPRA 1. It does not either correspond to XLPRA2 since, in XLPRA2, retinal development is aberrant with ERG abnormalities at as early as 5–6 weeks of age. At the age of 4 months retinal degeneration rapidly progresses and by 2 years, retina is fully degenerated in all affected dogs [9] whereas in the Border Collie PRA, the clinical signs are at their beginning when observed.
Pedigree
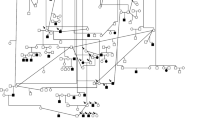
Out of the 487 clinically examined dogs, a subset of 80 Border Collies (33 affected, 30 males and 3 females) belonging to a large pedigree were sampled for the genetic analysis. By retrieving the genealogical data of these 80 dogs, a pedigree of 375 individuals was constituted using the Cyrillic 2.1 software but no common ancestor could yet be defined (Figure 5). As previously stated, all dogs are working Border Collies sampled from all over France and no bias is suspected in the sampling.

Illustration of a Border Collie pedigree segregating PRA constructed by the Cyrillic 2.1 software. This pedigree is constituted of 80 dogs, 33 dogs are affected (30 males and 3 females).
Segregation analysis
For inference of the genetic model best accounting for the observed segregation of the disease in the 375-dog pedigree, we performed a segregation analysis using the Generalized Linear Model approach implemented in Mendel 6.1. We chose this relatively simple segregation approach because a highly penetrant X-linked model was suspected in the family and because the use of Mendel6.1 made it possible to analyze the pedigree in all its complexity. The results are presented in Table 2. We first compared the likelihood of the general autosomal model with that of the X-linked model using the AIC criterion, as these two models are not nested. The general X-linked model fitted the data better (AICX-link = 88.4 vs AICautos = 110.6). If we constrained the X-linked model to either a general dominant (f12 = f22) or a general recessive model (f11 = f12), the parameter estimates converged to give a fully penetrant recessive model with a risk allele frequency = 0.46. The chi2 statistic for comparison of the fully penetrant X-linked recessive model with the general X-linked model was not statistically significant (pval = 0.10). Consequently, the fully penetrant X-linked recessive model with a risk allele frequency of 0.46 is the most parsimonious model best fitting the data.
Mutation screening
XLPRA1 and XLPRA2
In order to check if the PRA in the Border Collie could be due to the XLPRA1 or XLPRA2 mutations of the RPGR gene, we tested 36 PRA affected Border Collie for XLPRA1 and 34 affected Border Collie for XLPRA2. None of those mutations were detected in any dog.
CEA mutation
The Border Collie is one of about a dozen breeds of the Collie lineage [29], susceptible to be affected by the Collie Eye Anomaly (CEA) disease [30]. The frequency of this disease depends on the breeds and the countries [24], being rarely detected in France, clinical signs of CEA were found in only one dog of 1000 examined (Chaudieu, personal communication). Clinical investigations were not suggestive of CEA in any of the dogs, but 24 dogs, including 20 affected dogs, were nonetheless screened for the CEA mutation in the NHEJ1 gene [31] to exclude the possible co-segregation of CEA and PRA at the molecular level. This mutation was not detected in any of the 24 dogs tested.
PRA-prcd
The PRA-prcd mutation segregates in a large number of breeds, indeed, it has been found in 22 breeds [7]. To exclude the co-segregation of this mutation in Border Collies, we tested 55 dogs including 34 affected dogs for the prcd mutation in the PRCD gene [32]. This mutation was not detected in any of the 55 dogs tested.
As no known mutations for retinopathies likely to affect Border Collies were found in any of the dogs tested, we suggest that the PRA segregating in this breed corresponds to a new XLPRA (XLPRA3).
Discussion
In the present study, we described a new canine retinopathy. The clinical form described here is clearly characteristic of a progressive retinal atrophy (PRA): the onset of symptoms occurred at around three years of age, and peripheral vision was then progressively lost over the next years. The appearance of the fundus was compatible with PRA with distinct peripheral pigmentary changes, optic pallor, and attenuated retinal vessels. ERG responses were suggestive of the disease in two-year-old dogs. The predominant symptoms of night blindness, peripheral field defects, and absence of light sensitivity strongly suggest that this is a form of PRA [25, 26].
