
Abstract
Background
In China, primary EBV infection occurs during childhood with seroprevalence reaching about 100% by 10 years of age. There are few studies on EBV variants in diseases associated with EBV infection in Chinese children. In this study, we investigated the diversity of the EBV genes (EBNA-1 and LMP-1) and the relationship between EBV variants and the clinical phenotypes in diseases associated with EBV infections in Chinese pediatric cases.
Results
The frequencies of EBV type I in the IM, HLH and HL samples were 98.4%, 100% and 95.8%, respectively. Three known EBNA-1 variants were identified, including V-val (all were V-val-v1 sub-variant), P-thr' and V-Leu (MT). The frequency of V-val-v1 was 98.6% in the IM samples, 100% in the HLH samples and 97.1% in the HL samples. There were no significant differences of the distribution of EBNA-1 variants between IM, HLH and HL samples (P > 0.05). Three known LMP-1 variants, including China 1, China 2 and Med, were identified and China 1 was predominant in all groups (IM 88.6%, HLH 100% and HL 100%). The frequency of del-LMP-1 was 88.6% in the IM samples, 100% in the HLH samples and 96.0% in the HL samples. There were no significant differences in the frequency of del-LMP-1 between the IM, HLH and HL samples (P > 0.05). The frequency of Xho I loss was 90.6% in the IM samples, 100% in the HLH samples and 100% in the HL samples, with no significant difference in frequency (P > 0.05). In the EBV type I strain, V-val-v1 variant (EBNA-1) was linked with China1 variant (LMP-1) in 88.9% of the IM samples, 100% of the HLH samples and 80.0% of the HL samples in this study.
Conclusions
Type I EBV was the most prevalent subtype EBV in Chinese pediatric cases and V-val-v1 (EBNA-1) and China1 (LMP-1) variants were the most dominant variants. There was a strong linkage between V-val-v1 (EBNA-1) variant and China1 (LMP-1) variant in type I EBV. The sequence variation in EBV genes may represent a geographic polymorphism since no preferential associations were found between specific EBV variants and specific diseases in this study.
Similar content being viewed by others

Background
Epstein-Barr virus (EBV) is a lymphotrophic human gamma-1 herpes virus with a double stranded DNA genome comprised of approximately 170-kilobases. It is transmitted primarily through saliva and infects over 95% of the world's population [1]. In developing countries, primary EBV infection typically occurs in early childhood and is asymptomatic; in developed countries, infection occurs in later childhood or young adulthood and can manifest as infectious mononucleosis (IM), which is self-limiting. EBV is associated with not only nonmalignant diseases but also a number of malignant diseases, including Burkitt's lymphoma, nasopharyngeal carcinoma and Hodgkin lymphoma.
After primary infection, EBV establishes a lifelong latent infection in B lymphocytes [2]. During latent infection, EBV expresses a restricted set of genes, including two EBV-encoded RNAs (EBER-1 and EBER-2), six EBV nuclear antigens (EBNA-1, EBNA-2, EBNA-3A, -3B, -3C, and leader protein EBNA-LP), and three integral membrane proteins (LMP-1, LMP-2A and -2B).
Based on the sequence divergence of nuclear antigens (EBNA-2, -3A, -3B, and -3C) EBV can be divided into two types of EBV: type I and type II [3, 4]. Detailed sequence analysis has also been done with the EBNA-1, LMP-1 and BZLF1-genes, which are not type-specific in their sequence divergence among EBV strains.
EBNA-1 sequence variation has been described by comparing the sequence of several EBV isolates from tumors and cell lines with the sequence of prototype B95-8 [5]. EBV has been classified into five EBNA-1 variants based on polymorphism of the C-terminal region and the signature changes at amino acid residue 487 of EBNA-1, including the prototype P-ala and P-thr variants and the V-val, V-leu and V-pro variants [6].
LMP-1 gene polymorphisms were identified in several studies [7–11]. The most common polymorphisms contain substitutions of the nucleotides or amino acids including loss of restriction sites (such as Xho I in the N-terminus), variations of the number of 33 bp repeats, and a 30 bp deletion in the C-terminus (del-LMP-1). Ten major variants of LMP-1 (China 1, China 2, China 3, NC (North Carolina), Mediterranean+ (Med+), Med-, GD1, SEA 1, and SEA 2) have been described from different regions and diseases [8–10].
