
Abstract
Background
Interstitial pneumonias (IP) cover a broad spectrum of diseases. Open lung biopsies reveal histological patterns and suggest possible diagnoses. Complete clinical records are necessary for final diagnoses. Especially idiopathic interstitial pneumonias (IIP) according to the ATS/ERS classification can only be diagnosed under these predictions. The aim of this study was to compare the results of histological evaluations with the final diagnosis after interdisciplinary case evaluation.
Patients and methods
88 patients with interstitial pneumonia that underwent open lung biopsies were investigated. Histology and clinical records were available for review. Diagnosis was made in three steps: first on the sole basis of histology, second with clinical information given initially and third, on the basis of an interdisciplinary case evaluation.
Results
63 patients (72%) were diagnosed as idiopathic interstitial pneumonias according to ATS/ERS criteria. Further 10 (11%) cases of hypersensitivity pneumonitis, 7 (8%) Langerhans cell histiocytosis and 8 (9%) interstitial pneumonias of other known causes or associations were detected. Histological patterns alone agreed with the final diagnosis in 67%. In 82% histology and clinical information given to the pathologist could provide correct diagnosis. In the rest of cases, especially in non idiopathic interstitial pneumonias, an interdisciplinary case evaluation was needed.
Conclusions
Diagnosis of interstitial pneumonias by open lung biopsies needs sufficient clinical information. Because of the overlap of histological patterns, an interdisciplinary case evaluation that includes at least one clinical expert and one pathologist with excellent expertise and the follow-up of the patients is necessary to find correct diagnosis in all cases.
Virtual slides
The virtual slides for this article can be found here:http://www.diagnosticpathology.diagnomx.eu/vs/5031706258025129
Similar content being viewed by others


Background
Diffuse interstitial lung diseases (ILD) are disorders with a large spectrum of possible underlying causes. Most of ILD belong to the group of idiopathic interstitial pneumonias (IIP). But these diagnoses can only be made after exclusion of known etiological factors or associations.
Pulmonary fibrosis was first described by VON BÜHL in 1872[1]. The first generally accepted classification of idiopathic interstitial pneumonia was introduced by LIEBOW in 1975[2]. He distinguished usual interstitial pneumonia (UIP), bronchiolitis obliterans with interstitial pneumonia (BIP), desquamative interstitial pneumonia (DIP), lymphocytic interstitial pneumonia (LIP) and interstitial giant cell pneumonia (GIP). In 1990, KITAICHI described a further group designated as “unclassified interstitial pneumonia”[3]. This led to a revision of the Liebow classification by A. KATZENSTEIN[4]. The categories UIP and DIP remained, but LIP and GIP were abandoned, because they were no longer regarded as idiopathic disease. Respiratory bronchiolitis with interstitial lung disease (RBILD), acute interstitial pneumonia (AIP) and the non specific interstitial pneumonia (NSIP) were introduced as new entities.
The ATS/ERS (American Thoracic Society/European Respiratory Society) international multidisciplinary consensus classification of idiopathic interstitial pneumonias was developed in 2002 by a team of clinicians, pathologists and radiologists in order to standardize classification and achieve a broad acceptance among the participating disciplines[5]. By this classification LIP was reintroduced, but non idiopathic cases have to be excluded faithfully[6]. In clinical practise overlap to follicular bronchiolitis has been found[7].
Revised evidence-based guidelines for diagnosis and management of IPF as a collaborative effort between the ATS, ERS, Japanese Respiratory Society (JRS), and Latin American Thoracic Association (ALAT) have been published in 2011[8]. Non idiopathic origin of IIP has to be evaluated carefully, pulmonary symptoms can even proceed manifestations of connective tissue diseases[9].
Interobserver variability in the diagnosis of ILD is a problem for chest physicians, radiologists and pathologists[10–13]. Discordances also exist between general and pulmonary pathologists in the diagnosis of interstitial lung disease[14]. Therefore, standardization of the diagnostic process and quality assessment are necessary.
The aim of this study was the evaluation of patients with IIP undergoing open lung biopsies in three diagnostic steps to investigate the benefit of clinical information and final interdisciplinary case evaluation.
Patients and methods
Selection and categorization of the patients
All cases with histological diagnosis of interstitial pneumonia or pulmonary fibrosis involving patients of the Ruhrlandklinik Essen – West German Lung Center at the University Hospital Essen between 1993 and 2000 were retrospectively selected from the archive of the Department of Pathology at the Ruhr-University Bochum. The indication to perform open lung biopsies had been made by the clinicians on the basis of the available guidelines at that time. All patients have given written consent to surgical procedures und scientific evaluation of data. Data protection was done according to legal foundations. Patients without open lung biopsy (OLB) were excluded. Clinical records were reviewed in each case, follow up was requested by contacting local practitioners. Digital images or photographs of thoracic imaging (CXR, CT and HRCT) were only partially available for diagnostic review and written descriptions of radiological findings were often of poor quality. Therefore results of thoracic imaging were not included in the evaluation process.