All the clinical signs that we observed (menace responses, pupillary light reflexes, fundus examination) were evocating a PRA:
- focal lesions were evoluting from one eye to both in cases of unilateral lesions at the first examination;
- either focal or diffuse bilateral lesions were evolving towards a generalized disease of he fundus in re-examined dogs, with PRA compatible signs: tapetal hyperreflexion, vascular attenuation, palor and enlargement of the optic disc;
- vacuolar subcapsular equatorial cortical cataracts observed in some dogs with severe fundic lesions were described in other canine breeds as secondary to PRA [6, 27];
- the most important was midriasis, the most incomplete, unstable and the slower was the pupillary light reflex, with disappearing of the menace response in cases with severe fundic lesions.
If only focal isolated lesions were observed, there was no modification of the ERG. In cases of multiple or generalized fundic lesions, the hypovolted traces (sometimes non recordable in blue stimulation) were in favour of a progressive rod and cone degeneration.
We never found either ophthalmoscopic, functional or angiographic signs which correlated with CPRA: this disease was first described in several breeds, considered as recessive autosomal in the Briard shepherd dog, dominant autosomal with incomplete penetrance in the Labrador Retriever, the Shetland sheepdog [25–27], dominant autosomal with incomplete penetrance [26] or polygenic [25] in the Border Collie. Second, it appears that environmental factors were involved in CPRA: experimental Vitamin E deficiency exactly mimicked the same fundic lesions in the dog [33]. Nevertheless, in Border Collies, the results of eye examination scheme in the United Kingdom remained in favor of an hereditary origin: in 1965, 12% of the dogs were affected of CPRA; in 1989, only 2% were exhibiting ophthalmoscopic changes characteristic of the disease [25, 26]. All the findings of FA were demonstrating that the primary changes in the fundus were not related to a pigment epithelium dystrophy: we never found the specific lesions of fluorescence mottled masking by lipofuscinic like pigment deposits [34]. We showed that this PRA segregates according to an X-linked mode of transmission. This is compatible with the status of the few affected females identified. Indeed, the six females found affected, after clinical examination, were diagnosed between 18 months and 3 years old and clinical symptoms were very similar to those observed in males. Moreover, all females presented similar clinical features. For three of them, we do not have any genealogical data and thus we do not know the status of the parents. For the other three affected females, which are in the pedigree, they were all born from an affected father and an obligate carrier female. Considering the high level of inbreeding in the breed, we assume that affected females come from obligate carriers and affected males, supporting a fully penetrant recessive inheritance mode. Genetic tests carried out on subsets of dogs excluded the involvement of known mutations.
We excluded the XLPRA1 and XLPRA2 mutations as the possible causal mutation for this X-linked PRA. However, owing to differences in the clinical signs and age-of-onset observed with XLPRA2, the second ORF15 mutation in RPGR could likely be excluded [9]. The CEA mutation is frequent in the Collie lineage, but occurs at only low frequency in the Border Collie breed in France (Chaudieu, personal communication). We nonetheless tested for the presence of the CEA mutation that could co-segregate. As expected, the dogs analyzed did not carry the mutation, consistent with the low prevalence of CEA in the Border Collie. Finally, we also tested for the presence of the mutation for PRCD, which has been involved in many dog breeds displaying PRA-prcd, while these breeds have no evident common origin [7]. Neither CEA, nor prcd were found to co-segregate in affected Border Collies.
In human, retinitis pigmentosa occurs and presents clinical features similar to canine PRA. Human cases experience a decrease in peripheral and night vision in their twenties, then night blindness and diminution of the visual field in their thirties, but visual acuity and color vision may remain normal until advanced stages of the disease [13]. Clinically, X-linked RP are the most severe forms of RP [13] but only 2 genes have yet been identified on the X chromosome. Human families are often too small for the identification of the causal loci by linkage studies and families cannot be grouped for genetic linkage analyses because of possible genetic heterogeneity. Indeed, several causal genes have already been identified in inbred families [12]. As dog pedigrees are highly inbred, it may be a useful model to identify new candidate genes for X linked RP.