In China, primary EBV infection occurs during childhood with seroprevalence reaching about 100% by 10 years of age. However there are few studies on EBV variants in diseases associated with EBV infection in Chinese pediatric population [12]. So in this study, we investigated the diversity of the EBV genes EBNA-1 and LMP-1 in IM (a self-limiting disease), EBV associated Hemophagocytic lymphohistiocytosis (EBV-HLH) (a severe hematological disease triggered by EBV) and Hodgkin lymphoma (HL) in Chinese children.
Results
EBV types I and II in EBV associated diseases in children
The frequency of EBV type I or type II infection was determined in 107 samples. Type I EBV was present in 105 samples (98.1%, 105/107) and type II EBV in 2 samples (1.9%, 2/107) (Table 1).
EBNA-1 variants in EBV associated diseases in children
The C-terminal EBNA-1 fragment was amplified from 128 samples (Table 2). In this study, three known variants of the EBNA-1 including V-val (all were V-val-v1 Sub-variant), P-thr' and V-Leu (MT) [13–15] were observed (Table 2). Sub-variant V-val-v1 of EBNA-1 first reported by Do et.al [13] was the most common in the three groups (IM 98.6%, HLH 100%, HL 97.1%). There were no significant differences in the incidence of EBNA-1 variants between the IM, HLH and HL samples (P > 0.05).
LMP-1 variants in EBV associated diseases in children
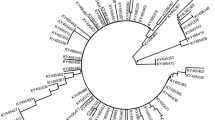
A total of 74 LMP-1 nucleotide sequences corresponding to the carboxyl-terminal were obtained and translated into amino acid for the phylogenetic analysis (Figure 1). Three known variants of LMP-1 including China 1, China 2 and Med were identified (Table 3). Among them China 1 was predominant in all groups. The frequency of China 1 in the IM, HLH and HL samples was 88.6% (39/44), 100% (25/25) and 100% (5/5), respectively.

Phylogenetic tree of the C-terminus of LMP-1. B95-8 variant according to Genebank: V01555.2. China 1 (Genebank: AY337723.1), China 2 (Genebank: AY337724.1), Alaskan (Genebank: AY337725.1), Med- (Genebank: AY337721.2), Med+ (Genebank: AY337722.2) and North Carolina (NC) (Genebank: AY337726.2) are the LMP-1 variants according to Miller et al.(1994), Sung et al.(1998) and Edwards et al (1999). IM: infectious mononucleosis; HLH: hemophagocytic lymphohistiocytosis. 'm' indicated the outpatient and the figure means the mumber of the sample.
A short fragment of LMP-1 containing the 30 bp deletion region was amplified from 20 paraffin-embedded tumors biopsies. Nineteen of them contained the del-LMP-1, and 1 specimen had wild type LMP-1 (wt-LMP-1). In total, the del-LMP-1 was detected in 88.6% (39/44) of IM samples, 100% (25/25) HLH samples and 96.0% (24/25) HL samples. There were no significant differences of the incidence of del-LMP-1 in three groups (P > 0.05).
The full length of LMP-1 was amplified in 54 samples. LMP-1 variant with Xho I loss was detected in 90.6% (29/32) of the IM, 100% (17/17) of the HLH and 100% (5/5) of the HL samples, respectively. There was no significant difference in the frequency of Xho I loss between the IM and HLH samples, or between the IM and HL samples (P > 0.05).
Linkage analysis
A combination of more than one polymorphic site in the EBV genome was investigated in 60 samples for this study. All 60 samples were EBV type I strains. The frequency of V-val-v1 (EBNA-1) variant lingking China1 (LMP-1) variant in the IM, HLH and HL samples was 88.9% (32/36), 100% (19/19) and 80.0% (4/5), respectively (Table 4). Preferential linkages of certain EBNA-1 variants to distinct LMP-1 diversity were found to exist. However, there were no significant differences between the distribution of linked EBV genes in the IM, HLH and HL samples (P > 0.05).
Discussion
EBV is a lymphotrophic human gamma-1 herpes virus. It is classified as EBV types I and II based on sequence variation in the EBNA2 and EBNA3 genes [3, 4]. Previous studies of healthy Asian populations showed that 85% of individuals were infected with EBV type I, 4% with EBV type II and 11% with both types [16–18]. In this study, the frequencies of EBV type I in IM, HLH and HL were 98.4%, 100% and 95.8%, respectively. No co-infection with both viral types was detected. This difference can be explained by the small number of samples analyzed in this work, or alternatively, that pediatric patients are still not subjected to subsequent re-infections as adults [1].
