
Abstract
Background
Dengue fever is perhaps the most important viral re-emergent disease especially in tropical and sub-tropical countries, affecting about 50 million people around the world yearly. In Colombia, dengue virus was first detected in 1971 and still remains as a major public health issue. Although four viral serotypes have been recurrently identified, dengue virus type 2 (DENV-2) has been involved in the most important outbreaks during the last 20 years, including 2010 when the fatality rate highly increased. As there are no major studies reviewing virus origin and genotype distribution in this country, the present study attempts to reconstruct the phylogenetic history of DENV-2 using a sequence analysis from a 224 bp PCR-amplified product corresponding to the carboxyl terminus of the envelope (E) gene from 48 Colombian isolates.
Results
As expected, the oldest isolates belonged to the American genotype (subtype V), but the strains collected since 1990 represent the American/Asian genotype (subtype IIIb) as previously reported in different American countries. Interestingly, the introduction of this genotype coincides with the first report of dengue hemorrhagic fever in Colombia at the end of 1989 and the increase of cases during the next years.
Conclusion
After replacement of the American genotype, several lineages of American/Asian subtype have rapidly spread all over the country evolving in new clades. Nevertheless, the direct association of these new variants in the raise of lethality rate observed during the last outbreak has to be demonstrated.
Similar content being viewed by others


Background
During the last few decades, the whole world has faced the re-emerging of different infectious diseases, being dengue one of the most important in terms of morbidity and mortality [1–5]. Dengue virus (DENV) is an arbovirus belonging to family flaviviridae and is responsible of a wide range of clinical manifestations in humans, including an acute self-limited flu-like illness known as dengue fever (DF) or a severe illness known as dengue hemorrhagic fever (DHF) characterized by a marked plasma leakage, which may progress to hypovolemic shock (dengue shock syndrome, DSS) with circulatory failure [1, 3, 4, 6–8]. Nevertheless, changes observed in clinical manifestations (in terms of severity) during the few last years have obliged to redefine this classification according to the presence of alarm signs [4].
As usual in flavivirus, DENV is an enveloped virus with a positive sense ssRNA of about 11 kb coding a single open reading frame for three structural proteins, core (C), membrane (M) and envelope (E), and seven non-structural proteins (NS1, NS2A, NS2B, NS3, NS4A, NS4B, NS5). Based on serological analysis, DENV can be differentiated as four distinct serotypes (DENV-1, DENV-2, DENV-3 and DENV-4), which are capable of causing the disease. Although the exact mechanisms for DHF remains unclear, it is generally accepted that a secondary infection with a heterologous serotype increases the risk of severe manifestations as a consequence of the antibody dependent enhancement (ADE) mechanism proposed by Halsted in the 80's [7]. However, an alternative explanation to patho-physiology of DHF is the emergence and spread of virulent strains originated as part of an evolutionary process [2, 9, 10]. In fact, molecular epidemiology using nucleotide sequence analysis from the DENV genome has demonstrated the occurrence of genotype clades within each serotype [11–29], which highlights the important role of DENV itself in disease severity rather than immune enhancement [2, 9, 10].
The four serotypes of DENV have been circulating in the Americas since the early 1900's, generating only slight cases of DF and sporadic cases of severe disease [1, 5, 6, 30]. It was not until 1981 when the first large epidemic of DHF occurred in Cuba and rapidly spread to Jamaica (1981-1982), Brazil (1986), and Venezuela (1989-1990) [1, 2, 5, 8, 9, 12, 23]. In Colombia, the first case of DHF was officially notified in December of 1989 from the village of Puerto Berrio (Antioquia department) [25, 31]. Since then, DHF became endemic and lethal cases rapidly increased during the next years. Although co-circulation of serotypes was common in different countries, samples from these major outbreaks confirmed DENV-2 as the main responsible of DHF cases. In 1997, Rico-Hesse et al., demonstrated that DENV-2 isolated from DHF outbreaks in Jamaica and the Caribbean islands (and possibly Cuba) in 1981-1982 belonged to a new clade formerly named "Asian genotype", probably introduced from South Asia, where severe infection has been persistent since the middle of the past century [2, 23]. To date, DENV-2 falls into seven subtypes (or genotypes) designed as Subtype I (Asian II), Subtype II, Subtype IIIa (Asian I), Subtype IIIb (American/Asian), Subtype IV (Cosmopolitan), Subtype V (American) and Sylvatic genotype [2, 12, 13]. Additionally, the existence of clades with distinctive geographical and temporal relationships has been suggested [23].
Historically, Colombia has been one of the most affected countries in the Americas with dengue epidemics [6, 30, 31]. In fact, during the last year it went through the largest dengue epidemic occurred in decades, with 157,152 cases notified (mostly DENV-2) and 217 deaths confirmed [32]. Nevertheless, there are no major studies regarding DENV-2 phylogenetic origin or genotype circulation and distribution [25]. Consequently, the present study tries to reconstruct phylogenetics of DENV-2 virus that has been circulating in Colombia during the 1980's in comparisons to the strains isolated since the emergence of DHF. In addition, this work describes the evolution of new clades during the last decade based on a partial nucleotide sequence of the envelope (E) gene.
Results
Virus recovery and confirmation
Forty-eight viruses obtained from symptomatic patients were isolated in mosquito cell culture and subsequently identified as DENV-2 serotype by monoclonal antibodies and confirmed by RT-PCR methods [33]. Isolates are listed in Table 1 indicating locality, isolation year, genotype and accession number.