The pedigree used for the segregation analysis is currently being completed for genetic linkage analysis to search for the genetic locus involved in the disease. The XLPRA1 and XLPRA2 mutations are not involved in this XLPRA, but we cannot exclude the involvement of other mutation in the RPGR gene. Indeed, this is the only gene involved in canine XLPRA with two mutations presently described in exon 15. In human, RPGR is frequently and differently mutated in XLRP. Other human candidates, such as the XLRP gene (RP2) or loci (RP6, 23, 24, 34) [14–19] may be analysed. We expect linkage analysis to be more efficient to test the involvement of genes and loci, as gene sequencing cannot unequivocally exclude the involvement of a gene if no mutation is found especially as some regions are usually unscreened, such as introns, 5' and 3' UTR or other regulatory regions.
Conclusion
Studies of this new XLPRA should lead to the identification of a new mutation in one of the previously identified genes or in a new gene on the X chromosome in dog. This disease could constitute a model for X-linked human RP. As 20% of XLRP genes remain to be identified in human [13], the Border Collie provides an opportunity to identify a new causal gene thus representing a potentially pertinent model for studying the genetic bases of XLRP in human.
Methods
All experiments have been performed under agreement reference number A35-238-13 delivered in July 2003 by the animal healthcare and protection service of the French government.
Clinical investigation/ophthalmologic examination
None of the dogs was kept for research purposes. All were privately owned sheep dogs.
The 487 dogs were examined both by evaluating pupillary light reflexes and menace response and by indirect ophthalmoscopy (30δ & 20δ lenses) after pupillary dilation (0.5% Tropicamide drops, Mydriaticum®, Théa, Clermont-Ferrand Cedex, France).
Electroretinography (ERG)
ERG included the following procedures: pupillary dilatation (Mydriaticum®); dark adaptation for 15 min; general anesthetic (2% xylazine, Rompun®, Bayer Pharma, Puteaux cedex – France: 0,05 mL/kg and 10% ketamine chlorhydrate, Imalgene 1000®, Merial, Lyon – France: 0,05 mL/kg) administered IV; 10 achromatic stimulations for each eye with a white standard flash (2,5 cd/s/m2), at a 1 Hz frequency; 10 chromatic stimulations for each eye with a blue filter (440 nm) at a scotopic level and a 1 Hz frequency; signals recorded with needle electrodes (active and reference electrodes for each eye).
Fluorescein Angiography (FA)
FA was performed without sedation after pupillary dilatation (Mydriaticum®) using a Kowa – RC fundus camera (Kowa Company, Tokyo, Japan) fitted with a specially adapted blue stimulation blue filter, yellow filters and loaded with Ilford HP5 ISO 400 black-and-white film (Ilford Imaging, Mobberley, UK); 10% Sodium fluoresceinate (2 mL/10 kg bodyweight, Ciba Vision Ophthalmics, Toulouse – France) was injected intravenously, when starting the examination of the fundus.
Pedigree analysis
A 375-dog multigeneration pedigree, connecting 87 dogs with known phenotypes was constructed. 33 dogs are affected (30 males and 3 females). This pedigree is highly inbred with frequent marriage loops. We used the Cyrillic software v2.1 [35] (© CyrillicSoftware) for genealogic and genetic data management.
All dogs subjected to genetic analysis for which parental DNA was available, were tested for parental compatibility using the ISAG reference markers.
Genomic DNA extraction
Blood samples and the accompanying pedigree were collected by DVM veterinaries or based on information obtained directly from the owners. All data were entered into a database. Genomic DNA was extracted from 5 ml of blood collected into EDTA, using the nucleon BACC 3 kit (GE Healthcare Bio-Sciences Corp, Piscataway, NJ, USA). The extracted DNA was "Whole Genome Amplified" using the V1 Genomiphi kit (GE Healthcare).