EBNA-1 is the only latent protein that is consistently expressed in all EBV-positive malignancies. EBNA-1 is critical in maintaining EBV in the infected cells and facilitating episomal replication [19, 20]. In replicating latently infected EBV cells, EBNA-1 dictates equal partitioning of EBV genome, thus reducing the loss of EBV infected cells [21]. EBNA-1 is also a transcriptional activator and activates expression of EBV transcripts via the latent C promoter [22]. EBNA-1 carboxy (C)-terminal sequence variants have been described based on the amino acid signature at codon 487, and designated prototype including P-ala (identical to prototype B95.8 strain), P-thr, and variant including V-val, V-leu, and V-pro [6]. New variants and subvariants have more recently been characterized [1, 13, 23].
In Asian, most studies of the EBNA-1 variations have focused on nasopharyngeal carcinoma (NPC). Mai et al. suggested that V-val EBNA-1 with the functional advantage compared with prototype shown in their study might contribute to the tumorigenesis of NPC by increasing the expression of itself or other viral or cellular genes [24]. Zhang et al. indicate that V-val variant infects NPC preferentially and that a susceptibility to a particular EBV isolate in the nasopharynx may exist during development of NPC [25]. However, Sandvej et al. found that V-val is the dominant variant in Asian regions and appear to be associated with Asian NPC patients but not found in Danish NPC biopsies [23]. Do et al. reported that V-val-v1 (EBNA-1) is predominant in NPC as well as in healthy EBV-carriers from an Asian population [13]. Wang et al. observed that V-val was the most common variant not only in NPC but also in EBV associated gastric carcinoma and healthy donors in Northern China [26]. Moreover, a review about genetic diversity of EBV showed that V-val was detected in both cases and controls almost exclusively in China [27].
IM is a self-limiting disease caused by primary EBV infection. EBV-HLH is a severe hematological disease triggered by EBV infection and HL is a malignant disease. In this study, the percentage of V-val-v1 (EBNA-1) in IM, HLH and HL were 98.6%, 100% and 97.1%, respectively. No significant differences were found in the incidence of V-val-v1 subvariant in IM, HLH and HL groups (P > 0.05). The results indicated that the V-val-v1 is the dominant variant in Chinese pediatric cases, and EBNA-1 polymorphism may represent a geographic polymorphism. The data from Argentinean pediatric population showed that V-leu sub-variants preferentially circulate in this region. Interestingly, they also favor the geographical association hypothesis since no evidence for a preferential compartment distribution of EBNA-1 variants and sub-variants was found [1].
LMP-1 is considered the EBV oncogene [28]. It encompasses a 25 aa N-terminus, six predicted transmembrane domains (aa: 26-196), and a C-terminus (aa: 197-386). Both the N- and C-termini are located in the cytoplasm of the cell [29, 30]. The C-terminus interacts with cellular proteins through the C-Terminal Activation Region 1 (CTAR1) and CTAR2 and activates several signaling pathways, including the transcriptional nuclear factor-kB (NF-kB) whose activation is linked to the inhibition of apoptosis [31, 32]. The most commonly reported LMP-1 gene polymorphisms was a 30 bp deletion in the C-terminus. The 30 bp deletion variant (del-LMP-1) was first detected in EBV isolated from cell lines derived from NPC patients from Southern China [33]. China 1 and Med+ variants have this 30 bp deletion.
A number of reports have focused on the del-LMP-1 variant in malignant diseases. Hadhri et al. found that del-LMP-1 variant was significantly (P = 0.006) more frequent in NPC (71.42%) than in control biopsies (52%) in Tunisia [34]. Tiwawech et al. also reported that a significant association between the del-LMP-1 variant and NPC susceptibility (P = 0.023) was exist in Thais. Moreover, the frequency of del-LMP-1 in NPC patients was associated with the clinical stage of NPC [16]. Research from South America (35 Brazilian HD patients and 27 Argentine pediatric patients of HD) show that del-LMP-1 was more frequently detected in Hodgkin's lymphoma than in non-neoplastic controls (P = 0.005) suggesting a pathogenic role of del-LMP-1 variant in Hodgkin's lymphoma in this region [35]. These results supporting the idea that EBV del-LMP-1 may be a disease-associated variant.