Phylogenetic reconstruction of DENV-2
Sequences from the carboxyl terminus of the envelope (E) gene from the 48 Colombian DENV-2 isolates were aligned in CLUSTAL W [34, 35] and compared with 28 previously reported sequences elsewhere, resulting in a trivial alignment as long as there were not insertions or deletions (INDELS). Although the use of the whole E protein gene is highly recommended to reconstruct DENV phylogenies, the 224 bp sequence used in our study has been demonstrated to be useful to infer phylogenetic relationships while the topology is fully maintained [26, 27, 29].
The Maximum Likelihood (ML) approach using only Colombian isolates and one sylvatic strain to root the tree (Figure 1), clearly shows two major clades, one involved mainly viruses isolated between years 1982 to 1988 and the other one mostly viruses isolated since 1990.

Evolutionary relationships of Colombian DENV-2. Phylogenetic tree of 52 DENV-2 Colombian isolates was computed using the Maximum Likelihood method. Nodes with significant Neighbor- Joining bootstrap values (≥ 50) are indicated. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. Two well defined clades are shown in red and blue. The tree is rooted using one Sylvatic sequence.
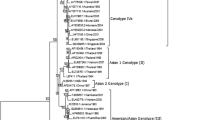
In order to associate those clades, the ML tree was reconstructed using all previously defined DENV-2 subtypes [Subtype I (Asian II), Subtype II, Subtype IIIa (Asian I), Subtype IIIb (American/Asian), Subtype IV (Cosmopolitan), Subtype V (American) and Sylvatic genotype] (Figure 2). According to the Akaike Information Criterion (AIC), the model that better fitted the data was the Tamura-Nei equal transversion frequencies (TrNef) model. Fourteen Colombian isolates (13 reported for the first time and 1 previously described) grouped into the Subtype V clade (American genotype) close to the oldest strains from Tahiti (DENV-2/PF/Tahiti/1971; DENV-2/PF/Tahiti/1973), Trinidad (DENV-2/TT/257/1972; DENV-2/TT/565/1972; DENV-2/TT/572_CDC/1983) and the latest detected in Central (DENV-2/HN/1991; DENV-2/CR/CRA_94/1994; DENV-2/CR/CRB_94/1994) and North America (DENV-2/MX/1995). Thirteen out of these Colombian viruses were isolated between 1982 and 1988. Interestingly, although the most recent detection of American genotype was in Peru in 1996, we found one virus isolated in 2002 (DENV-2/CO/355_Guaviare/2002) belonging to this subtype. To discard cross contamination, RNA of the sample was newly obtained and re-amplified.

Evolutionary relationships of DENV-2. Phylogenetic tree of 52 DENV-2 Colombian isolates and 24 worldwide previously reported was computed using the Maximum Likelihood method. Nodes with significant Neighbor-Joining bootstrap values (≥ 5)are indicated. The tree is drawn to scale, with branch lengths in the same units as those of the evolutionary distances used to infer the phylogenetic tree. American genotype (Subtype V) and Colombian isolates belonging to this clade are show in red. American/Asian genotype (Subtype IIIb) and Colombian isolates belonging to this clade are show in blue. The tree is rooted using one sylvatic sequence.
On the other hand, 35 Colombian sequences (34 novel and 1 previously reported) isolated between 1992 and 2010 belong to Subtype IIIb (American/Asian genotype) near to DENV-2/JM/Jamaica/1983, the putative first virus of this genotype introduced into the Americas. Subtype IIIb is divided in two clades, one representing the Asian viruses and the other one comprising the American isolates. Interestingly, one Colombian strain (DENV-2/CO/351861_Bolivar/1988) grouped into the Asian clade, separated from the remaining isolates. In order to confirm this result, RNA was extracted and amplified again. Once reconstructed, the tree showed the same topology. Nevertheless, the bootstrap support for this node is under 50%.
Two isolates from 1961 and 1971 (DENV-2/CO/V-163/1961; DENV-2/CO/256/1971) match to the 1944 New Guinea strain (Subtype I/Asian II genotype). However, it was not possible to establish the origin of those viruses.
The fragment analyzed in our study corresponded to the carboxyl terminus of envelope (E) gene, covering amino acid 422 to 495 of the E protein. As previously reported, all the American isolates have Valine (V) at the position 485, whereas Asian strains have Isoleucine at the same position [23]. On the other hand, all the sequences belonging to the Genotype IIIb have Valine at the position 484 and Alanine at the 491 (only one Colombian isolate has Valine at this position), while Genotype V isolates (all of them) have Isoleucine and Valine respectively.
Using Bayesian inference and according to the 95% highest posterior density (HPD) under the strict molecular clock model, the root of the tree including sylvatic strain is placed around 270 years ago and the substitution rate was 6.6 × 104 substitutions per site per year, close to the previously reported [20, 36–38]. Topology of the tree (Figure 3) shows two well supported clades representing American genotype (PP = 0.99) and the Asian/American genotype (PP = 0.99). Again, all but one (DENV-2/CO/355_Guaviare/2002) of the Colombian strains isolated between 1982 and 1988 fell into the subtype V, while those further isolated until 2010 get into the subtype IIIb. The phylogeny demonstrates a huge sustained spread of viruses all over the country, especially during the last 10 years. In fact since the year 2000, Colombian strains have been evolving in different clades, mostly clustered by the time of isolation. Interestingly, during the last epidemic in 2010, at least two different lineages had been circulating one in different localities and one almost exclusively at the Amazonas department.