Segregation analysis
Segregation analysis was performed on a 375-dog pedigree, that corresponds partially to the diagnosed dogs, using the generalized linear model-based approach implemented in Mendel 6.1 [36]. We analyzed the qualitative phenotype under study, by considering the logit function as a link function in the model. Briefly, let p be the probability of being affected, conditional on the genotype and let X11, X12, X22 be binary variables describing the genotype at a biallelic disease locus (X11 = 1 and X12 = X22 = 0 for genotype 11). The general model is . The different segregation models to be compared in the analysis are expressed as constraints on (β1, β2, β3). Likelihoods of the models are compared with likelihood ratio statistics when models are nested and using the AIC criterion (best model minimizes (-2 lnL + 2 No), where L is the Likelihood of the model and No the number of independent parameters estimated in the model) for non nested models.
Mutation testing
Dogs of the pedigree were screened for the known mutations, XLPRA1 [9], prcd [7, 32] and the mutation for Colley Eye Anomaly (CEA) [31] as described. For the XLPRA2 mutation, primers were designed using the RPGR ORF15 sequence (AF385629) [9]. The PCR were performed with DNA amplified by Genomiphi V2 (GE Healthcare).
Primers:
For XLPRA1 mutation [9]:
RGF14: AAGGGGAGGAGAAAGGGGAGGCT
RGR13: TCCCTCTTCCTCCTCCCCTTCATA
For XLPRA2 mutation:
XLPRA2U: AGAGGCAGAATGGGAAGGAA
XLPRA2L: CCGTCTTCCCTGTTTTTCAC
For CEA mutation [31]:
aF-D GGAGGAGTCATCATGACTTGC
aR-D GACTGGTATTATCAAAGGTCAC
For PRA-prcd mutation [32]:
prcd1f: CCAGTGGCAGCAGGAACC
prcd2r: CCGACCTGCTGCCCACGACTG
References
Patterson DF: Companion animal medicine in the age of medical genetics. Journal of veterinary internal medicine/American College of Veterinary Internal Medicine. 2000, 14 (1): 1-9. 10.1892/0891-6640(2000)14[1:CAMITA]2.0.CO;2.
Petersen-Jones S: Advances in the molecular understanding of canine retinal diseases. The Journal of small animal practice. 2005, 46 (8): 371-380. 10.1111/j.1748-5827.2005.tb00333.x.
Aguirre GD, Acland GM: Models, Mutants, and Man: Searching for Unique Phenotypes and Genes in the Dog Model of Inherited Retinal Degeneration. The Dog and its Genome. Edited by: Ostrander EA, Giger U, Lindblad-Toh K. New York: Cold Spring Harbor Laboratory Press; 2006.
Acland GM, Aguirre GD, Bennett J, Aleman TS, Cideciyan AV, Bennicelli J, Dejneka NS, Pearce-Kelling SE, Maguire AM, Palczewski K, et al: Long-term restoration of rod and cone vision by single dose rAAV-mediated gene transfer to the retina in a canine model of childhood blindness. Mol Ther. 2005, 12 (6): 1072-1082. 10.1016/j.ymthe.2005.08.008.
Le Meur G, Stieger K, Smith AJ, Weber M, Deschamps JY, Nivard D, Mendes-Madeira A, Provost N, Pereon Y, Cherel Y, et al: Restoration of vision in RPE65-deficient Briard dogs using an AAV serotype 4 vector that specifically targets the retinal pigmented epithelium. Gene therapy. 2007, 14 (4): 292-303. 10.1038/sj.gt.3302861.
Chaudieu G: Affections oculaires héréditaires ou à prédisposition raciale chez le chien. Edited by: Vétérinaire EdP. 2004.