Bronchoalveolar lavage
BAL was performed during local anesthesia using fiberoptic bronchoscopy according to established guidelines[15]. In brief, a flexible bronchoscope was wedged into a segmental bronchus of the middle lobe or the lingula. Sterile isotonic saline was instilled in five to ten 20 ml aliquots up to a total volume of 100–200 ml, with immediate aspiration by gentle suction after each aliquot. The recovered BAL fluid was immediately processed in the laboratory. The fluid was pooled, filtered through two layers of gauze, and centrifuged at 500 g for 10 min at room temperature. The cells were counted in a haemocytometer. Slides were stained with May-Grünwald-Giemsa stain (Merck, Germany) and a total of 600 cells were counted for the cell differentials. A trypan blue exclusion test was performed for evaluating cell viability.
Histology and immunohistochemistry
Slides were stained with hematoxylin and eosin in all cases, further slides with Elastica van Gieson, Periodic Acid Schiff and Prussian Blue stains were available in parts. In cases of suspected Langerhans cell histiocytosis immunohistochemistry with monoclonal antibodies against S-100 Protein and CD1a was done (ABC method, DAKO Hamburg, Germany). Additionally in one case of LIP staining for lymphocyte subtypes (CD 3 and CD 20, ABC method, DAKO Hamburg, Germany) was performed.
Diagnostic evaluation process
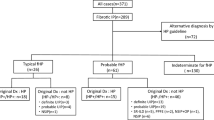
Diagnostic evaluation was done in three steps. Firstly, diagnosis was made as a slide review on the basis of histological patterns found by the pathologist (DT), blinded to the BAL results and other clinical data. Confidence of histological diagnosis was estimated as certain, probable or possible. If no clear cut diagnosis could be made, the most probable diagnosis was listed. Secondly, histological re-evaluation was done on the basis of clinical diagnoses made by the clinician given on the pathology request form. Thirdly, final diagnosis was made by the pulmonologist (UC) and the pathologist (DT) on the light of all clinical, radiological and histological data as interdisciplinary case evaluation (Figure1). The results of these three diagnostic steps were compared.

Diagnostic steps in interstitial lung diseases . Different steps necessary for sufficient diagnosis.
Statistics
Data are expressed as mean ± standard deviation. We used t-student test and ANOVA, for two or multiple groups respectively, to compare normally distributed variables. Comparison of non-normally distributed variables between two groups was done with the Mann–Whitney U test. Comparison of categorical variables between two groups, including the results of the single diagnostic steps vs interdisciplinary case evaluation, was done by the Fisher’s exact probability test. All statistical analyses were done using SPSS version 17.0 for Windows (SPSS Inc., Chicago, IL). Differences were considered statistically significant when the p value was < 0.05.
Results
A total of 92 cases with interstitial pneumonia underwent diagnostical evaluation. 3 patients with putride pneumonia and one patient with bronchiolitis were excluded from further analysis. Overall confidence of histological evaluation was estimated as certain in 67% of cases (N = 62), probable in 24% of cases (N = 22), and possible in 9% (N = 8). Histological patterns alone agreed with the final diagnosis in 67% of cases and in 82% histology and clinical information provided the diagnosis, compared with interdisciplinary case evaluation (Table1, p < 0.037 exact Fisher’s test).
Idiopathic interstitial pneumonia (IIP)
63 patients (72%) were diagnosed as idiopathic interstitial pneumonias by interdisciplinary case evaluation according to ATS/ERS criteria (Table 1). There were 27 (43%) patients with idiopathic pulmonary fibrosis (IPF), 14 (22%) with non specific interstitial pneumonia (NSIP), 6 (9.5%) with respiratory bronchiolitis with interstitial lung disease (RBILD), 3 (5%) with desquamative interstitial pneumonia (DIP), 10 (16%) with cryptogenic organizing pneumonia (COP) and 3 (5%) with lymphocytic interstitial pneumonia (LIP).
Usual interstitial pneumonia (UIP)/Idiopathic pulmonary fibrosis (IPF)
Male patients were dominant (Table2). The patients’ mean age was 60 years, the oldest among all ILDs. Clinically, dyspnea (96%) and cough (78%) were present. There were 67% smokers with a mean of 17.2 pack years (Table2). Bronchoalveolar lavage (BAL) showed a lower percentage of lymphocytes (18%) than NSIP, COP, or HP cases (Table3).
Microscopically a UIP pattern with heterogene changes, honeycombing, a variable fibrosis, mucus plugging, lymphoplasmacytic infiltrates of variable densities and fibroblastic foci was seen (Figure2).