Molecular Clock of DENV-2. DENV-2 divergence time was estimated using year of isolation (scale used in the tree) as calibration points under the strict molecular clock model using GTR + Γ + I parameters. Posterior Probability (PP) values are indicated for each node, and the blue bars represent the extent of the 95% highest posterior density (HPD) intervals for each divergence time. American genotype (Subtype V) and Colombian isolates belonging to this clade are show in red. American/Asian genotype (Subtype IIIb) and Colombian isolates belonging to this clade are show in blue.
Discussion
Between 1950 and 1960, the Pan American Health Organization (PAHO) Aedes aegypti eradication program to fight urban yellow fever was successful to suppress dengue transmission [5]. By the year 1952 Aedes aegypti was virtually eradicated from Colombia, and only few cases of Dengue were reported on the Magdalena valley [5, 30, 31]. Unfortunately, predictions made by Dr. Hernando Groot about the real impact of dengue in the Americas were ignored and the implementation of these eradication campaigns were abandoned by the late 60's and the subsequent decades, leading the mosquitoes to proliferate and spread all over the American continent [30]. Dengue syndrome re-emerged and rapidly became the most important infectious viral disease in the Americas [1, 4, 5, 30, 31]. Since then, all DENV serotypes have been detected, being DENV-2 perhaps the most important in terms of morbidity and mortality [1, 4, 5, 30, 31].
We have reconstructed the phylogenetic history of DENV-2 in Colombia and reported for the first time the distribution of genotypes across time. Large epidemics of DENV-2 were first occurred in the Caribbean Islands, starting in Trinidad & Tobago (1953), following by Curação and Haití (1968) [1, 5]. First outbreaks of DENV-2 reported in mainland, probably as a spillover from the islands, occurred in French Guiana (1970) and Colombia (1971) [1, 5, 30, 31]. For about 10 years, the virus was reported only in Colombia where it was generating DF until 1981, when this serotype was first reported in Cuba and Jamaica [1, 5, 8]. Our study clearly demonstrates that Colombian DENV-2 isolated up to 1988 belongs to a well supported clade, grouped with strains previously defined as Subtype V (American genotype) [2, 12, 13]. One of the most significant issues of dengue history in the Americas is perhaps the first DHF outbreak occurred simultaneously in Cuba and Jamaica in 1981 [1, 8, 23, 39, 40]. Further studies demonstrated that DENV-2 involved in this severe epidemic belonged to a different genotype very close to previously characterized Asian strains [2, 12, 23, 39, 40]. This new Asian-American virus (currently known as Subtype IIIb) generates a well supported clade, nested Jamaica strains above (DENV-2/JM/Jamaica/1983) and Vietnam and China as the origin of subclade (DENV-2/CN/1985; DENV-2/VN/CTD44/1988; DENV-2/VN/CTD28/1997). Thirty-five (35) out of the 36 Colombian viruses isolated after year 1990 fell into this clade, demonstrating the spread of the Asian- American genotype all over the country during the last 20 years. Interestingly, the introduction of this subtype clearly coincides with the first official report of DHF at the end of 1989 (Puerto Berrio, Antioquia) and the sustained increase of severe cases observed during the next years [30]. Two major explanations have been suggested for DHF to occur. The antibody dependent enhancement (ADE) theory proposes the rise of severity as a result of a secondary heterologous infection, essentially in hyperendemic areas [7]. However, in Colombia the 4 serotypes were circulating already (DENV-3 in 1975, DENV-1 since 1978, DENV-4 since 1983) and yet, there were not DHF cases reported even in those localities were co-circulation of at least 2 serotypes was noted. On the other hand, the sudden increase in DHF cases after the introduction of the Subtype IIIb in the Americas (probably in Jamaica in 1981) supports the idea of the emerging of virulent strains (hemorrhagic strains) and replace of the less aggressive native American genotypes [2, 9, 10, 12]. The marked split showed in our study between the isolates obtained before and after the appearance of DHF, clearly agrees with the second hypothesis, although the first one can explain the high incidence of severe dengue currently observed in some hyperendemic localities with co-circulation of serotypes others than DENV-2. In fact, during the last epidemic in Colombia (2010) DENV-1 and DENV-2 were isolated in high proportion equally in both DF and DHF cases but secondary infection was not demonstrated. Moreover, all four serotypes were detected in fatal cases, even though DENV-2 was the most frequent [32]. All together, these findings suggest that hyperendemicity summed to increased virulence are both decisive for DHF maintenance, more than two separate factors [2, 9, 10, 12].
The introduction of the DENV-2 American genotype in Colombia is easy to explain, considering that the A. aegypti eradication programs failed in the Caribbean coast, leading the mosquito spread from Maracaibo (Venezuela) to Maicao (La Guajira, Colombia) in 1968 [30, 31]. By the year 1971, the entire Colombian Atlantic coast was re-infested, including most of the important ports located in Barranquilla (Atlántico) and Cartagena (Bolivar), which maintained major commercial trades with the Caribbean islands where the virus was already established.