However, there were some controversial findings. See et al. concluded that the presence of del-LMP-1 was associated with the Chinese race [36]. Saechan et al. found a higher prevalence (P = 0.026) of del-LMP-1 variant in Southern Thais than in Central Thais [9]. In this study, there was no significant difference in the frequency of del-LMP-1 between the IM, HLH and HL samples (P > 0.05) in Chinese children. And the same trend was also observed in another study in China, with 83% of Hodgkin's lymphoma and 86% of healthy controls showing del-LMP-1 variant [37]. Thus, the del-LMP-1 variant may represent a geographic or race associated polymorphism.
The loss of Xho I restriction site in the N-terminus of the LMP-1 gene is another commonly reported LMP-1 gene polymorphism. Hu et al. first described the loss of the Xho I site from exon 1 of the LMP-1 gene in the CAO cell line derived from a Chinese NPC case [28]. The loss of the Xho I site is caused by a single base substitution from G to T at the nucleotide position 169425. It has been reported that the Xho I loss was significantly associated with Asian NPC patients and the Xho I loss has been considered as a specific HL marker [28, 38, 39]. However, See et al. found that the Xho I loss was not only associated with type III NPC, but also with the Chinese race [36]. In this study the frequency of Xho I loss was 90.6% in the IM samples, 100% in the HLH samples and 100% in the HL samples. There was no significant difference in the frequency of Xho I loss between the IM and HLH samples or the IM and HL samples (P > 0.05). These findings suggest that the loss of Xho I site may not be a polymorphism associated with disease phenotypes.
Studies conducted to date have focused on EBV within different polymorphic regions, making comparisons across studies difficult and limiting the ability to define the full spectrum of diversity within the EBV genome [27]. Thus, a combination of more than one polymorphic site in the EBV genome might be involved in determining disease characteristics. This study showed a high linkage between V-val-v1 variant (EBNA-1) and China1 variant (LMP-1) in type I EBV strains. There were no significant differences between the distribution of the linkage in the IM, HLH and HL samples (P > 0.05).
There were some limitations of this study. Because of relatively small size of samples, some variants, such as P-thr, was detected with very low positive rate compared with previous study in this region. Some blood samples may harbor a lower level of viral load, and PCR products were not obtained from all IM and HLH samples.
Conclusions
In conclusion, this study described the diversity of the EBV genes and investigated the association between EBV genotypes and the clinical phenotypes of EBV-related diseases in Chinese pediatric cases. Type I EBV was the most prevalent subtype EBV in Chinese pediatric cases and V-val-v1 (EBNA-1) and China1 (LMP-1) variants were the most dominant variants. There was a strong linkage between V-val-v1 variant (EBNA-1) and China1 variant (LMP-1) in type I EBV. The sequence variation in EBV genes may represent a geographic polymorphism since no preferential associations were found between specific EBV variants and specific diseases. A more comprehensive exploration of EBV diversity across its entire genome might allow for the development of a consensus classification system of EBV variants into variant classes that could guide subsequent studies aimed at understanding EBV geographic and disease associations [27].
Methods
Patients and Samples
One hundrard and sixteen peripheral blood specimens were obtained from pediatric patients in Beijing Children's Hospital Affiliated Capital Medical University. Eighty four were IM and 32 were EBV-HLH. IM and HLH were diagnosed according to their respective diagnostic criteria [40, 41]. All procedures were reviewed and approved by the Committee of Human Studies at the Beijing Children's Hospital affiliated the Capital Medical University. A written informed consent was obtained from all patients' parents.
Thirty four paraffin-embedded tissue samples and five fresh tissue samples from HL cases were also enrolled into this study.
DNA extraction
Genomic DNA was extracted from 2 ml whole blood samples by using a blood genomic DNA isolation kit (Cat#RT403, Tiangen, China) according to manufacturer's instructions. A QIAamp DNA FFPE Tissue kit (Cat.No.56404, QIAGEN GmbH, Hilden, Germany) was used to extract the DNA from the paraffin-embedded HL tissues following manufacturer's instructions. The isolation of DNA from the fresh HL biopsies was performed using a phenol-chloroform extraction method.
