
Abstract
Background
The city of Sao Paulo has the highest AIDS case rate, with nearly 60% in Brazil. Despite, several studies involving molecular epidemiology, lack of data regarding a large cohort study has not been published from this city.
Objectives
This study aimed to describe the HIV-1 subtypes, recombinant forms and drug resistance mutations, according to subtype, with emphasis on subtype C and BC recombinants in the city of São Paulo, Brazil.
Study design
RNA was extracted from the plasma samples of 302 HIV-1-seropositive subjects, of which 211 were drug-naive and 82 were exposed to ART. HIV-1 partial pol region sequences were used in phylogenetic analyses for subtyping and identification of drug resistance mutations. The envelope gene of subtype C and BC samples was also sequenced.
Results
From partial pol gene analyses, 239 samples (79.1%) were assigned as subtype B, 23 (7.6%) were F1, 16 (5.3%) were subtype C and 24 (8%) were mosaics (3 CRF28/CRF29-like). The subtype C and BC recombinants were mainly identified in drug-naïve patients (72.7%) and the heterosexual risk exposure category (86.3%), whereas for subtype B, these values were 69.9% and 57.3%, respectively (p = 0.97 and p = 0.015, respectively). An increasing trend of subtype C and BC recombinants was observed (p < 0.01).
Conclusion
The HIV-1 subtype C and CRFs seem to have emerged over the last few years in the city of São Paulo, principally among the heterosexual population. These findings may have an impact on preventive measures and vaccine development in Brazil.
Similar content being viewed by others


Background
The huge genetic variability of HIV-1 results in a complex and dynamic molecular classification of types (HIV-1 and 2), groups (M,N,O,P), and the pandemic group M could be divided into subtypes (A-D,F-H,J,K) and recombinant forms, such as circulating recombinant forms (CRF) and unique recombinant forms (URF). Such HIV-1 variation has an important impact on diagnosis, viral load measurement and the performance of HIV-1 genotyping systems [1–3]. Thus, HIV-1 subtypes also contribute to the capacity of HIV-1 to evade the host immune response, [4] which can affect the response to antiretroviral treatment and, consequently, to the emergence of drug resistance [5]. Some studies have suggested that coreceptor switching from CCR5 to CXCR4 is less common in HIV-1 subtype C, [6] showing a lower rate of accumulation of mutations that confer resistance than subtype B [5]. Concerning the replication fitness of subtype C, the studies were controversial, [7–9] and in relation to transmission in utero, this subtype presented high efficiency compared to subtypes A or D [8, 10, 11]. These properties suggest an overall transmission advantage, in part, possibly related to efficient replication in dendritic cells, the targets of the onset of HIV-1 infection [11].
Although HIV-1 subtype B is the most studied, HIV-1 C predominates globally and is responsible for approximately 50% of infections [12]. This subtype is not widespread, but it is the most prevalent in sub-Saharan Africa and in the populous countries, such as India and China, where HIV infection rates are highest among heterosexuals [12, 13]. Up to now, five circulating recombinant forms (CRFs) involving this subtype have been detected, three of them presenting recombinations between subtypes B and C, CRF07, CRF08, described in China, [14, 15] and CRF31_BC, described in Brazil [16]. The other two present recombinations between subtypes C and D, CRF10_CD and CRF41_CD [17, 18]. Overall, more than 18% of new infections have been attributed to HIV-1 recombinants [19].
The Brazilian AIDS epidemic is mainly driven by subtypes B, F1, and BF1 recombinants; however, in the Southern region, subtype C and BC recombinants can represent up to 50% of cases, depending of the geographical studied region [20–27]. Despite the documented early circulation of HIV-1 subtype C outside of the southern region in 1992, one case from the city of São Paulo [28] and another in 1986 in Santos, [29] only recently has a continuous increase in cases been observed [30–32].
The current study was conducted to determine HIV-1 genetic diversity and PI/RTI resistance associated mutations among HIV-1 infected drug naïve and HAART patients in a cohort in the city of São Paulo. Moreover, patients classified as HIV-1 subtype C and BC recombinants were analyzed in more details.
Objectives
The present study aimed to describe HIV-1 subtypes, CRFs and drug resistance mutations in drug-naïve and failing HAART individuals living in the city of São Paulo, in particular attention to those classified as subtype C and BC.