More difficult to explain is the replacement event of the American genotype by the Asian- American [2, 12]. Since the introduction of Subtype IIIb, American had been detected only in few cases during the middle 90's in Central America and as late as 1996 in Peru [41]. The replacement and extinction of genotypes have been described as a stochastic event occurring during periods of depletion in mosquitoes population or low number of susceptible hosts [20, 38, 42–46]. In the present study we found one virus isolated in 2002 (DENV-2/CO/355_Guaviare/2002) placed inside the Subtype V (American genotype), indicating perhaps that the genotype is not extinct. Although differences in fitness have not been surely demonstrated (see below), it is possible for the Asian genotypes to hold a higher transmission pattern, restricting the "native" virus to low circulation dynamics and probably causing only subclinical (undetectable) infections. On this matter, it is important to notice that the samples collected come from the surveillance system and belong to symptomatic patients. Therefore, the opportunity to isolate this genotype again is even lower.
There are two major pressures affecting DENV evolution process. One is the attachment to a susceptible cell, leading to entry by membrane fusion and the other is the host immune response [18, 20, 38, 47–51]. The envelope (E) protein is involved in both processes and therefore the most representative to infer adaptation patterns. In fact, Weaver et. al. had demonstrated the constrained effect occurring in virus obligated to alternate between invertebrate vector and vertebrate host [52, 53]. Nevertheless, this effect is possible reduced when transmission rates are very high (hyperendemic areas) in human hosts [29]. On the other hand, positive selection on some DENV-2 genotypes had been previously inferred into immunogenic zones of E protein, specifically in amino acids 91, 129, 131 and 491, indicating perhaps a way for immune response evasion [18, 20, 23, 49]. According to our results and as previously reported, all the American isolates have Valine at the position 485, whereas Asian strains have Isoleucine at the same position [23]. Interestingly, Valine at 484 and Alanine at 491 were conserved all over the Genotype IIIb, while Genotype V isolates have Isoleucine and Valine at the same positions clearly resembling the ancestral state observed in sylvatic strains (Malaysia, DENV-2/MY/Sylvatic/1970) [54]. Although the impact of this phenotypic change (if any) remains to be determined, but it strongly suggests a positive selection process acting over the E protein [49, 50].
Evolution dynamics of DENV-2 is affected by several factors. Because of the lack of proof-reading activity of RNA-dependent RNA-polymerase, RNA viruses usually present higher mutation rates than DNA viruses [47, 55–57]. Nevertheless, arboviruses (as mentioned above) are subject to a trade-off effect when they alternatively replicate in humans and mosquitoes [52, 53]. In fact, Holmes had demonstrated that arboviruses (in general) generate more deleterious mutation than other RNA viruses. [51]. As a consequence, susceptible human populations together with vector densities might lead different evolution patterns in distinct geographic areas. Colombia is perhaps one of the most highly endemic countries in the Americas region, with a current co-circulation of the four DENV serotypes and 75% of the territory having elevated rates of A. aegypti infestation [31, 32]. Moreover, by the year 2010, 157,152 cases of dengue were confirmed including 9.482 corresponding to DHF with 2.28% of lethality [32]. During this time, 662 viruses were isolated and 40.4% were identified as DENV-2. In spite of the constrained effect, our results of the Bayesian analysis clearly show an intense evolution process, supported by the different clades generated since the first circulation. According to the tree, Subtype IIIb Colombian isolates fall into at least 3 clades or "lineages" especially well defined after the year 2000. One clade put together most of the samples collected between 2001 and 2004. The second clade groups mainly viruses isolated in 2005 (DENV-2/CO/241_Guaviare/2005), 2007 (DENV-2/CO/V3374/2007) and 2010 (DENV-2/CO/410149_Cesar/2010; DENV-2/CO/408243_Risaralda/2010; DENV-2/CO/408339_Valle/2010; DENV-2/CO/408338_Valle/2010). Interestingly, there is a third clade clustering almost exclusively with strains from Amazonas department (only one from Santander) isolated during the last epidemic (2010). Until 2009, there was not dengue transmission in the Amazonas because of the small A. aegipty population, and only sporadic cases of imported infection had been notified [30]. Nevertheless, in 2010 the number of dengue patients significantly increased in Leticia, the capital city of Amazonas [32]. Epidemiological surveillance system let us confirm that most of that reported cases came from the neighbor Peruvian city of Iquitos, where a dengue outbreak was already taking place. Together, these results demonstrate the establishment and co-circulation of different lineages of the Asian/American genotype during the last decade, and the entrance of a new one during the last Colombian epidemic.
In conclusion, our phylogenetic reconstruction suggests the circulation of DENV-2 American Subtype V in Colombia for about 20 years, until the early 90's when the Asian/American Subtype IIIb replaced it. Although the first entry and subsequent establishment of this new genotype clearly coincide with the emerging and increase of severe DHF, there is no formal evidence of enhanced virulence on this genotype. On the other hand, during the last 20 years Subtype IIIb has been evolving locally and co-circulation of different clades is observed. In fact, introduction of a new "lineage" probably from Peru to the Colombian Amazon region is strongly supported. Even with the lack of viral pathogenic markers certainly documented, it is compelling that the clinical manifestation of dengue infection has changed. Atypical signs such as viscerotropism or encephalitis are becoming more recurrent and lethality rates are increasing in hyperendemic countries including Colombia. Therefore, control programs should include the surveillance of potentially pathogenic DENV genotypes together with mosquito control and people education campaigns.