PCR amplification and sequence analysis
Definition of EBV type I/II
EBV type was determined by PCR with specific primers for EBNA3C. The sequences and positions of these primers are as follows: EBNA3C-F, 5'-AGA AGG GGA GCG TGT GTT GT-3' (B95-8 coordinate 87651-87670); EBNA3C-R, 5'-GGC TCG TTT TTG ACG TCG GC-3' (B95-8 coordinate 87803-87784), which yield an amplification product of 153 bp for EBV-I and a product of 246 bp for EBV-II, as previously described [3]. PCR was performed in 50 ul using 1.5 ul of 10 pM of the forward and reverse primers, 25 ul 2 × Hotsart Taq PCR Master Mix (Tiangen, China), 5 ul genomic DNA. The reaction mixture was initially denatured at 95°C for 5 min followed by 35 cycles including denaturation at 95°C for 45 s, annealing at 56°C for 45 s, extension at 72°C for 1 min, and finally elongation at 72°C for 10 min. After PCR assay, the amplified products were subjected to electrophoresis in 2% agarose gel and visualized under ultraviolet light.
EBNA-1 amplification
EBNA-1 C-ter region was amplified by nested PCR. The primers were from a previous study [1]. PCR was performed in 50 ul contained 1.5 ul of 10 pM of the forward and reverse primers, 25 ul 2 × Hotsart Taq PCR Master Mix (Tiangen, China), and 1.5 ul genomic DNA. PCR conditions were the same to the previous study [1]. The amplified fragments were 329 bp.
LMP-1 amplification
The oligonucleotide primers were designed to amplify the full length of LMP-1. The sequences and positions of these primers are as follows: LMP-1-F1, 5'-AGG GAG TGT GTG CCA GTT AAG-3' (B95-8 coordinate 168053-168073); LMP-1-R1, 5'-CAA ACA CAC GCT TTC TAC TTC C-3' (B95-8 coordinate 169679-169700); LMP-1-FS1, 5'-AGG TTA GTC ATA GTA GCT TAG CTG-3' (B95-8 coordinate 168157-168180); LMP-1-RS1, 5'-TCA ACG CAG TCT TAG GTA TCT G-3' (B95-8 coordinate 168953-168974); LMP-1-FS2, 5'-AGG GAG TCA TCG TGG TGG TGT-3' (B95-8 coordinate 168917-168739); LMP-1-RS2, 5'-ACT GCC CTG AGG ATG GAA CAC-3' (B95-8 coordinate 169466-169486).
The first round polymerase chain reaction (PCR) was performed with LMP-1-F1 and LMP-1-R1. 1 ul of 10 pM of the forward and reverse primers, 12.5 ul 2 × Hotsart Taq PCR Master Mix (Tiangen, China), and 2 ul genomic DNA were mixed in a 25 ul reaction. The reaction mixture was initially denatured at 94°C for 5 min followed by 30 cycles including denaturation at 94°C for 45 s, annealing at 53°C for 45 s, extension at 72°C for 90 s, and finally elongation at 72°C for 5 min. The products of the first round PCR were amplified in the second round PCR with LMP-1-FS1 and LMP-1-RS2 or with LMP-1-FS1, LMP-1-RS1 and LMP-1-FS2, LMP-1-RS2 as described above. In the second round PCR the reaction volume was increased to 50 ul. The amplified fragments were 1648 and 1330 (or 818 and 786) bp, respectively.
For the paraffin-embedded HL tissues, a short fragment of LMP-1 containing the 30 bp deletion region was amplified also using the nested PCR. The sequences and positions of the primers are as follows: del-LMP-1F1, 5'-CTA GCG ACT CTG CTG GAA AT-3' (B95-8 coordinate 167934-167915); del-LMP-1R1, 5'-GAG TGT GTG CCA GTT AAG GT-3' (B95-8 coordinate 167598-167617); del-LMP-1F2, 5'- TGG AGG GAG AGT CAG TCA GGC-3' (B95-8 coordinate 167643-167663); del-LMP-1R2, 5'-ATT GAC GGA AGA GGT TGA AAA-3' (B95-8 coordinate 167897-167877). The first round polymerase chain reaction (PCR) was performed with del-LMP-1F1 and del-LMP-1R1. 1 ul of 10 pM of the forward and reverse primers, 12.5 ul 2 × Hotsart Taq PCR Master Mix (Tiangen, China), and 6 ul genomic DNA were mixed in a 25 ul reaction. The reaction mixture was initially denatured at 95°C for 5 min followed by 30 cycles including denaturation at 94°C for 45 s, annealing at 56°C for 45 s, extension at 72°C for 1 min, and finally elongation at 72°C for 10 min. The products of the first round PCR were amplified in the second round PCR with del-LMP-1F2 and del-LMP-1R2. In the second round PCR the reaction volume was increased to 50 ul and the genomic DNA was decreased to 4 ul. The amplified fragments were 337 and 255 bp, respectively.