Study design
Study population
The study population was composed by HIV-1-infected patients who have been followed-up at the Ambulatory Service of the Department of Secondary Immunodeficiency Clinic of the Clinical Hospital, University of São Paulo Medical School (HC/FMUSP), São Paulo, SP, Brazil, one of the largest teaching and research hospitals in Brazil.
According to the Brazilian Ministry of Health, HIV genotyping test should be routinely performed in patients under antiretroviral therapy who present a plasma viral load over 2000 copies/mL [33]. Following this directive, a total of 82 patients were genotyped at HC/FMUSP from March 2002 to June 2010. During the same period, 211 drug-naive individuals and nine individuals who had unknown status in relation to antiretroviral therapy (ART) were also genotyped. Demographic and clinical data were obtained from clinical charts or direct interview and the patients individually allow us for publication, as shown at Table 1. This study protocol was approved by the Research Ethics Committee of HC/FMUSP, under protocol number 774/99, and written informed consent was obtained from all patients. During the follow-up, CD4+T cells and HIV-1 viral load were determined using the Brazilian Ministry of Health guidelines.
RNA isolation, RT-PCR and DNA sequencing
HIV-1 viral RNA was isolated from plasma samples using the QIAamp® Viral RNA Mini Kit (Qiagen, Hilden, Germany) extraction method, in accordance with the manufacturer’s protocol. Complementary DNA was performed by RT-PCR protocol with random primers and using the PR/RT PCR strategy previously described by Gonzales et al. [34, 35]. The PCR products were purified using QIAquick® (Qiagen, Hilden, Germany), in accordance with the manufacturer’s protocol. The PCR purified products were sequenced utilizing ABI Prism Big Dye Terminator Ready Reaction Kit version 3.0 (Applied Biosystems®, Foster City, CA), in accordance with their respective protocols. The reactions were analyzed using the ABI Prism 3100 Genetic Analyzer (Applied Biosystems®, Foster City, CA).
Sequence analysis
The chromatograms of all sequenced DNA were visually using the Sequencher program version 4.0.5 (Gene Codes) and manually edited. PR/RT sequencing was conducted to detect drug resistance mutations and HIV-1 subtypes. Major antiretroviral drug resistance mutations were classified according to Stanford University online HIV Drug Resistance Database, [36]. The International AIDS Society-USA Panel and Update of the Drug Resistance Mutations in HIV-1, Spring 2008 [37–39].
Nucleotide sequences were aligned using the Clustal X program [40] and later hand edited for minor adjustments and gap-stripped. An alignment of 860 bp that partially covered the PR/RT region (nucleotides 2343-3203) was used for phylogenetic inferences under Neighbor-Joining (NJ) algorithm in Mega 4.0.2 program [41]. In order to confirm the classification of the non-B sequences, Bayesian analysis was performed. The jModeltest 0.1.1 program [42] was used to select the best-fit model of nucleotide substitution under the Akaike information criteria, resulting in the choice of the GTR + I + G model, [43] as implemented in MrBayes v3.1.2 [44]. For each data set, two runs of 4 chains each (one cold and three heated, temp = 0.20) were run for 4x107 generations, with a burn-in of 4x106 generations. Convergence of parameters was assessed by calculating the effective sample size (ESS) using TRACER v1.4 (http://beast.bio.ed.ac.uk/Tracer./) excluding an initial 10% for each run. All parameter estimates for each run showed an ESS values >100. A final Bayesian majority-rule consensus tree was obtained for the data set. The SplitsTree program version 4 [42] was used to confirm the phylogenetic relationship of the recombinant samples using the NeighborNet, based on the pairwise distance estimated by the F84 parameter model, as GTR + I + G model was not available in this program. Recombination analysis was performed by bootscan analysis as implemented in the Simplot version 3.5.1, [44] using group M reference sequences representative of the HIV-1 subtypes. Bootstrap values (>70) supporting branching with reference sequences were determined in NJ trees constructed using the K2-parameter model, [45] based on 100 resamplings, with a 200 nt sliding window moving in steps of 20 bases. In order to confirm the genetic structure of putative recombinant viruses, NJ phylogenetic analyses were conducted using the fragments of sequences assigned to specific HIV-1 subtypes according to the proposed breakpoint position by the bootscanning analysis.