Usual interstitial pneumonia . The UIP pattern shows honey combing and fibroblastic foci (haematoxylin eosin stain (HE), original magnification 100x).
Non specific interstitial pneumonia (NSIP)
In this group, most patients were female (Table2). The mean age was 49.2 years and dyspnea (93%) and cough (64%) were found present since 1.7 years (Table2). Moreover, 50% of the patients were suffering from clear expectorations and chronic fatigue (data not shown). There were 57% smokers with a mean of 14.5 pack years. Bronchoalveolar lavage showed a mild lymphocytosis (36%) (Table3).
Histologically, interstitial pneumonia without any diagnostic criteria of other pulmonary diseases was found. The alterations consisted primarily of a mild to moderate interstitial chronic inflammation, usually with lymphocytes and a few plasma cells. A mesh-like or bronchiolocentric fibrosis sometimes also with honeycombing, but without fibroblastic foci was demonstrated (Figure3). All cases showed fibrotic changes, however, a prominent inflammation (cellular pattern) could not be noted. NSIP patterns were also found in cases of EAA and interstitial pneumonias of other known causes and associations (Table1).

Non specific interstitial pneumonia . A mesh-like fibrosis of the alveolar septa is seen. Fibroblastic foci are not found. Aggregates of lymphocytes are sometimes found (HE, original magnification 40x).
Respiratory bronchiolitis with interstitial lung disease (RBILD)
Male gender was dominant, in this group (Table2). Beside dyspnea and cough, expectoration and fatigue were also present (both in 67% of patients). All patients were smokers with a mean of 27 pack years (Table2). Bronchoalveolar lavage showed a prevalence of macrophages (77%) (Table3).
Morphologically a bronchiolocentric aggregation of macrophages in combination with slight fibrosis and some interstitial infiltrates were seen (Figure4). Respiratory bronchiolitis was also found in EAA and LH, which could be clarified after immunohistochemistry or case evaluation (Table1).

Respiratory bronchiolitis with interstiatial lung disease . In RBILD bronchiolocentric aggregates of macrophages and an associated interstitial fibrosis are seen (HE, original magnification 40x).
Desquamative interstitial pneumonia (DIP)
Patients were mostly male (5/6), the mean age was 50.5 years (Table2). Two patients were active smokers with a mean consumption of 8 pack years. Bronchoalveolar lavage showed an increase of neutrophils (34%) and eosinophils (4%).
Biopsies revealed a massive intraalveolar aggregation of macrophages. Besides a low grade interstitial fibrosis without honeycombing, some lymphocytes were seen (Figure5). All cases were diagnosed already without clinical data (Table1).

Desquamative interstitial pneumonia . A pronounced diffuse intraalveolar aggregation of macrophages and an interstitial fibrosis can be found in DIP (HE. original magnification 40x).
Cryptogenic organizing pneumonia (COP)
Patients were mainly male and smokers, with a mean of 21.4 pack years (Table2). Half of the patients had a history of pneumonia (data not shown). In the BAL fluid, lymphocytosis was seen (40%) (Table3).
Morphologically granulation tissue was found within the bronchioles, alveolar ducts and the alveolar spaces, which was accompanied by an interstitial inflammatory reaction (Figure6). Organizing pneumonia (OP) was also seen in cases of NSIP and interstitial pneumonias of other known causes and associations, which caused initial misdiagnoses before case evaluation (Table1).

Cryptogenic organizing pneumonia. In alveolar ducts and bronchioles buds of granulation tissue are detected (HE, original magnification 100x).
Lymphocytic interstitial pneumonia (LIP)
All patients (n = 3) were male and in the mean 55.9 years old. There were no smokers among the patients (Table2). In BAL, lymphocytosis was seen (45.5%).
Microscopically, a dense lympho-plasmacellular infiltration with germinal centres but without blast proliferations were seen (Figure7). There were no findings suggesting mucosa associated lymphoma. No evidence of clinical or subclinical connective tissue diseases or immunodeficiency was given in these patients. All cases could already be diagnosed without any clinical information (Table1). Because of the small number of patients enrolled, no statistical calculation could be performed.

Lymphocytic interstitial pneumonia . Prominent infiltrates of lymphocytes and plasmacells and some germinal centers are seen (HE, original magnification 40x).
Non idiopathic interstitial pneumonia
25 patients (28%) had interstitial pneumonia of non idiopathic origin. Among these were 10 cases of hypersensitivity pneumonitis (11%), 7 of Langerhans cell histiocytosis (8%), and 8 of interstitial pneumonias of other known causes and associations (9%). Non idiopathic interstitial pneumonias could not be diagnosed without interdisciplinary case evaluation (first step: 12% vs. second step: 44% correctness).