Sequence analysis
Sequence analysis was performed on an ABI3730XL Genetic Analyzer. Sequences were aligned and analyzed with Bioedit V7.0.9 software. Sequences were compared with B95.8 reference sequence (GenBank V01555) and with other isolates from GenBank database. Phylogenetic tree were made by MEGA V4.0.2 software.
Statistical analysis
SPSS 13.0 for windows was used for statistical elaboration. A Chi-square test or a Fisher's exact test was used to compare the distribution of EBV type I and II, EBNA-1 and LMP-1 variants between the IM, HLH and HL samples. P-values less than 0.05 were considered to be significant differences.
References
Lorenzetti MA, Altcheh J, Moroni S, Moscatelli G, Chabay PA, Preciado MV: EBNA1 sequences in Argentinean pediatric acute and latent Epstein-Barr virus infection reflect circulation of novel South American variants. J Med Virol 2010, 82: 1730-1738. 10.1002/jmv.21871
Young LS, Rickinson AB: Epstein-Barr virus: 40 years on. Nat Rev Cancer 2004, 4: 757-768. 10.1038/nrc1452
Sample J, Young L, Martin B, Chatman T, Kieff E, Rickinson A, Kieff E: Epstein-Barr virus types 1 and 2 differ in their EBNA-3A, EBNA-3B, and EBNA-3C genes. J Virol 1990, 64: 4084-4092.
Adldinger HK, Delius H, Freese UK, Clarke J, Bornkamm GW: A putative transforming gene of Jijoye virus differs from that of Epstein-Barr virus prototypes. Virology 1985, 141: 221-234. 10.1016/0042-6822(85)90253-3
Robertson ES, Ooka T, Kieff ED: Epstein-Barr virus vectors for gene delivery to B lymphocytes. Proceedings of the national academy of sciences of the United States of Aamerica 1996, 93: 11334-11340. 10.1073/pnas.93.21.11334
Bhatia K, Raj A, Guitierrez MI, Judde JG, Spangler G, Venkatesh H, Magrath IT: Variation in the sequence of Epstein Barr virus nuclear antigen 1 in normal peripheral blood lymphocytes and in Burkitt's lymphomas. Oncogene 1996, 13: 177-181.
Lin SX, Zong YS, Zhang M, Han AJ, Zhong BL, Liang YJ: Study of sequence variations of Epstein-Barr virus LMP1 gene in nasopharyngeal carcinoma. Zhonghua Bing Li Xue Za Zhi 2005, 34: 791-795.
Edwards RH, Seillier-Moiseiwitsch F, Raab-Traub N: Signature amino acid changes in latent membrane protein 1 distinguish Epstein-Barr virus strains. Virology 1999, 261: 79-95. 10.1006/viro.1999.9855
Saechan V, Mori A, Mitarnun W, Settheetham-Ishida W, Ishida T: Analysis of LMP1 variants of EBV in Southern Thailand: evidence for strain-associated T-cell tropism and pathogenicity. J Clin Virol 2006, 36: 119-125. 10.1016/j.jcv.2006.01.018
Sung NS, Edwards RH, Seillier-Moiseiwitsch F, Perkins AG, Zeng Y, Raab-Traub N: Epstein-Barr virus strain variation in nasopharyngeal carcinoma from the endemic and non-endemic regions of China. Int J Cancer 1998, 76: 207-215. 10.1002/(SICI)1097-0215(19980413)76:2<207::AID-IJC7>3.0.CO;2-0
Nagamine M, Takahara M, Kishibe K, Nagato T, Ishii H, Bandoh N, Ogino T, Harabuchi Y: Sequence variations of Epstein-Barr virus LMP1 gene in nasal NK/T-cell lymphoma. VIRUS GENES 2007, 34: 47-54. 10.1007/s11262-006-0008-5
Jin Y, Xie Z, Lu G, Yang S, Shen K: Characterization of variants in the promoter of BZLF1 gene of EBV in nonmalignant EBV-associated diseases in Chinese children. Virol J 2010, 7: 92. 10.1186/1743-422X-7-92
Do NV, Ingemar E, Phi PT, Jenny A, Chinh TT, Zeng Y, Hu L: A major EBNA1 variant from Asian EBV isolates shows enhanced transcriptional activity compared to prototype B95.8. Virus Res 2008, 132: 15-24. 10.1016/j.virusres.2007.10.020
Chen YY, Chang KL, Chen WG, Shibata D, Hayashi K, Weiss LM: Epstein-Barr virus-associated nuclear antigen-1 carboxy-terminal gene sequences in Japanese and American patients with gastric carcinoma. Lab Invest 1998, 78: 877-882.