HIV-1 subtype C and BC env sequences
The envelope gene was also sequenced when subtype C and BC PR/RT sequences were identified. A fragment of 0.6 kb of the envelope region of the HIV-1 was amplified by nested-PCR primers LB1 (TAGAATGTACACATGGAATT)/, LB2 (GCCCATAGTGCTTCCTGCTGCT) as outer primers and LB3 (GCAGTCTAGCAGAAGAAGA)/, LB4 (CTTCTCCAATTGTCCCTCATA) as inner primers. The sequence analysis of the env region was performed as previously described for the PR/RT region (data not shown).
Sequence data
All the sequences generated were submitted to the GenBank database and the assigned accession numbers were: pol region: GU288708-GU288746, GU288748-GU288754, GU288756-GU288776, GU288778-GU288786, GU288788-GU288792, GU288794-GU288807, GU288809-GU288813, JN195817-JN196018 and env region: JN196019-JN196040.
Results
Demographic and clinical data
A total of 302 HIV-1-infected patients were analyzed, of these, 225 (75%) were men and 77 (25%) were women, with a mean age of 36 years-old. The distribution by the exposure categories were as follows: 61% heterosexual, 23% men who have sex with men, 9% bisexual and 7% other. According to the clinical status, 153 patients (72.5%) were asymptomatic, 55 (26.1%) were symptomatic and for 3 (1.4%), no information was obtained. The mean RNA plasma viral load was 5.28 log10/mL and the CD4+T cell count was 350 cells/mm3 for naïve patients. For treated patients, 38 (46.3%) were asymptomatic, 27 (33%) were symptomatic and for 17 (20.7%), no information was obtained. The mean RNA plasma viral load was 5.0 log10/mL and the CD4+T cell count was 246 cells/mm3.
HIV-1 PR/RT subtype classification
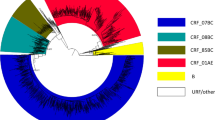
According to the phylogenetic and bootscan analyses, 239 patients (79.1%) were assigned to subtype B (167 naïve, 66 treated and 6 with no information concerning treatment-ND), 23 (7.6%) were assigned to subtype F1 (13 naïve, 8 treated and 2 ND), 16 (5.3%) were subtype C (12 naïve, 3 treated and 1 ND), and 24 (8%) were recombinant forms (19 naïve and 5 treated). Among the recombinant forms, 14 were BF recombinants (11URF_BF1 and 3 CRF28/29), 1 BU, 1 FD, 2 FU and 6 BC recombinants (5 BC with the same PR/RT recombinant patterns, in which only two of them were related and 1 CRF31/D). The Bayesian tree of the non-subtype B sequences is depicted in Figure 1. Interestingly, a group of four BC sequences presenting a well supported clustering and presenting the same recombinant pattern was detected; further full genomic sequencing is required in order to describe a new CRF_BC.

Majority-rule Bayesian consensus tree of the pr / rt region (860nt) from non subtype B samples collected in Sao Paulo city from 2002 to 2010. Posterior probability values superior to 0.80 are indicated. The sequences described in the present study were star marked.
Primary and secondary resistance
HIV-1 primary resistance mutations were detected in 42 (20%) out of 211 naive individuals, among these, 8 (3.8%) presented major PI resistance mutations, 29 (13.7%) presented NRTI resistance mutations and 27 (12.8%) presented NNRTI resistance mutations. Overall, 20% of individuals presented resistance to one antiretroviral class, 7.6% presented resistance to two classes, and 2.8% presented resistance to all three classes. HIV-1 secondary resistance mutations were investigated in 82 patients receiving HAART regimen, among these, 30 (36.6%) presented major PI resistance mutations, 51 (62.2%) NRTI resistance mutations and 42 (51.2%) NNRTI resistance mutations. Overall, 17.1% of individuals presented resistance to one antiretroviral class, 56.1% presented resistance to two classes, and 76.8% presented resistance to all three classes.
Comparing the resistance levels in two distinct periods of the study, 2002–2006 and 2007–2010, the following were verified: PI resistance mutations (5.3% vs 2.6%; p = 0.5), NRTI (12% vs 18%, p = 0.3), NNRTI (10.5% vs 18%; p = 0.18), showing an increasing trend of resistance levels, except for PI resistance mutations. Among the 82 treated patients, the following were verified: PI resistance mutations (46.2% vs 20%; p = 0.03), NRTI (42.3% vs 56.7%; p = 0.3), NNRTI (28.8% vs 46.7%; p = 0.17), similar results to naïve patients were observed (data not shown). The major resistance mutations detected in PR and RT regions in drug naïve or HAART individuals, independent of HIV-1 subtype, were: I93L, L10I/F/V, L63P, M36I/L/V, V118I, V179D/I, as presented in Figure 2.