Hypersensitivity pneumonitis (HP)
There was a prevalence of females in this group (Table2). Dyspnea was seen in all patients; cough (80%) and expectorations (70%) were also reported (Table2). All patients were positive for precipitins to several different avian serum antigens (data not shown). In the BAL 60% lymphocytes, 33% macrophages, 5.5% neutrophils, 1.3% eosinophils and 0.2% mast cells were seen (Table3).
Cases were classified according to dominating pattern as UIP (n = 2), RB (n = 2), NSIP (n = 5) and OP (n = 1). In three cases histiocytic aggregates and granulomas could be found in the second look, which allowed histopathological diagnosis of HP (Figure8, Table1). Only case specific evaluation allowed diagnosis in all patients.

Hypersensitivity pneumonitis. Interstitial infiltrates of lymphocytes and plasmacells and some aggregates of histiocytic cells are found (HE, original magnification 100x).
Langerhans cell histiocytosis (LCH)
These patients were younger than those with IPF, NSIP, and the other ILD (p < 0.05) and were 100% smokers, with a mean consumption of 22.5 pack years. In the BAL 4.5% lymphocytes, 86% macrophages, 7.5% neutrophils, 1.5% eosinophils and 0.5% mast cells were seen (Table3).
Initially without clinical information four cases were diagnosed as RBILD (Table1). Morphologically definitive diagnosis could be made in all cases by immunohistochemical demonstration of Langerhans cells (positive reaction for CD1a and S100 protein, Figures9,10).

Langerhans cell histiocytosis . Patchy infiltrates of histiocytes, lymphocytes and plasmacells are seen (HE, original magnification 100x).

Immunohistochemistry of Langerhans cell histiocytosis. Aggregates of Langerhans cells are detectable by monoclonal antibodies against CD1a (ABC-method, original magnification 200x).
Interstitial pneumonias of other known causes and associations
There were 8 patients in this group, mostly male (Table2), their mean age was 55.4 years. Dyspnea was present in all patients, and cough in 62.5% and fatigue in 62.5%. Most patients were smokers (62.5%, mean 13.1 pack years).
Morphologically OP (n = 3) and NSIP-like (n = 2), UIP-like (n = 2) or DAD-like (n = 1) patterns were seen. OP was found in IP with concomitant giant bullous emphysema, colitis ulcerosa and alcoholic cirrhosis. NSIP-like histology was seen in asbestosis and bone marrow transplantation, UIP-like changes in Jo-1 syndrome and after ACE inhibitor therapy. A DAD-like pattern was found in a patient with rheumatoid arthritis (Table4).
Asbestosis was diagnosed after detection of asbestos body fragments in the tissue and of asbestos bodies in BAL. But all other cases were only detected after case specific evaluation (Table1).
Discussion
In general, the diagnostic criteria defined by the ATS/ERS classification of IP were applicable in all cases. Distributions of subgroups of IIP in this study were in accordance with the existing literature (Table5). Some small differences might be explained by selection of patients for open lung biopsies. The number of LIP cases seems to be higher than in other studies, where it is described as rare disease with a frequency below 2%[16, 17].
With regards to demographics and patients’ characteristics, male predominance in IPF was also found in other studies[22–25], and also in RBILD and DIP[26–28] as well as in COP[29–32]. Concerning NSIP, some studies found a female predominance[33, 34], while others noted a male predominance[18, 25, 35]. In comparison to other studies the clinical history, main clinical symptoms and the rate of smokers in the different subgroups of IP showed only small deviations (Table6).
Differences in the three different diagnostic steps
Histological patterns alone agreed with the final diagnosis in 67% of the cases. In 82% histology and clinical information could provide correct clinical diagnosis compared with final case evaluation. This provides a significant improvement of correct diagnosis, but only interdisciplinary case evaluation clarified all cases adequately. Strong distinctions are found in the different diseases.
Diagnosis of IPF/UIP based on the histological pattern alone was withdrawn in 2 cases after receiving clinical information and in further 2 cases after interdisciplinary case evaluation. UIP pattern is compatible with the clinical diagnosis of IPF if other etiological factors can be excluded. A questionnaire of 11 centres for pulmonary fibrosis published in Germany 2003, involving 62 patients revealed that IPF had been diagnosed usually 21 months after the beginning of symptoms[40]. Open lung biopsies were taken in 34%, high resolution CT was done in 71% and precipitins were analysed only in 33%. These data point to a relatively poor standard in the diagnosis of IPF and excluding interstitial pneumonias of known origin and association with UIP pattern before the publication of the ATS/ERS statement. Limitations of the clinical criteria for diagnosis of IPF have been demonstrated in other studies[41]. In HRCT honeycombing was seen in 44.4% of our cases with IPF. According to the actual guideline for IPF OLB is no longer necessary in cases with proven IPF in the appropriate clinical and radiological setting (HRCT with UIP pattern). Therefore OLB will now only be done in cases with possible IPF by history and HRCT[8].