Methods
Virus strains
DENV-2 strains used in this study were obtained from the virus collection of the National Health Institute (INS, Virology Lab, Bogotá, Colombia), and comprised 48 isolates from different outbreaks, epidemics and routine epidemiological surveillance. Clinical samples were collected between 1982 and 2010 from different localities all around the country, so they represent most viruses circulating in Colombia during the last 30 years (Table 1). All viral stocks were inoculated on C6/36 Aedes albopictus cells growing in Eagle's minimal essential medium (E-MEM) supplemented with 2% fetal calf serum (FSC). After 10 days of incubation at 28°C, monolayer was disrupted and supernatant was then recovered by centrifugation and stored at -80°C until use. The remained cells were washed with Phosphate Buffer Saline (PBS) and dripped on slides; after fixed in cool acetone, slides were incubated with monoclonal antibodies (anti-DENV-1 to anti-DENV-4, kindly donated by CDC, Puerto Rico) for one hour, washed with PBS and incubated again with a fluorescent conjugated antibody. Additionally, DENV-2 serotype confirmation was done by reverse transcription polymerase chain reaction (RT-PCR) using specific primers [33].
Viral RNA extraction, RT-PCR and sequencing
Cell culture supernatants were used to extract viral RNA using QIAamp Viral RNA Minikit (Qiagen, Germany) following manufacturer's instructions. Briefly, 140 μl of each supernatant was placed into 560 μl of AVL buffer with 5.6 μl of carrier RNA and mixed with ethanol (96-100%) before passed through a column by centrifugation. After washing with buffers AW1 and AW2 RNA was finally eluted with 60 μl of AVE buffer and stored at -80°C until use. Five micro liters from each RNA extraction were used as template in a one step RT-PCR reaction (Qiagen, One-Step RT-PCR kit) as previously described [33]. Primers used [DEN2S1871 (5'-TAGCAGAAACACARCATGGNAC-3') and DEN2AS2622 (5'-CAATTCTGGTGTTATTTGYTTCCAC-3')] were designated to amplify 751 bp from the joining region E/NS1. Reactions were evaluated in 1% agarose gels stained with ethidum bromide and negative reactions were subjected to nested PCR as previously reported [29, 33], using specific nested primers DEN2S2042 (5'-CAGTCAACATAGAAGCAGAACC-3') and DEN2AS2549 (5'-GCYGAAGCTAGTTTTGAAGGGG-3')]. Nested PCR was evaluated in 1% agarose gel stained with ethidium bromide.
Amplified products (from RT-PCR or nested PCR) were purified using QIAquick PCR Purification Kit (QIAGEN, Germany) and then used as template for sequencing reactions using the ABI Prism Dye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Foster City, CA) [29, 33]. A total of 224 bp [corresponding to carboxyl terminus of envelope (E) gene] from 48 new sequences were compared with 28 previously sequenced strains from all over the world, available in GenBank. Consensus sequences were aligned using the program CLUSTAL W included in MEGA package version 4.0 [34, 35].
Phylogenetic analyses
Phylogenetic trees were reconstructed with the Maximum Likelihood (ML) methods incorporated in the Paralleled and Integrated Framework for Phylogenetic Inference with Automatic Likelihood Model Selector (PALM) program, which combines Clustal W, PhyML, MODELTEST, ProtTest and others in one interface. [34, 35, 58–60]. Statistical significance of tree topology was assessed with a bootstraping with 1000 replicates. Obtained trees were visualized using the FigTree 1.2.2. program. All the ML and MODELTEST parameters obtained are available upon request.
Substitution rates and molecular clock
In addition, estimated rate of evolutionary change (nucleotide substitutions per site per year) and tree root age was obtained with the program BEAST (Bayesian Evolutionary Analysis by Sampling Trees) [61], which uses Bayesian Markov Chain Montecarlo (MCMC) algorithms combined with the chosen model and prior knowledge of sequence data to infer the posterior probability distribution of phylogenies [61–65]. We analyze the data using the year of isolation as calibration points to estimate divergence time in years. Rate variation among branches was inferred under the strict molecular clock model, whereas substitution rate among sites was calculated with the General Time-Reversible model (GTR) combined with the gamma parameter and proportion of invariant sites (GTR + Γ + I) model. MCMC was run for 10,000,000 steps and sampled every 500 steps and the 10,000 first steps of each run were discarded. BEAST format files were obtained in the provided BEAUti graphical interface and the trees were visualized with the FigTree 1.2.2. program. Finally, statistical analyze were carried out in the Tracer package [61].
References
Gubler D: Dengue and dengue hemorrhagic fever: its history and resurgence as a global public health problem. In Dengue and dengue hemorrhagic feve. Edited by: Gubler DJ, Kuno G. Oxford: CAB International; 1997.
Rico-Hesse R, Harrison LM, Salas RA, Tovar D, Nisalak A, Ramos C, Boshell J, de Mesa MT, Nogueira RMR, da Rosa AT: Origins of dengue type 2 viruses associated with increased pathogenicity in the Americas. Virology 1997, 230: 244-251. 10.1006/viro.1997.8504
McBride WJ, Bielefeldt-Ohmann H: Dengue viral infections: pathogenesis and epidemiology. Microbes Infect 2000, 2: 1041-1050. 10.1016/S1286-4579(00)01258-2
World Health Organization: Dengue guidelines for diagnosis, treatment, prevention and control. In Special Program for Research and Training in Tropical Diseases. France, World Health Organization; 2009.