Frequency of the main associated resistance mutations in naïve and HAART patients according to the viral subtype.
Secondary resistance mutations in the PR region were detected in individuals infected with subtypes B and F1 (G73S, I50L/V, I54L, I84V, L33F, L89V, V11I, V32I), and in the RT region (K101E, L100I, P225H, V108I, Y181C, Y188L). The mutations K65R, Q151M, Y115F were identified only in subtype B samples.
Demographical and clinical data from individuals classified as subtype C and BC recombinants
Since very little data is available concerning subtype C individuals outside the southern region of Brazil, in the present work, this group was the focus of more specific analysis. Thus, comparisons between the 22 HIV-1 individuals classified as subtype C and BC recombinants and subtype B individuals verified that the most common transmission mode was sexual and mean age in both groups was approximately 36 years-old. Interestingly, in the subtype C and BC recombinants group, 7 (31.8%) were women and 16 (72.7%) were asymptomatic. The mean viral load and CD4+ T cells counts were 4.64 log10/mL and 281 cells/mm3, respectively, in naive patients, while for patients receiving HAART treatment, the means were 4.23 log10/mL and 34 cells/mm3, respectively. Among HIV-1-infected individuals in the subtype B group, 47 (19.7%) were women and 155 (64.9%) were asymptomatic (p = 0.97 and p = 0.76; respectively). The means for viral load and CD4+ T-cells counts were 5.33 log10/mL and 349 cells/mm3, respectively, for naive patients, while for patients receiving HAART, the means were 5.02 log10/mL and 249 cells/mm3, respectively.
An increasing trend of subtype C and BC was observed, increasing from two cases from 2003 to 2006, to 20 cases from 2007 and 2010 (p < 0.05).
Sixteen out of 22 (72.7%) subtype C or BC recombinant individuals were naïve and the main mutations identified in the PR/RT region were G333A, I93L, L63P, M36I/T, V118I. Five (22.7%) were receiving treatment and the main mutations identified were I93L, L63P, M36I/T, M41L, M184V, T215F (to a lesser extent) and for 1 (4.6%) subtype C and BC individual, no information concerning treatment was available. For subtype B individuals, 167 (69.9%) were naïve patients and the main mutations identified were: D67N, K70R, G190A/S, L90M, T69A/D/N/S. Sixty six (27.6%) were receiving treatment and the main mutations identified were A98G, D30N, L74I/V, L210I/M/W, N88D, T69A/D/N/S, V90I, V106I/M, and for 6 (2.5%) individuals, no information concerning treatment was available. These mutations were verified only in the subtype B group.
Only two cases (2/16) showed non-analogue nucleotide drug resistance mutation for subtype C, while four cases (4/6) showed non-analogue nucleotide drugs resistance mutation for subtype BC in naïve patients.
The demographical and clinical data for subtype C and BC recombinant patients with the mutations identified in this cohort are listed in Table 1A and B.
Discussion
As expect, the HIV-1 B was the most common subtype in the cohort (79.1%) followed by subtype F1 (7.6%). A relevant presence of subtype C (5.3%) was observed and an increase in the number of mosaics (8%), of which 2% were BC recombinants. The first identified case of HIV-1 subtype C in this cohort from the city of São Paulo was from 2003, even though this cohort has been ollowed since 1989. In fact, this subtype predominates in South, where the majority of counties present the highest rates of HIV-1 infection in Brazil, [46] in contrast with Southeastern, where a small incidence of around 3% of subtype C is observed [16, 27]. However, the present study and other more recent studies have documented an increased prevalence of this subtype outside of the Southern region [30, 47].
The present cohort was predominantly naive heterosexual patients for all HIV-1 subtypes. The mode of transmission for HIV-1 C was clearly associated with heterosexual contact, similar to the major source for this subtype in Africa and Asia [27]. Thus, its presence in the city of São Paulo and the increasing number of cases seem to indicate that subtype C has increased transmission capacity, as occurred in Asia and Africa, the epicenter of the pandemic [30]. It is too early to determine whether the presence of viruses with molecular characteristics of other subtypes has clinical or epidemiological importance, but it certainly indicates the dynamics of the epidemic, with the number of new infections and high prevalence of various subtypes circulating in the Brazilian population.