Zangerl B, Goldstein O, Philp AR, Lindauer SJ, Pearce-Kelling SE, Mullins RF, Graphodatsky AS, Ripoll D, Felix JS, Stone EM, et al: Identical mutation in a novel retinal gene causes progressive rod-cone degeneration in dogs and retinitis pigmentosa in humans. Genomics. 2006, 88 (5): 551-563. 10.1016/j.ygeno.2006.07.007.
Goldstein O, Zangerl B, Pearce-Kelling S, Sidjanin DJ, Kijas JW, Felix J, Acland GM, Aguirre GD: Linkage disequilibrium mapping in domestic dog breeds narrows the progressive rod-cone degeneration interval and identifies ancestral disease-transmitting chromosome. Genomics. 2006, 88 (5): 541-550. 10.1016/j.ygeno.2006.05.013.
Zhang Q, Acland GM, Wu WX, Johnson JL, Pearce-Kelling S, Tulloch B, Vervoort R, Wright AF, Aguirre GD: Different RPGR exon ORF15 mutations in Canids provide insights into photoreceptor cell degeneration. Hum Mol Genet. 2002, 11 (9): 993-1003. 10.1093/hmg/11.9.993.
Orphanet: Prévalence des maladie rares: Une enquète bibliohraphique Janvier 2007. Edited by: d'Orphanet LC. 2007, [http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases.pdf]
Maubaret C, Hamel C: [Genetics of retinitis pigmentosa: metabolic classification and phenotype/genotype correlations]. Journal francais d'ophtalmologie. 2005, 28 (1): 71-92. 10.1016/S0181-5512(05)81029-0.
RetNet. [http://www.sph.uth.tmc.edu/Retnet/]
Hamel C: Retinitis pigmentosa. Orphanet J Rare Dis. 2006, 1: 40-10.1186/1750-1172-1-40.
Hardcastle AJ, Thiselton DL, Zito I, Ebenezer N, Mah TS, Gorin MB, Bhattacharya SS: Evidence for a new locus for X-linked retinitis pigmentosa (RP23). Investigative ophthalmology & visual science. 2000, 41 (8): 2080-2086.
Ott J, Bhattacharya S, Chen JD, Denton MJ, Donald J, Dubay C, Farrar GJ, Fishman GA, Frey D, Gal A, et al: Localizing multiple X chromosome-linked retinitis pigmentosa loci using multilocus homogeneity tests. Proc Natl Acad Sci USA. 1990, 87 (2): 701-704. 10.1073/pnas.87.2.701.
Meindl A, Dry K, Herrmann K, Manson F, Ciccodicola A, Edgar A, Carvalho MR, Achatz H, Hellebrand H, Lennon A, et al: A gene (RPGR) with homology to the RCC1 guanine nucleotide exchange factor is mutated in X-linked retinitis pigmentosa (RP3). Nature genetics. 1996, 13 (1): 35-42. 10.1038/ng0596-35.
Schwahn U, Lenzner S, Dong J, Feil S, Hinzmann B, van Duijnhoven G, Kirschner R, Hemberger M, Bergen AA, Rosenberg T, et al: Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nature genetics. 1998, 19 (4): 327-332. 10.1038/1214.
Gieser L, Fujita R, Goring HH, Ott J, Hoffman DR, Cideciyan AV, Birch DG, Jacobson SG, Swaroop A: A novel locus (RP24) for X-linked retinitis pigmentosa maps to Xq26–27. Am J Hum Genet. 1998, 63 (5): 1439-1447. 10.1086/302121.
Melamud A, Shen GQ, Chung D, Xi Q, Simpson E, Li L, Peachey NS, Zegarra H, Hagstrom SA, Wang QK, et al: Mapping a new genetic locus for X linked retinitis pigmentosa to Xq28. Journal of medical genetics. 2006, 43 (6): e27-10.1136/jmg.2005.031518.
Tong Z, Yang Z, Meyer JJ, McInnes AW, Xue L, Azimi AM, Baird J, Zhao Y, Pearson E, Wang C, et al: A novel locus for X-linked retinitis pigmentosa. Annals of the Academy of Medicine, Singapore. 2006, 35 (7): 476-478.