Schneider JMPH, Droll D: A timeline for dengue in the Americas to December 31, 2000 and noted first occurrences. In Division of Disease Prevention and Control. Pan American Health Organization; 2001.
Boshell J: Dengue en Colombia. Biomedica 1986, 6: 101-106.
Halstead SB: Pathogenesis of Dengue: Challenges to Molecular Biology. Sci 1988, 239: 476-481. 10.1126/science.3277268
Guzman MG, Kouri DJ, Bravo J, Soler M, Vasquez S, Santos M, Villaescusa R, Basanta P, Indan G, Ballester JM: Dengue hemorrhagic fever in Cuba. II. Clinical Investigations. Trans Royal Soc Trop Med Hyg 1984, 78: 239-241. 10.1016/0035-9203(84)90286-4
Barcelos L, Batista A, Portela G, Paiva B, Germano J, Bonjardim CA, Peregrino PC, Geessien E: Dengue virus 3 genotype 1 assosiated with dengue fever and dengue hemorrhagic fever, Brazil. Emerg Infect Dis 2008, 14: 314-316. 10.3201/eid1402.070278
Leitmeyer KC: Dengue virus structural differences that correlate with pathogenesis. J Virol 1999, 73: 4738-4747.
Trent DW, Grant JA, Monath TP, Manske CL, Corina M, Fox GE: Genetic variation and microevolution of dengue 2 virus in Southeast Asia. Virol 1989, 172: 523-535. 10.1016/0042-6822(89)90195-5
Rico-Hesse R: Molecular evolution and distribution of dengue viruses type 1 and 2 in nature. Virol 1990, 174: 479-493. 10.1016/0042-6822(90)90102-W
Lewis JA, Chang GJ, Lanciotti RS, Kinney RM, Mayer LW, Trent DW: Phylogenetic relationships of dengue-2 viruses. Virol 1993, 197: 216-224. 10.1006/viro.1993.1582
Chunge E, Cassar O, Drouet MT, Guzman MG, Laille M, Rosen L, Deubel V: Molecular epidemiology of dengue-1 and dengue-4 viruses. J Gen Virol 1995, 76: 1877-1884. 10.1099/0022-1317-76-7-1877
Lanciotti RS, Gubler DJ, Trent DW: Molecular evolution and phylogeny of dengue-4 viruses. J Gen Virol 1997, 78: 2279-84.
Rico-Hesse R, Harrison LM, Nisalak A, Vaughn DW, Kalayanarooj S, Green S, Rothman AL, Ennis FA: Molecular evolution of dengue type 2 virus in Thailand. AmJTrop Med Hyg 1998, 58: 96-101.
Nogueira RM, Miagostovich MP, Goncalvez H: Molecular epidemiology of dengue viruses in Brazil. Cad Saude Publica 2000, 16: 205-211. 10.1590/S0102-311X2000000100021
Twiddy SS, Farrar JJ, Chau NV, Wills B, Gould EA, Gritsun T, Lloyd G, Holmes EC: Phylogenetic relationships and differential selection pressures among genotypes of dengue-2 virus. Virol 2002, 298: 63-72. 10.1006/viro.2002.1447
Foster JE, Bennett SN, Vaughan H, Vorndam V, McMillan WO, Carrington CVF: Molecular evolution and phylogeny of dengue type 4 virus in the Caribbean. Virol 2003, 306: 126-34. 10.1016/S0042-6822(02)00033-8
Holmes EC, Twiddy SS: The origin, emergence and evolutionary genetics of dengue virus. Infect Genet Evol 2003, 3: 19-28. 10.1016/S1567-1348(03)00004-2
Hwang KP, Chu PY, Tung YC, Wang HL, Yueh YY, Wu YC, Chin C, Lin KH: Molecular epidemiological study of dengue virus type 1 in Taiwan. J Med Virol 2003, 70: 404-409. 10.1002/jmv.10410
Uzcategui NY, Comach G, Camacho D, Salcedo M, de Quintana MC, Jimenez M, Siera G, Cuello R, James WS, Turner S, Holmes EC, Gould EA: Molecular epidemiology of dengue virus type 3 in Venezuela. J Gen Virol 2003, 84: 1569-1575. 10.1099/vir.0.18807-0
Foster JE, Bennett SN, Carrington CVF, Vaughan H, McMillan WO: Phylogeography and molecular evolution of dengue 2 in the Caribbean basin, 1981-2000. Virol 2004, 324: 48-59. 10.1016/j.virol.2004.03.020
Loroño-Pino MA, Farfan-Ale JA, Zapata-Peraza AL, Rosado-Paredes EP, Flores-Flores LF, García-Rejón JE: Introduction of the American/Asian genotype of dengue 2 virus into the Yucatan State of Mexico. AmJTrop Med Hyg 2004, 71: 485-492.
Méndez JA, Bernal MP, Calvache D, Boshell J: Genotipificación y análisis filogenético de cepas colombianas del virus dengue tipo 2. NOVA 2003, 1: 37-43.