RBILD was overdiagnosed by histology plus clinical information compared with final evaluation (10 vs. 6 cases). A variable amount of RB can be found in smokers, which might explain overdiagnosis in those cases. Also in smokers with Langerhans cell histiocytosis RB is usually seen.
Idiopathic NSIP was overdiagnosed by histology plus clinical information compared with final evaluation (20 vs. 16 cases). Diagnosis of non-specific changes is difficult, therefore further evaluation is particularly crucial.
COP was nearly correctly diagnosed by histology plus clinical information compared with final evaluation (12 vs. 13 cases). In one case diagnosis was withdrawn in step 2 because changes were no longer evaluated as adequately expressed. The diagnosis of OP can be difficult if changes are only seen in small parts of the tissue. In this entity, correlation with radiomorphology (patchy consolidation) is essential for the correct diagnosis.
Microscopical diagnosis of DIP and LIP was done without problems and remained the final diagnosis in all steps. The DAD pattern could be diagnosed histologically as well, but proved to be associated with rheumatoid arthritis after case evaluation.
Diagnosis of non IIP without clinical information is difficult. Only 3 of 7 cases with LCH were diagnosed by histology alone. In clinically suspected LCH immunohistochemistry has to be done, which achieved correct diagnosis in all cases. In our cases HP was not suspected by histology alone. With clinical information, histological features of HP were recognized in 3 of 10 patients. In chronic HP a variety of changes ranging from UIP or NSIP patterns to OP is known[42]. In the smoking related diseases RBILD and LCH an overlap of patterns is seen regularly, which is reported in the literature as well[43].
Interstitial pneumonias of other known causes and associations are characterized by morphological patterns also seen in IIP, the only difference is the detection of a known etiological factor[44]. Without carefully evaluated clinical history and laboratory examinations these diseases cannot be diagnosed.
With regards to the BAL findings, we found that differential cytology is of additional value in the diagnostic process. This was also shown in other studies (Table7). Highest degree of lymphocytosis in IIP is seen in COP. A percentage of over 60% is suggestive for HP. Entities like bronchiolocentric interstitial pneumonia[45], idiopathic bronchiolitis[46–48] or inhaled drug induced ILD[49], have to be excluded histologically and clinically. Further aspects in diagnosis, prognosis and therapy of IP will be offered by systemic biology[50], surfactant expression and immunohistochemistry[51, 52], and genetics[53].
Open lung biopsies are only undertaken in otherwise unclear cases. Histology may show only non characteristic features. Case specific evaluation of clinical and pathological data is therefore a necessity for the exact diagnosis of IP, which was demonstrated by other studies[10, 13] and recommended by other experts[17]. Major limitations of our study were the incompleteness of reviewable HRCT data for all patients and the lack of a second pathologist to calculate the level of agreement.
Conclusions
Diagnosis of interstitial pneumonias, a group of quite rare diseases, requires sufficient clinical information and the knowledge of possible clinical, radiological and histological patterns.
An interdisciplinary case evaluation that includes at least one clinical expert and one pathologist with excellent expertise and the follow-up data of the patients is necessary to find correct diagnoses.
Abbreviations
- ATS:
-
American thoracic society
- COP:
-
Cryptogenic organizing pneumonia
- CT:
-
Computer Tomogram
- CXR:
-
Chest X rays
- DAD:
-
Diffuse alveolar damage
- DIP:
-
Diffuse interstitial pneumonia
- ERS:
-
European respiratory society
- FEV1:
-
Forced expiratory volume in 1 second
- FVC:
-
Forced vital capacity
- HP:
-
Hypersensitivity pneumonia
- HRCT:
-
High resolution computer tomography
- IP:
-
Interstitial pneumonia
- IIP:
-
Idiopathic interstitial pneumonia
- ILD:
-
Interstitial lung disease
- IPF:
-
Idiopathic pulmonary fibrosis
- LCH:
-
Langerhans cell histiocytosis
- LIP:
-
Lymphocytic interstitial pneumonia
- NSIP:
-
Nonspecific interstitial pneumonia
- OLB:
-
Open lung biopsy
- OP:
-
Organizing pneumonia
- UIP:
-
Usual interstitial pneumonia
- TLC:
-
Total lung capacity.
References
Homolka J: Idiopathic pulmonary fibrosis: a historical review. CMAJ. 1987, 137: 1003-1005.
Liebow AA: Definition and classification of interstitial pneumonias in human pathology. Prog Respir Res. 1975, 8: 1-31.