Recombinations between subtypes B and C have been identified among the HIV-1 samples obtained in Southern Brazil, [22] and now in this cohort. Although subtype C is responsible for more than 56% of HIV-1 infections worldwide, up to now, only four out of the 49 known CRFs involving recombinations with subtype C have been described worldwide: CRF07_BC, CRF08_BC, CRF10_CD and CRF41_CD (CRF41_CD, has not yet been published) [14, 15, 17]. So far, only one of these is present in Brazil, [18] CRF31_BC [16]. The five BC recombinants identified in this work could represent a new CRF_BC, but full genomic analysis is required to elucidate this possibility.
As previously described, the prevalence of resistance associated mutations appear to differ between subtype B and others (non-B), selected mutations in codons 41, 210 and 215 are more frequent in the former, whereas mutations at codon 67, 70 and 219 are more common in subtype C [31]. The L210W mutation appeared in subtypes B and F, while the mutation L210I/M appeared only in subtype BU. The common resistance associated mutations for all HIV-1 subtypes are 118, 184 and 215. Mutations M36I, L63P, L89M, and I93L are related to antiretroviral resistance in subtype B and have also been identified in subtype C as polymorphisms, [48] these mutations were also verified in patients from the present cohort. In the samples obtained here, the most common resistance associated mutations detected in all HIV-1 subtypes were: PI codons 10, 36, 63 and 93 and NNRTI codons 179 and 190. The K103N mutation was not observed in the mosaics samples and the V90I mutation was not verified in subtype F.
The mutation V106M was previously described as occurring rapidly in subtype C virus, leading to high level of resistance, [47] even though it was not verified in the samples obtained here. This mutation may be a signature in subtype C patients treated with efavirenz and may have the potential to confer high-level multi-NNRTI resistance [19, 48]. Differential drug resistance acquisition was verified among subtypes B and C, in which subtype C viruses apparently acquired a lower number of mutations than subtype B for PI and NRTI, but not for NNRTI [7]. As previously described, our group also observed that the proportion of subtype C or BC mosaics among naive cases is significantly higher than among treated individuals, corroborating the hypothesis of more recent introduction of this subtype and recombinants in the Southeastern region [30].
In conclusion, the percentage of subtype C, BC and CRFs could be increasing in the city of São Paulo in recent years. Molecular epidemiological information concerning HIV-1 strains is proving to be important in elucidating the dynamics of HIV spread and the formulation of future vaccine strategies.
References
Aghokeng AF, Vergne L, Mpoudi-Ngole E, Mbangue M, Deoudje N, Mokondji E, Nambei WS, Peyou-Ndi MM, Moka JJL, Delaporte E, Peeters M: Evaluation of transmitted HIV drug resistance among recently-infected antenatal clinic attendees in four Central African countries. Antivir Ther 2009, 14: 401-411.
Kim JE, Beckthold B, Chen Z, Mihowich J, Malloch L, Gill MJ: Identification of a novel HIV type 1 subtype H/J recombinant in Canada with discordant HIV viral load (RNA) values in three different commercial assays. AIDS Res Hum Retrovir 2007,23(11):1309-1313. 10.1089/aid.2007.0080
Peeters M, Aghokeng AF, Delaporte E: Genetic diversity among human immunodeficiency virus-1 non-B subtypes in viral load and drug resistance assays. Clin Microbiol Infect 2010, 16: 1525-1531. 10.1111/j.1469-0691.2010.03300.x
Pinto ME, Struchiner CJ: HIV-1 diversity: a tool for studying the pandemic. Cad Saúde Pública 2006,22(3):473-484.