Domingo C, Palacios G, Jabado O, Reyes N, Niedrig M, Gascón J, Cabrerizo M, Lipkin WI, Tenorio A: Use of a Short Fragment of the C-Terminal E Gene for Detection and Characterization of Two New Lineages of Dengue Virus 1 in India. J Clin Microbiol 2006, 44: 1519-1529. 10.1128/JCM.44.4.1519-1529.2006
Usme-Ciro JA, Mendez JA, Tenorio A, Rey Gloria J, Domingo C, Gallego-Gomez JC: Simultaneous circulation of genotypes I and III of dengue virus 3 in Colombia. Virol J 2008, 5: 101-110. 10.1186/1743-422X-5-101
Kukreti H, Dash PK, Parida M, Chaudhary A, Saxena P, Rautela RS, Mittal V, Chhabra M, Bhattacharya D, Lal S, Rao PVL, Rai A: Phylogenetic studies reveal existence of multiple lineages of a single genotype of DENV-1 (genotype III) in India during 1956-2007. Virol J 2009, 6: 1. 10.1186/1743-422X-6-1
Mendez JA, Usme-Ciro JA, Domingo C, Rey GJ, Sanchez JA, Tenorio A, Gallego-Gomez JC: Phylogenetic history demonstrates two different lineages of dengue type 1 virus in Colombia. Virol J 2010, 7: 226. 10.1186/1743-422X-7-226
Groot H: The reinvasion of Colombia by Aedes aegypti: aspects to remember. AmJTrop Med Hyg 1980, 29: 330-338.
Padilla JC, Rojas D, Sáenz R, Rojas D, Bello B, Méndez JA: Dengue en Colombia: epidemiologia, estrategias de intervención y perspectivas del problema. In Ministerio de la Protección Social-Instituto Nacional de Salud. Colombia; 2010:in press.
Instituto Nacional de Salud: Boletín de vigilancia de Dengue en Colombia. 2011.
Domingo C, Palacios G, Niedrig M, Cabrerizo M, Jabado O, Reyes N, Lipkin WI, Tenorio A: A New Tool for the Diagnosis and Molecular Surveillance of Dengue Infections in Clinical Samples. Dengue Bulletin 2004, 28: 87-95.
Thompson JD, Higgins DG, Gibson TJ: CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, positions specific gap penalties and weight matrix choice. Nucleic Acids Res 1994, 22: 4673-4680. 10.1093/nar/22.22.4673
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: Molecuar Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 2007, 24: 1596-1599. 10.1093/molbev/msm092
Zanotto PM, Gould EA, Gao GF, Harvey PH, Holemes EC: Population dynamics of flaviviruses revealed by molecular phylogenies. Proc Natl Acad Sci USA 1996, 93: 548-53. 10.1073/pnas.93.2.548
Twiddy SS, Holmes EC, Rambaut A: Inferring the rate and time-scale of dengue virus evolution. Mol Biol Evol 2003, 20: 122-129. 10.1093/molbev/msg010
Weaver SC, Vasilakis N: Molecular evolution of dengue viruses: Contributions of phylogenetics to understanding the history and epidemiology of the preeminent arboviral disease. Inf Gen & Evol 2009, 9: 523-540. 10.1016/j.meegid.2009.02.003
Sariol CA, Pelegrino JL, Martinez A, Arteaga E, Kouri G, Guzman MG: Detection and genetic relationship of dengue virus sequences in seventeen-year-old paraffin-embedded samples from Cuba. AmJTrop Med Hyg 1999, 61: 994-1000.
Guzman MG, Deubel V, Pelegrino JL, Rosario D, Marrero M, Sariol C, Kouri G: Partial nucleotide and amino acid sequences of the envelope and the envelope/nonstructural protein-1 gene junction of four dengue-2 virus strains isolated during the 1981 Cuban epidemic. AmJTrop Med Hyg 1995, 52: 241-246.
Watts DM, Porter KR, Putvatana P, Vasquez B, Calampa C, Hayes CG, Halstead SB: Failure of secondary infection eith American genotype dengue 2 to cause dengue hemorrhagic fever. Lancet 1999, 354: 1431-1434. 10.1016/S0140-6736(99)04015-5
Holmes EC: The phylogeography of human viruses. Mol Ecol 2004, 13: 745-756. 10.1046/j.1365-294X.2003.02051.x
Myat TH, Lowry K, Jiang L, Hlaing T, Holmes EC, Aascov J: Lineage extinction and replacement in dengue type 1 virus populations are due to stochastic events rather than to natural selection. Virol 2005, 336: 163-172. 10.1016/j.virol.2005.03.018
Carrington CVF, Foster JE, Pybus OG, Bennett SN, Holmes EC: Invasion and maintenance of dengue virus type 2 and type 4 in the Americas. J Virol 2005, 79: 14680-14687. 10.1128/JVI.79.23.14680-14687.2005
Salda LTD, Parquet MDC, Matias RR, Natividad FF, Kobayashi N, Morita K: Molecular epidemiology of dengue 2 viruses inthe Philippines: genotype shift and local evolution. AmJTrop Med Hyg 2005, 73: 796-802.
Bennett SN, Holmes EC, Chirivella M, Rodríguez DM, Beltrán M, Vorndam V, Gubler DJ, McMillan WO: Molecular evolution of dengue 2 virus in Puerto Rico: Positive selection in the viral envelope accompanies clade reintroduction. J Gen Virol 2006, 87: 885-893. 131 10.1099/vir.0.81309-0
Domingo E, Holland JJ: RNA virus mutations and fitness for survival. Annu Rev Microbiol 1997, 51: 151-178. 10.1146/annurev.micro.51.1.151
Holmes EC, Burch SS: The causes and consequences of genetic variation in dengue virus. Trends Microbiol 2000, 8: 74-7. 10.1016/S0966-842X(99)01669-8
Twiddy SS, Woelk CH, Holmes EC: Phylogenetic evidence for adaptive evolution of dengue viruses in nature. J Gen Virol 2002, 83: 1679-1689.