Kitaichi M: Pathologic features and the classification of interstitial pneumonia of unknown etiology. Bull Chest Dis Res Inst Kyoto Univ. 1990, 23: 1-18.
Katzenstein AL: Idiopathic interstitial pneumonia: classification and diagnosis. Monogr Pathol. 1993, 36: 1-31.
American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus: Classification of the Idiopathic Interstitial Pneumonias. Am J Respir Crit Care Med. 2002, 165: 277-304.
Swigris JJ, Berry GJ, Raffin TA, Kuschner WG: Lymphoid interstitial pneumonia: a narrative review. Chest. 2002, 122: 2150-2164. 10.1378/chest.122.6.2150.
Terada T: Follicular bronchiolitis and lymphocytic interstitial pneumonia in a Japanese man. Diagn Pathol. 2011, 6: 85-10.1186/1746-1596-6-85.
Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE, Kondoh Y, Myers J, Muller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schunemann HJ: An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med. 2011, 183: 788-824. 10.1164/rccm.2009-040GL.
Lunardi F, Balestro E, Nordio B, Cozzi F, Polverosi R, Sfriso P, Braccioni F, Calabrese F: Undifferentiated connective tissue disease presenting with prevalent interstitial lung disease: case report and review of literature. Diagn Pathol. 2011, 6: 50-10.1186/1746-1596-6-50.
Fishbein MC: Diagnosis: to biopsy or not to biopsy: assessing the role of surgical lung biopsy in the diagnosis of idiopathic pulmonary fibrosis. Chest. 2005, 128: 520S-525S. 10.1378/chest.128.5_suppl_1.520S.
Hunninghake GW, Zimmerman MB, Schwartz DA, King TE, Lynch J, Hegele R, Waldron J, Colby T, Muller N, Lynch D, Galvin J, Gross B, Hogg J, Toews G, Helmers R, Cooper JA, Baughman R, Strange C, Millard M: Utility of a lung biopsy for the diagnosis of idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2001, 164: 193-196.
Nicholson AG, Addis BJ, Bharucha H, Clelland CA, Corrin B, Gibbs AR, Hasleton PS, Kerr KM, Ibrahim NB, Stewart S, Wallace WA, Wells AU: Inter-observer variation between pathologists in diffuse parenchymal lung disease. Thorax. 2004, 59: 500-505. 10.1136/thx.2003.011734.
Thomeer M, Demedts M, Behr J, Buhl R, Costabel U, Flower CD, Verschakelen J, Laurent F, Nicholson AG, Verbeken EK, Capron F, Sardina M, Corvasce G, Lankhorst I: Multidisciplinary interobserver agreement in the diagnosis of idiopathic pulmonary fibrosis. Eur Respir J. 2008, 31: 585-591. 10.1183/09031936.00063706.
Lettieri CJ, Veerappan GR, Parker JM, Franks TJ, Hayden D, Travis WD, Shorr AF: Discordance between general and pulmonary pathologists in the diagnosis of interstitial lung disease. Respir Med. 2005, 99: 1425-1430. 10.1016/j.rmed.2005.03.008.
Technical recommendations and guidelines for bronchoalveolar lavage (BAL): Report of the European Society of Pneumology Task Group. Eur Respir J. 1989, 2: 561-585.
Kim DS, Collard HR, King TE: Classification and natural history of the idiopathic interstitial pneumonias. Proc Am Thorac Soc. 2006, 3: 285-292. 10.1513/pats.200601-005TK.
Larsen BT, Colby TV: Update for pathologists on idiopathic interstitial pneumonias. Arch Pathol Lab Med. 2012, 136: 1234-1241. 10.5858/arpa.2012-0225-RA.
Bjoraker JA, Ryu JH, Edwin MK, Myers JL, Tazelaar HD, Schroeder DR, Offord KP: Prognostic significance of histopathologic subsets in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 1998, 157: 199-203.
Nagai S, Kitaichi M, Itoh H, Nishimura K, Izumi T, Colby TV: Idiopathic nonspecific interstitial pneumonia/fibrosis: comparison with idiopathic pulmonary fibrosis and BOOP. Eur Respir J. 1998, 12: 1010-1019. 10.1183/09031936.98.12051010.
Travis WD, Matsui K, Moss J, Ferrans VJ: Idiopathic nonspecific interstitial pneumonia: prognostic significance of cellular and fibrosing patterns: survival comparison with usual interstitial pneumonia and desquamative interstitial pneumonia. Am J Surg Pathol. 2000, 24: 19-33. 10.1097/00000478-200001000-00003.
Nicholson AG, Colby TV, du Bois RM, Hansell DM, Wells AU: The prognostic significance of the histologic pattern of interstitial pneumonia in patients presenting with the clinical entity of cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med. 2000, 162: 2213-2217.