Wrightham MN, Stewart JP, Janjua NJ, Pepper SD, Sample C, Rooney CM, Arrand JR: Antigenic and sequence variation in the C-terminal unique domain of the Epstein-Barr virus nuclear antigen EBNA-1. Virology 1995, 208: 521-530. 10.1006/viro.1995.1183
Tiwawech D, Srivatanakul P, Karalak A, Ishida T: Association between EBNA2 and LMP1 subtypes of Epstein-Barr virus and nasopharyngeal carcinoma in Thais. J Clin Virol 2008, 42: 1-6. 10.1016/j.jcv.2007.11.011
Zhou XG, Sandvej K, Li PJ, Ji XL, Yan QH, Zhang XP, Da JP, Hamilton-Dutoit SJ: Epstein--Barr virus gene polymorphisms in Chinese Hodgkin's disease cases and healthy donors: identification of three distinct virus variants. J Gen Virol 2001, 82: 1157-1167.
Srivastava WKCAG: Coinfection of multiple strains of Epstein-Barr virus in immunocompetent normal individuals: reassessment of the viral carrier state. BLOOD 2000, 95: 2443-2445.
Yates J, Warren N, Reisman D, Sugden B: A cis-acting element from the Epstein-Barr viral genome that permits stable replication of recombinant plasmids in latently infected cells. Proc Natl Acad Sci USA 1984, 81: 3806-3810. 10.1073/pnas.81.12.3806
Yates JL, Camiolo SM, Ali S, Ying A: Comparison of the EBNA1 proteins of Epstein-Barr virus and herpesvirus papio in sequence and function. Virology 1996, 222: 1-13. 10.1006/viro.1996.0392
Aiyar A, Tyree C, Sugden B: The plasmid replicon of EBV consists of multiple cis-acting elements that facilitate DNA synthesis by the cell and a viral maintenance element. EMBO J 1998, 17: 6394-6403. 10.1093/emboj/17.21.6394
Sugden B, Warren N: A promoter of Epstein-Barr virus that can function during latent infection can be transactivated by EBNA-1, a viral protein required for viral DNA replication during latent infection. J Virol 1989, 63: 2644-2649.
Sandvej K, Zhou XG, Hamilton-Dutoit S: EBNA-1 sequence variation in Danish and Chinese EBV-associated tumours: evidence for geographical polymorphism but not for tumour-specific subtype restriction. J Pathol 2000, 191: 127-131. 10.1002/(SICI)1096-9896(200006)191:2<127::AID-PATH614>3.0.CO;2-E
Mai SJ, Ooka T, Li DJ, Zeng MS, Jiang RC, Yu XJ, Zhang RH, Chen SP, Zeng YX: Functional advantage of NPC-related V-val subtype of Epstein-Barr virus nuclear antigen 1 compared with prototype in epithelial cell line. Oncol Rep 2007, 17: 141-146.
Zhang XS, Wang HH, Hu LF, Li A, Zhang RH, Mai HQ, Xia JC, Chen LZ, Zeng YX: V-val subtype of Epstein-Barr virus nuclear antigen 1 preferentially exists in biopsies of nasopharyngeal carcinoma. Cancer Lett 2004, 211: 11-18. 10.1016/j.canlet.2004.01.035
Wang Y, Liu X, Xing X, Cui Y, Zhao C, Luo B: Variations of Epstein-Barr virus nuclear antigen 1 gene in gastric carcinomas and nasopharyngeal carcinomas from Northern China. Virus Res 2010, 147: 258-264. 10.1016/j.virusres.2009.11.010
Chang CM, Yu KJ, Mbulaiteye SM, Hildesheim A, Bhatia K: The extent of genetic diversity of Epstein-Barr virus and its geographic and disease patterns: a need for reappraisal. Virus Res 2009, 143: 209-221. 10.1016/j.virusres.2009.07.005
Hu LF, Zabarovsky ER, Chen F, Cao SL, Ernberg I, Klein G, Winberg G: Isolation and sequencing of the Epstein-Barr virus BNLF-1 gene (LMP1) from a Chinese nasopharyngeal carcinoma. J Gen Virol 1991,72(Pt 10):2399-2409.