Bennett SN, Holmes EC, Chirivella M, Rodríguez DM, Beltrán M, Vorndam V, Gubler DJ, McMillan WO: Selection-driven evolution of emergent dengue virus. Mol Biol Evol 2003, 20: 1650-1658. 10.1093/molbev/msg182
Holmes EC: Patterns of intra- and interhost nonsynonymous variation reveal strong purifying selection in dengue virus. J Virol 2003, 77: 11296-11298. 10.1128/JVI.77.20.11296-11298.2003
Coffey LL, Vasilakis N, Brault AC, Powers AM, Tripet F, Weaver SC: Arbovirus evolution in viv is constrained by host alternation. PNAS 2008, 105: 6970-6975. 10.1073/pnas.0712130105
Vasilakis N, Deardorff ER, Kenney JL, Rossi S, Hanley K, Weaver S: Mosquitoes put the brake on arbovirus evolution: Experimental evolution reveals slower mutation accumulation in Mosquito than vertebrate cells. PLoS Pathogens 2009, 5: 1-18.
Wang E, Ni H, Xu R, Barrett ADT, Watowich SJ, Gubler DJ, Weaver SC: Evolutionary relationships of endemic/epidemic and sylvatic dengue viruses. J Virol 2000, 74: 3227-3234. 10.1128/JVI.74.7.3227-3234.2000
Domingo E, Escarmis C, Sevilla N, Moya A, Elena SF, Quer J, Novella IS, Holland JJ: Basic concepts in RNA virus evolution. FASEB J 1996, 10: 859-864.
Drake JW, Holland JJ: Mutation rates among RNA viruses. Proc Natl Acad Sci USA 1999, 96: 13910-13913. 10.1073/pnas.96.24.13910
Shubert AM, Putoni C: Evolution of the sequence composition of flaviviruses. Inf Gen & Evol 2010, 10: 129-136. 10.1016/j.meegid.2009.11.004
Posada D, Crandall KA: Modeltest: testing the model of DNA substitution. Bioinformatics 1998, 14: 817-818. 10.1093/bioinformatics/14.9.817
Gilbert DG: Convert sequence formats using ReadSeq.[http://iubio.bio.indiana.edu/soft/molbio/readseq/java/]
Chen SH, Su SY, Lo CZ, Chen KH, Huang TJ, Kuo BH, Lin CY: PALM: A paralleled and integrated framework for Phylogenetic inference with Automatic Likelihood Model selectors. PLoS One 2009,4(12):e8116. 10.1371/journal.pone.0008116
Drummond A, Rambaut A: BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 2007, 7: 214-221. 10.1186/1471-2148-7-214
Archibald JK, Mort ME, Crawford DJ: Bayesian inference of phylogeny: a non-technical primer. Taxon 2003, 52: 187-191. 10.2307/3647388
Rambaut A: Estimating the rate of molecular evolution: incorporating non-contemporaneous sequences into maximum likelihood phylogenies. Bioinformatics 2000, 16: 395-399. 10.1093/bioinformatics/16.4.395
Drummond A, Ho SYW, Phillips MJ, Rambaut A: Relaxed phylogenetics and dating with confidence. PLoS Biology 2006, 4: 1-11. 10.1371/journal.pbio.0040001
Lemey P, Rambaut A, Drummond A, Suchard MA: Bayesian phylogeography finds its roots. PLoS Computational Biology 2009, 5: e1000520. 10.1371/journal.pcbi.1000520
Acknowledgements
We thank the Red Nacional de Laboratorios-Instituto Nacional de Salud, Colombia. We are grateful to Pablo Martínez and Noelia Reyes for technical assistance in amplifying and sequencing at the ISCIII; Mary Luz Gunturiz for sequencing at the Instituto Nacional de Salud; Luis Angel Murillo from the University of California for reviewing the manuscript; Lissethe Pardo for technical assistance at the Instituo Nacional de Salud; RIVE/CYTED (Red Iberoamericana de Virosis Emergentes) allowed the authors to meet with several other researchers in the field.
This research was supported by Instituto Colombiano para el Desarrollo de la Ciencia y la Tecnología Francisco José de Caldas-COLCIENCIAS grants 11150416336 CT 234-2004, 11150418079 and 111540820511 from the Colombian government and the Instituto Nacional de Salud resources.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JAM contributed to the experimental design, carried out the experiments and phylogenetic and molecular clock analysis, and wrote the manuscript. JAUC contributed to the experimental design, carried out the experiments and provided a critical review of the manuscript. CD participated in the experimental design, contributed to the interpretation of data and the critical review of the manuscript. GJR contributed to the experimental design and provided a critical review of the manuscript. JAS contributed with phylogenetic and molecular clock analysis and BEAST running and provided a critical review of the manuscript. AT conceived the experimental design and provided a critical review of the manuscript. JCGG conceived the study, participated in its design and coordination and provide a final review of the manuscript. All authors read and approved the final version of the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Méndez, J.A., Usme-Ciro, J.A., Domingo, C. et al. Phylogenetic reconstruction of dengue virus type 2 in Colombia. Virol J 9, 64 (2012). https://doi.org/10.1186/1743-422X-9-64
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-9-64