Martinez-Cajas JL, Pai NP, Klein MB, Wainberg MA: Differences in resistance mutations among HIV-1 non-subtype B infections: a systematic review of evidence (1996–2008). J Int AIDS Soc 2009, 12: 1-11. 10.1186/1758-2652-12-1
Tscherning C, Alaeus A, Fredriksson R, Björndal A, Deng H, Littman DR, Fenyö EM, Albert J: Differences in chemokine coreceptor usage between genetic subtypes of HIV-1. Virology 1998, 241: 181-188. 10.1006/viro.1997.8980
Ping LH, Nelson JA, Hoffman IF, Schock J, Lamers SL, Goodman M, Vernazza P, Kazembe P, Maida M, Zimba D, Goodenow MM, Eron JJ, Fiscus SA, Cohen MS, Swanstrom R: Characterization of V3 sequence heterogeneity in subtype C human immunodeficiency virus type 1 isolates from Malawi: underrepresentation of X4 variants. J Virol 1999, 73: 6271-6281.
Rodriguez MA, Ding M, Ratner D, Chen Y, Tripathy SP, Kulkarni SS, Chatterjee R, Tarwater PM, Gupta P: High replication fitness and transmission efficiency of HIV-1 subtype C from India: implications for subtype C predominance. Virology 2009, 385: 416-424. 10.1016/j.virol.2008.12.025
Ariën KK, Abraha A, Quiñones-Mateu ME, Kestens L, Vanham G, Arts EJ: The replicative fitness of primary human immunodeficiency virus type 1 (HIV-1) group M, HIV-1 group O, and HIV-2 isolates. J Virol 2005,79(14):8979-8990. 10.1128/JVI.79.14.8979-8990.2005
Sucupira MCA, Caseiro MM, Alves K, Tescarollo G, Janini LM, Sabino EC, Castelo A, Shafer KP, Diaz RS: High levels of primary antiretroviral resistance genotypic mutations and B/F recombinants in Santos, Brazil. AIDS Patient Care STDs 2007,21(2):116-128. 10.1089/apc.2006.0079
Geretti AM: HIV-1 subtypes: epidemiology and significance for HIV management. Curr Opin Infect Dis 2006,19(1):1-7. 10.1097/01.qco.0000200293.45532.68
Hemelaar J, Gouws E, Ghys PD, Osmanov S, WHO-UNAIDS Network for HIV Isolation and Characterisation: Global trends in molecular epidemiology of HIV-1 during 2000–2007. AIDS 2011,25(5):679-689. 10.1097/QAD.0b013e328342ff93
Hemelaar J, Gouws E, Ghys PD, Osmanov S: Global and regional distribution of HIV-1 genetic subtypes and recombinants in 2004. AIDS 2006,20(16):W13-W23. 10.1097/01.aids.0000247564.73009.bc
McCutchan FE: Understanding the genetic diversity of HIV-1. AIDS 2000,14(Suppl 3):S31-S44.
Su L, Graf M, Zhang Y, Briesen HV, Xing H, Kostler J, Melzl H, Wolf H, Shao Y, Wagner R: Characterization of a virtually full-length human immunodeficiency virus type 1 genome of a prevalent intersubtype (C/B’) recombinant strain in China. J Virol 2000,74(23):11367-11376. 10.1128/JVI.74.23.11367-11376.2000
Santos AF, Sousa TM, Soares EAJM, Sanabani S, Martinez AMB, Sprinz E, Silveira J, Sabino EC, Tanuri A, Soares MA: Characterization of a new circulating recombinant form comprising HIV-1 subtypes C and B in southern Brazil. AIDS 2006,20(16):2011-2019.
Koulinska IN, Ndung’u T, Mwakagile D, Msamanga G, Kagoma C, Fawzi W, Essex M, Renjifo B: A new human immunodeficiency virus type 1 circulating recombinant form from Tanzania. AIDS Res Hum Retrovir 2001,17(5):423-431. 10.1089/088922201750102508
The Circulating Recombinant Forms (CRFs). Los Alamos National Laboratory web site, , accessed 25 May 2011 http://www.hiv.lanl.gov/content/sequence/HIV/CRFs/CRFs.html Los Alamos National Laboratory web site, , accessed 25 May 2011
Jülg B, Goebel FD: What’s new in HIV/AIDS HIV genetic diversity: any implications for drug resistance? Infection 2005, 33: 299-301. 10.1007/s15010-005-6405-1
Silva MM, Telles FQ, da Cunha CA, Rhame FS: HIV subtype, epidemiological and mutational correlations in patients from Paraná, Brazil. Braz J Infect Dis 2010,14(5):495-501.