Monaghan H, Wells AU, Colby TV, du Bois RM, Hansell DM, Nicholson AG: Prognostic implications of histologic patterns in multiple surgical lung biopsies from patients with idiopathic interstitial pneumonias. Chest. 2004, 125: 522-526. 10.1378/chest.125.2.522.
Carrington CB, Gaensler EA, Coutu RE, FitzGerald MX, Gupta RG: Natural history and treated course of usual and desquamative interstitial pneumonia. N Engl J Med. 1978, 298: 801-809. 10.1056/NEJM197804132981501.
Matsuo K, Tada S, Shibayama T, Ueno Y, Miyake T, Takehara H, Kataoka M, Harada M, Kimura I: Factors affecting prognosis of idiopathic interstitial pneumonia. Acta Med Okayama. 1996, 50: 37-46.
Daniil ZD, Gilchrist FC, Nicholson AG, Hansell DM, Harris J, Colby TV, du Bois RM: A histologic pattern of nonspecific interstitial pneumonia is associated with a better prognosis than usual interstitial pneumonia in patients with cryptogenic fibrosing alveolitis. Am J Respir Crit Care Med. 1999, 160: 899-905.
Myers JL, Veal CF, Shin MS, Katzenstein AL: Respiratory bronchiolitis causing interstitial lung disease. A clinicopathologic study of six cases. Am Rev Respir Dis. 1987, 135: 880-884.
Yousem SA, Colby TV, Gaensler EA: Respiratory bronchiolitis-associated interstitial lung disease and its relationship to desquamative interstitial pneumonia. Mayo Clin Proc. 1989, 64: 1373-1380.
Canessa PA, Prattico L, Bancalari L, Fedeli F, Bacigalupo B, Silvano S: Respiratory bronchiolitis associated with interstitial lung disease. Monaldi Arch Chest Dis. 2004, 61: 174-176.
Guerry-Force ML, Muller NL, Wright JL, Wiggs B, Coppin C, Pare PD, Hogg JC: A comparison of bronchiolitis obliterans with organizing pneumonia, usual interstitial pneumonia, and small airways disease. Am Rev Respir Dis. 1987, 135: 705-712.
Costabel U, Teschler H, Guzman J: Bronchiolitis obliterans organizing pneumonia (BOOP): the cytological and immunocytological profile of bronchoalveolar lavage. Eur Respir J. 1992, 5: 791-797.
Muller NL, Guerry-Force ML, Staples CA, Wright JL, Wiggs B, Coppin C, Pare P, Hogg JC: Differential diagnosis of bronchiolitis obliterans with organizing pneumonia and usual interstitial pneumonia: clinical, functional, and radiologic findings. Radiology. 1987, 162: 151-156.
King TE, Mortenson RL: Cryptogenic organizing pneumonitis. The North American experience. Chest. 1992, 102: 8S-13S.
Katzenstein AL, Fiorelli RF: Nonspecific interstitial pneumonia/fibrosis. Histologic features and clinical significance. Am J Surg Pathol. 1994, 18: 136-147. 10.1097/00000478-199402000-00003.
Park CS, Jeon JW, Park SW, Lim GI, Jeong SH, Uh ST, Park JS, Choi DL, Jin SY, Kang CH: Nonspecific interstitial pneumonia/fibrosis: clinical manifestations, histologic and radiologic features. Korean J Intern Med. 1996, 11: 122-132.
Cottin V, Donsbeck AV, Revel D, Loire R, Cordier JF: Nonspecific interstitial pneumonia. Individualization of a clinicopathologic entity in a series of 12 patients. Am J Respir Crit Care Med. 1998, 158: 1286-1293.
Meyers JL: Respiratory bronchiolitis with interstitial lung disease. Sem Respir Med. 1992, 13: 134-139. 10.1055/s-2007-1006265.
Moon J, du Bois RM, Colby TV, Hansell DM, Nicholson AG: Clinical significance of respiratory bronchiolitis on open lung biopsy and its relationship to smoking related interstitial lung disease. Thorax. 1999, 54: 1009-1014. 10.1136/thx.54.11.1009.
Costabel U: BOOP and COP. Thorax. 1992, 47: 207-
Izumi T, Kitaichi M, Nishimura K, Nagai S: Bronchiolitis obliterans organizing pneumonia. Clinical features and differential diagnosis. Chest. 1992, 102: 715-719. 10.1378/chest.102.3.715.
Günther A, Ermert L, Breithecker A, Hackstein N, Eickelberg O, Morr H, Grimminger F, Velcovsky H-G, Seeger W: Klassifikation, Diagnostik und Therapie der idiopathischen interstitiellen Pneumonien: Eine kritische Bestandsaufnahme der gegenwärtig in Deutschland geübten Praxis. Dtsch Arztebl. 2003, 100: 1676-1685.