Baer R, Bankier AT, Biggin MD, Deininger PL, Farrell PJ, Gibson TJ, Hatfull G, Hudson GS, Satchwell SC, Seguin C, et al.: DNA-sequence and expression of the B95-8 Epstein-Barr Virus genome. Nature 1984, 310: 207-211. 10.1038/310207a0
Fennewald S, Vansanten V, Kieff E: Nucleotide-sequence of an messenger-RNA transcribed in latent growth-transforming virus-infection indicates that it may encode a membrane-protein. Journal of Virology 1984, 51: 411-419.
Hatzivassiliou E, Mosialos G: Cellular signaling pathways engaged by the Epstein-Barr virus transforming protein LMP1. Front Biosci 2002, 7: d319-d329. 10.2741/hatziva
Izumi KM, Kieff ED: The Epstein-Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-kappaB. Proc Natl Acad Sci USA 1997, 94: 12592-12597. 10.1073/pnas.94.23.12592
Miller WE, Edwards RH, Walling DM, Raab-Traub N: Sequence variation in the Epstein-Barr virus latent membrane protein 1. J Gen Virol 1994,75(Pt 10):2729-2740.
Hadhri-Guiga B, Khabir AM, Mokdad-Gargouri R, Ghorbel AM, Drira M, Daoud J, Frikha M, Jlidi R, Gargouri A: Various 30 and 69 bp deletion variants of the Epstein-Barr virus LMP1 may arise by homologous recombination in nasopharyngeal carcinoma of Tunisian patients. Virus Res 2006, 115: 24-30. 10.1016/j.virusres.2005.07.002
Guiretti DM, Chabay PA, Valva P, Stefanoff CG, Barros MH, De Matteo E, Renault IZ, Preciado MV, Hassan R: Structural variability of the carboxy-terminus of Epstein-Barr virus encoded latent membrane protein 1 gene in Hodgkin's lymphomas. J Med Virol 2007, 79: 1722-1730.
See HS, Yap YY, Yip WK, Seow HF: Epstein-Barr virus latent membrane protein-1 (LMP-1) 30-bp deletion and Xho I-loss is associated with type III nasopharyngeal carcinoma in Malaysia. World J Surg Oncol 2008, 6: 18. 10.1186/1477-7819-6-18
Zhou XG, Sandvej K, Li PJ, Ji XL, Yan QH, Zhang XP, Da JP, Hamilton-Dutoit SJ: Epstein--Barr virus gene polymorphisms in Chinese Hodgkin's disease cases and healthy donors: identification of three distinct virus variants. J Gen Virol 2001, 82: 1157-1167.
Chen ML, Tsai CN, Liang CL, Shu CH, Huang CR, Sulitzeanu D, Liu ST, Chang YS: Cloning and characterization of the latent membrane protein (LMP) of a specific Epstein-Barr virus variant derived from the nasopharyngeal carcinoma in the Taiwanese population. Oncogene 1992, 7: 2131-2140.
Tan EL, Peh SC, Sam CK: Analyses of Epstein-Barr virus latent membrane protein-1 in Malaysian nasopharyngeal carcinoma: high prevalence of 30-bp deletion, Xho1 polymorphism and evidence of dual infections. J Med Virol 2003, 69: 251-257. 10.1002/jmv.10282
Henter JI, Horne A, Arico M, Egeler RM, Filipovich AH, Imashuku S, Ladisch S, McClain K, Webb D, Winiarski J, Janka G: HLH-2004: Diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer 2007, 48: 124-131. 10.1002/pbc.21039
Tsuchiya S: Diagnosis of Epstein-Barr virus-associated diseases. Crit Rev Oncol Hematol 2002, 44: 227-238. 10.1016/S1040-8428(02)00114-2
Acknowledgements
We wish to express our thanks to the research project sponsored by the Fund of Training Program of High Level Technological Personnel in Beijing Health Bureau (No. 2009-3-42) from the Beijing municipal government. We thank the staff of Virology laboratory in Beijing Children's Hospital for collecting the samples used in these studies.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JHA carried out most of the experiments and wrote the manuscript. CYL, ZZH and JMX helped with the experiments. ZDX designed the experiments and revised the manuscript. All of the authors read and approved the final version of this manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Ai, J., Xie, Z., Liu, C. et al. Analysis of EBNA-1 and LMP-1 variants in diseases associated with EBV infection in Chinese children. Virol J 9, 13 (2012). https://doi.org/10.1186/1743-422X-9-13
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1743-422X-9-13