Brindeiro RM, Diaz RS, Sabino EC, Morgado MG, Pires IL, Brigido L, Dantas MC, Barreira D, Teixeira PR, Tanuri A, Brazilian Network for Drug Resistance Surveillance: Brazilian network for HIV drug resistance surveillance (HIV-BResNet): a survey of chronically infected individuals. AIDS 2003,17(7):1063-1069. 10.1097/00002030-200305020-00016
Rodrigues R, Scherer LC, Oliveira CM, Franco HM, Sperhacke RD, Ferreira JL, Castro SM, Stella IM, Brigido LF: Low prevalence of primary antiretroviral resistance mutations and predominance of HIV-1 clade C at polymerase gene in newly diagnosed individuals from south Brazil. Virus Res 2006,116(1–2):201-207.
Brígido LF, Nunes CC, Oliveira CM, Knoll RK, Ferreira JL, Freitas CA, Alves MA, Dias C, Rodrigues R, Research Capacity Program: HIV type 1 subtype C and CB Pol recombinants prevail at the cities with the highest AIDS prevalence rate in Brazil. AIDS Res Hum Retrovir 2007, 23: 1579-1586. 10.1089/aid.2007.0102
Csillag C: HIV-1 subtype C in Brazil. Lancet 1994, 344: 1354.
Rossini MAA, Diaz RS, Caseiro M, Turcato G, Accetturi CA, Sabino EC: HIV-1 subtypes among intravenous drug users from two neighboring cities in São Paulo State, Brazil. Braz J Med Biol Res 2001, 34: 45-47. 10.1590/S0100-879X2001000100005
Brigido LFM, Ferreira JLP, Almeida VC, Rocha SQ, Ragazzo TG, Estevam DL, Rodrigues R, for the São Paulo HIV Salvage Workgroup: Southern Brazil HIV type 1 C expansion into the state of São Paulo, Brazil. AIDS Res Hum Retrovir 2011, 27: 339-344. 10.1089/aid.2010.0157
Munerato P, Sucupira MC, Oliveros MPR, Janini LM, de Souza DF, Pereira AA, Inocencio LA, Diaz RS: HIV type 1 antiretroviral resistance mutations in subtypes B, C, and F in the City of São Paulo, Brazil. AIDS Res Hum Retrovir 2010,26(3):265-273. 10.1089/aid.2008.0288
de Souza AC, de Oliveira CM, Rodrigues CL, Silva SA, Levi JE: Short communication: molecular characterization of HIV type 1 BF pol recombinants from São Paulo, Brazil. AIDS Res Hum Retrovir 2008, 24: 1521-1525. 10.1089/aid.2008.0089
: National Program on STD/AIDS, Brazilian Ministry of Health, Brazil. National Network of Laboratories for Genotyping–AIDS. ( ) http://www.aids.gov.br ()
Gonsalez CR, Alcalde R, Nishiya A, Barreto C, Silva FES, Almeida A, Mendonça M, Ferreira F, Fernandes SS, Casseb J, Duarte AJS: Drug resistance among chronic HIV-1-infected patients naïve for use of anti-retroviral therapy in Sao Paulo city. Virus Res 2007,129(2):87-90. 10.1016/j.virusres.2007.06.021
Gonsalez CR, Alcalde R: HIV/AIDS. In HIV resistance to ARVs: implications for new therapeutic options. Edited by: Casseb J. Atheneu: LIM/HC/FMUSP; 2010:71-83.
Stanford University online. HIV Drug Resistance Database web site, ( ) http://www.hivdb.Stanford.edu/index.html HIV Drug Resistance Database web site, ()
Hirsch MS, Günthard HF, Schapiro JM, Brun-Vézinet F, Clotet B, Hammer SM, Johnson VA, Kuritzkes DR, Mellors JW, Pillay D, Yeni PG, Jacobsen DM, Richman DD: Antiretroviral drug resistance testing in adult HIV-1 infection: 2008 recommendations of an International AIDS Society-USA Panel. Clin Infect Dis 2008, 47: 266-285. 10.1086/589297
Johnson VA, Brun-Vezinet F, Clotet B, Gunthard HF, Kuritzkes DR, Pillay D, Schapiro JM, Richman DD: Update of the drug resistance mutations in HIV-1: Spring 2008. Top HIV Med 2008,16(1):62-68.