Aziz ZA, Wells AU, Hansell DM, Bain GA, Copley SJ, Desai SR, Ellis SM, Gleeson FV, Grubnic S, Nicholson AG, Padley SP, Pointon KS, Reynolds JH, Robertson RJ, Rubens MB: HRCT diagnosis of diffuse parenchymal lung disease: inter-observer variation. Thorax. 2004, 59: 506-511. 10.1136/thx.2003.020396.
Churg A, Muller NL, Flint J, Wright JL: Chronic hypersensitivity pneumonitis. Am J Surg Pathol. 2006, 30: 201-208. 10.1097/01.pas.0000184806.38037.3c.
Caminati A, Harari S: Smoking-related interstitial pneumonias and pulmonary Langerhans cell histiocytosis. Proc Am Thorac Soc. 2006, 3: 299-306. 10.1513/pats.200512-135TK.
Camus P, Fanton A, Bonniaud P, Camus C, Foucher P: Interstitial lung disease induced by drugs and radiation. Respiration. 2004, 71: 301-326. 10.1159/000079633.
Yousem SA, Dacic S: Idiopathic bronchiolocentric interstitial pneumonia. Mod Pathol. 2002, 15: 1148-1153. 10.1097/01.MP.0000037309.04985.B4.
Muller NL, Miller RR: Diseases of the bronchioles: CT and histopathologic findings. Radiology. 1995, 196: 3-12.
Epler GR: Heterogeneity of bronchiolitis obliterans organizing pneumonia. Curr Opin Pulm Med. 1998, 4: 93-97. 10.1097/00063198-199803000-00006.
Epler GR: Diseases of the bronchioles. 1994, New York: Raven
Scheel AH, Krause D, Haars H, Schmitz I, Junker K: Talcum induced pneumoconiosis following inhalation of adulterated marijuana, a case report. Diagn Pathol. 2012, 7: 26-10.1186/1746-1596-7-26.
Peckham RM, Shorr AF, Helman DL: Potential limitations of clinical criteria for the diagnosis of idiopathic pulmonary fibrosis/cryptogenic fibrosing alveolitis. Respiration. 2004, 71: 165-169. 10.1159/000076678.
Nagata N, Kitasato Y, Wakamatsu K, Kawabata M, Fukushima K, Kajiki A, Kitahara Y, Watanabe K: Prognostic value of immunohistochemical surfactant protein A expression in regenerative/hyperplastic alveolar epithelial cells in idiopathic interstitial pneumonias. Diagn Pathol. 2011, 6: 25-10.1186/1746-1596-6-25.
Goldmann T, Kahler D, Schultz H, Abdullah M, Lang DS, Stellmacher F, Vollmer E: On the significance of Surfactant Protein-A within the human lungs. Diagn Pathol. 2009, 4: 8-10.1186/1746-1596-4-8.
Studer SM, Kaminski N: Towards systems biology of human pulmonary fibrosis. Proc Am Thorac Soc. 2007, 4: 85-91. 10.1513/pats.200607-139JG.
Haslam PL, Turton CW, Heard B, Lukoszek A, Collins JV, Salsbury AJ, Turner-Warwick M: Bronchoalveolar lavage in pulmonary fibrosis: comparison of cells obtained with lung biopsy and clinical features. Thorax. 1980, 35: 9-18. 10.1136/thx.35.1.9.
Shindoh Y, Shimura S, Tomioka M, Aikawa T, Sasaki H, Takishima T: Cellular analysis in bronchoalveolar lavage fluids in infiltrative and fibrotic stages of idiopathic pulmonary fibrosis. Tohoku J Exp Med. 1986, 149: 47-60. 10.1620/tjem.149.47.
Nagai S, Izumi T: Bronchiolitis obliterans with organizing pneumonia. Curr Opin Pulm Med. 1996, 2: 419-423. 10.1097/00063198-199609000-00012.
Acknowledgements
We thank the formerly head of the Department of Pathology at the Ruhr-University Bochum Prof. Dr. K. Morgenroth for making this study possible.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
DT designed the study, did all histological investigations (first step and second step) and wrote the manuscript. HM collected data from all patients and did statistical analysis. FB revised statistical analysis and the manuscript. JW reviewed the manuscript from the view of pathology. UC did review of clinical and radiological data and suggested final diagnosis. Interdisciplinary case evaluation was done by DT and UC. All authors revised the final manuscript and gave their consent for publication.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Theegarten, D., Müller, H.M., Bonella, F. et al. Diagnostic approach to interstitial pneumonias in a single centre: report on 88 cases. Diagn Pathol 7, 160 (2012). https://doi.org/10.1186/1746-1596-7-160
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1746-1596-7-160