HGMD®, Human Gene Mutation Database. [http://www.hgmd.cf.ac.uk/ac/index.php]
Sutter NB, Ostrander EA: Dog star rising: the canine genetic system. Nature reviews. 2004, 5 (12): 900-910. 10.1038/nrg1492.
Galibert F, Andre C, Hitte C: [Dog as a mammalian genetic model]. Med Sci (Paris). 2004, 20 (8–9): 761-766.
Lowe JK, Kukekova AV, Kirkness EF, Langlois MC, Aguirre GD, Acland GM, Ostrander EA: Linkage mapping of the primary disease locus for collie eye anomaly. Genomics. 2003, 82 (1): 86-95. 10.1016/S0888-7543(03)00078-8.
Parry HB: Degenerations of the dog retina. VI. Central progressive atrophy with pigment epithelial dystrophy. The British journal of ophthalmology. 1954, 38 (11): 653-668. 10.1136/bjo.38.11.653.
Barnett KC: Retinal atrophy. The Veterinary record. 1965, 77 (51): 1543-1560.
Rubin LF: Inherited eye disease in purebred dogs. Williams & Wilkins; 1989.
Chaudieu G: Etude de l'atrophie progressive de la rétine chez le Border Collie: Renseignements fournis par 213 examens pratiqués chez 161 chiens. Prat Méd Chir Anim Comp. 2001, 36: 55-73.
Neff MW, Robertson KR, Wong AK, Safra N, Broman KW, Slatkin M, Mealey KL, Pedersen NC: Breed distribution and history of canine mdr1-1Delta, a pharmacogenetic mutation that marks the emergence of breeds from the collie lineage. Proc Natl Acad Sci USA. 2004, 101 (32): 11725-11730. 10.1073/pnas.0402374101.
Bedford PG: Collie eye anomaly in the border collie. The Veterinary record. 1982, 111 (2): 34-35.
Acland GM, Kukekova AV, Aguirre GD, Ostrander EA, Akey D, Goldstein O: Diagnostic test to collie eye anomaly. Patent: EP 1 652 937. 2006, CORNELL RESEARCH FOUNDATION I, NY 14850 (US), 31-
Aguirre G, Acland G, Zangerl B, Goldstein O, Pearce-Kelling S, Felix J, Sidjanin DJ: Method for identifying progressive rod-cone degeneration in dogs. Patent: EP 1 798 295. CORNELL RESEARCH FOUNDATION I, NY 14850 (US). 2007, 22-
Riis RC, Sheffy BE, Loew E, Kern TJ, Smith JS: Vitamin E deficiency retinopathy in dogs. American journal of veterinary research. 1981, 42 (1): 74-86.
Lescure F: Fluorescein angiographic atlas of the small animal ocular fundus. Prat Méd Chir Anim Comp. 1998.
CyrillicSoftware. [http://www.cyrillicsoftware.com/]
Lange K, Cantor R, Horvath S, Perola M, Sabatti C, Sinsheimer J, Sobel E: Mendel version 4.0: A complete package for the exact genetic analysis of discrete traits in pedigree and population data sets. Amer J Hum Genetics. 2001, 69 (supplement):
Suber ML, Pittler SJ, Qin N, Wright GC, Holcombe V, Lee RH, Craft CM, Lolley RN, Baehr W, Hurwitz RL: Irish setter dogs affected with rod/cone dysplasia contain a nonsense mutation in the rod cGMP phosphodiesterase beta-subunit gene. Proc Natl Acad Sci USA. 1993, 90 (9): 3968-3972. 10.1073/pnas.90.9.3968.
Dekomien G, Runte M, Godde R, Epplen JT: Generalized progressive retinal atrophy of Sloughi dogs is due to an 8-bp insertion in exon 21 of the PDE6B gene. Cytogenetics and cell genetics. 2000, 90 (3–4): 261-267.