Shafer RW, Rhee SY, Pillay D, Miller V, Sandstrom P, Schapiro JM, Kuritzkes DR, Bennett D: HIV-1 protease and reverse transcriptase mutations for drug resistance surveillance. AIDS 2007,21(2):215-223. 10.1097/QAD.0b013e328011e691
Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG: The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 1997,25(24):4876-4882. 10.1093/nar/25.24.4876
Tavaré S: Some probabilistic and statistical problems in the analysis of DNA sequences. American Mathematical Society. Lect Math Life Sci 1986, 17: 57-86.
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 2007, 24: 1596-1599. 10.1093/molbev/msm092
Posada D: jModelTest: phylogenetic model averaging. Mol Biol Evol 2008, 25: 1253-1256. 10.1093/molbev/msn083
Ronquist F, Huelsenbeck JP: MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinform Appl Note 2003,19(12):1572-1574.
Ray S: Simplot v3.5.1. 2009. Available from: http://www.med.jhu.edu/deptmed/sray/download/
Kimura M: A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 1980,16(2):111-120. 10.1007/BF01731581
Huson DH, Bryant D: Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 2006,23(2):254-267.
Jones LR, Dilernia DA, Manrique JM, Moretti F, Salomon H, Carrillo MG: In-depth analysis of the origins of HIV type 1 subtype C in South America. AIDS Res Hum Retrovir 2009,25(10):951-959. 10.1089/aid.2008.0293
Carvalho BC, Cardoso LPV, Damasceno S, Stefani MMA: Moderate prevalence of transmitted drug resistance and interiorization of HIV type 1 subtype C in the Inland North State of Tocantins, Brazil. AIDS Res Hum Retrovir 2011, 27: 1-7. 10.1089/aid.2010.0156
Abreu CM, Brindeiro PA, Martins NA, Arruda MB, Bule E, Stakteas S, Tanuri A, Brindeiro RM: Genotypic and phenotypic characterization of human immunodeficiency virus type 1 isolates circulating in pregnant women from Mozambique. Arch Virol 2008,153(11):2013-2017. 10.1007/s00705-008-0215-6
Loemba H, Brenner B, Parniak MA, Maayan S, Spira B, Moisi D, Oliveira M, Detorio M, Wainberg MA: Genetic divergence of human immunodeficiency virus type 1 Ethiopian clade C reverse transcriptase (RT) and rapid development of resistance against non-nucleoside inhibitors of RT. Antimicrob Agents Chemother 2002,46(7):2087-2094. 10.1128/AAC.46.7.2087-2094.2002
Brenner B, Turner D, Oliveira M, Moisi D, Detorio M, Carobene M, Marlink RG, Schapiro J, Roger M, Wainberg MA: A V106M mutation in HIV-1 clade C viruses exposed to efavirens confers cross-resistance to non-nucleoside reverse transcriptase inhibitors. AIDS 2003,17(1):F1-F5. 10.1097/00002030-200301030-00001
Acknowledgements
The authors would like to thank all the patients who participated in this study, ADEE3002 Group (Ambulatory Service of the Secondary Immunodeficiency Clinic of Clinical Hospital-HC/FMUSP), particularly Claudio R. Gonsalez, Lucas A. Medeiros, Ana Paula R. Veiga, Marcelo Mendonça and Eduardo R. Lagonegro. They would also like to thank Rosangela M. Araujo and Noemia Orii for the flow cytometry experiments, Jose Eduardo Martins for his assistance in determining HIV-1 viral loads, Dr Shirley Komninakis for kindly providing the envelope region primers, Fernando L. Melo and Anna Nishiya for their technical assistance. Lucio Martins, Andre Seiji Goto and Demetrius Vignati Alves da Silva for IT assistance. Fapesp, CNPq, LIM56/HC/FMUSP and FFM for support.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that there are no competing interests.
Authors’ contributions
RA: Drafted the manuscript, analyzed data; MLG: Statistical analysis, phylogenetic analysis, Drafted the manuscript; ADJS: Revised the drafted the manuscript; JC: Designed the study, analyzed the data and drafted the final version of the manuscript. All authors read and approved the final manuscript.
Rosana Alcalde, Monick L Guimarães contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Alcalde, R., Guimarães, M.L., Duarte, A.J. et al. Clinical, epidemiological and molecular features of the HIV-1 subtype C and recombinant forms that are circulating in the city of São Paulo, Brazil. Virol J 9, 156 (2012). https://doi.org/10.1186/1743-422X-9-156
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-9-156