Petersen-Jones SM, Entz DD, Sargan DR: cGMP phosphodiesterase-alpha mutation causes progressive retinal atrophy in the Cardigan Welsh corgi dog. Investigative ophthalmology & visual science. 1999, 40 (8): 1637-1644.
Kijas JW, Cideciyan AV, Aleman TS, Pianta MJ, Pearce-Kelling SE, Miller BJ, Jacobson SG, Aguirre GD, Acland GM: Naturally occurring rhodopsin mutation in the dog causes retinal dysfunction and degeneration mimicking human dominant retinitis pigmentosa. Proc Natl Acad Sci USA. 2002, 99 (9): 6328-6333. 10.1073/pnas.082714499.
Kijas JW, Miller BJ, Pearce-Kelling SE, Aguirre GD, Acland GM: Canine models of ocular disease: outcross breedings define a dominant disorder present in the English mastiff and bull mastiff dog breeds. The Journal of heredity. 2003, 94 (1): 27-30. 10.1093/jhered/esg007.
Zhang Q, Acland GM, Parshall CJ, Haskell J, Ray K, Aguirre GD: Characterization of canine photoreceptor phosducin cDNA and identification of a sequence variant in dogs with photoreceptor dysplasia. Gene. 1998, 215 (2): 231-239. 10.1016/S0378-1119(98)00310-2.
Sidjanin DJ, Lowe JK, McElwee JL, Milne BS, Phippen TM, Sargan DR, Aguirre GD, Acland GM, Ostrander EA: Canine CNGB3 mutations establish cone degeneration as orthologous to the human achromatopsia locus ACHM3. Hum Mol Genet. 2002, 11 (16): 1823-1833. 10.1093/hmg/11.16.1823.
Veske A, Nilsson SE, Narfstrom K, Gal A: Retinal dystrophy of Swedish briard/briard-beagle dogs is due to a 4-bp deletion in RPE65. Genomics. 1999, 57 (1): 57-61. 10.1006/geno.1999.5754.
Mellersh CS, Boursnell ME, Pettitt L, Ryder EJ, Holmes NG, Grafham D, Forman OP, Sampson J, Barnett KC, Blanton S, et al: Canine RPGRIP1 mutation establishes cone-rod dystrophy in miniature longhaired dachshunds as a homologue of human Leber congenital amaurosis. Genomics. 2006, 88 (3): 293-301. 10.1016/j.ygeno.2006.05.004.
Guziewicz KE, Zangerl B, Lindauer SJ, Mullins RF, Sandmeyer LS, Grahn BH, Stone EM, Acland GM, Aguirre GD: Bestrophin gene mutations cause canine multifocal retinopathy: a novel animal model for best disease. Investigative ophthalmology & visual science. 2007, 48 (5): 1959-1967. 10.1167/iovs.06-1374.
Acknowledgements
We thank the CNRS and the ANRT, who helped to fund TV, through a CIFRE convention with Antagene, an "animal genomics" company. The authors also thank Dr. Cazelle, Dr. PF Isard and Dr. Peroux, DVM for clinical examinations and the AFBC, (the French Border Collie Association), especially Dominique Cornuet, Sandrine Chartier and Anne Moreillon, and the many breeders, trainers and owners for genealogical data and samples.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
TV collected samples, constructed the pedigrees and performed molecular analyses; he interpreted data and actively participated to the manuscript redaction. GC did all clinical diagnoses, collected samples and actively participated to the writing of the manuscript. PJ and CB performed the statistical analyses and interpreted the data; they critically revised the manuscript. DD performed genetic tests. BH extracted DNA from blood samples. GQ and FG gave critical advices on the work and manuscript. AT contributed to the conception and design of the work and to the writing of the manuscript. CA contributed to the conception and design of the work and drafted the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Vilboux, T., Chaudieu, G., Jeannin, P. et al. Progressive Retinal Atrophy in the Border Collie: A new XLPRA. BMC Vet Res 4, 10 (2008). https://doi.org/10.1186/1746-6148-4-10
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1746-6148-4-10