
Abstract
Background
The Falkland Islands and Patagonia are traditionally assigned to the Magellan Biogeographic Province. Most marine species in Falkland waters are also reported from southern Patagonia. It remains unclear if relatively immobile, marine benthic, shallow-water species maintain gene flow, and by what mechanism. Recurrent fluctuations in sea level during glacial cycles are regarded as a possible mechanism that might have allowed genetic exchange between the regions. However, the realized genetic exchange between the Falkland Islands and Patagonia has never been estimated.
Results
This study analyses the genetic structure of three populations of the marine shallow-water isopod Serolis paradoxa (Fabricius, 1775) from the Falkland Islands and southern Patagonia (central Strait of Magellan and the Atlantic opening) applying seven nuclear microsatellites and a fragment of the mitochondrial 16S rRNA gene. Both marker systems report highest genetic diversity for the population from the central Strait of Magellan and lowest for the Falkland Islands. The estimated effective population sizes were large for all populations studied. Significant differentiation was observed among all three populations. The magnitude of differentiation between Patagonia and the Falkland Islands (16S: uncorrected p-distance 2.1%; microsatellites: standardized F'ST > 0.86) was an order of magnitude higher than between populations from within Patagonia. This indicates that there is currently no effective gene flow for nominal S. paradoxa between these two regions and it has been absent for time exceeding the last glacial maximum. We argue that specimens from the Strait of Magellan and the Falkland Islands very likely represent two distinct species that separated in the mid-Pleistocene (about 1 MY BP).
Conclusion
The results of this study indicate limited gene flow between distant populations of the brooding isopod Serolis paradoxa. The patterns of genetic diversity suggest that the only recently inundated Strait of Magellan was colonized by different source populations, most likely from Atlantic and Pacific coastal waters. Our results demonstrate that more systematic testing of shared faunal inventory and realized genetic exchange between Patagonia and the Falkland Islands is needed before a consensus concerning the position of the Falkland Islands relative to the Magellan zoogeographic province can be reached.
Similar content being viewed by others

Background
Present-day distribution of a species is the result of a complex interplay between (1) extrinsic factors such as isolation of landmasses, climatic conditions and availability of niches and (2) intrinsic factors such as dispersal capability and physiological tolerance. Extrinsic factors typically influence the distribution of many species in the same way because they act on an ecosystem scale. Over time, this leads to a characteristic assemblage of species with similar distribution patterns within larger geographical areas, so called biogeographic provinces, and distinct gaps between them [1]. In the Southern Hemisphere, the Magellan Biogeographic Province has obvious close ties to the whole of South America to which it is connected today; some of its species inventory, however, stems from times before the Gondwana breakup [2, 3]. The Falkland Islands are connected to the South American shelf and located approximately 500 km to the east of Patagonia (Figure 1). Based on the current knowledge of their species inventory, the Falkland Islands are commonly assigned to the Magellan Biogeographic Province [4–7], sometimes as a more distinct 'subregion' of this province ([8] and references therein). The geologic history of the Falkland Islands is completely detached from continental South America as the islands drifted to their current position on a microplate that originally formed part of the African continental plate. Their current position was reached approximately 130 MY BP [9–11]. Close biogeographic ties between the Falkland Islands and Patagonia are widely accepted and even more plausible in the marine realm because here biotic exchange does not depend on the existence of land bridges [12–15]. Furthermore, major ocean current systems facilitate dispersal of specimens. This is generally supported by elevated levels of gene flow among the few species investigated so far in this region [16–19].

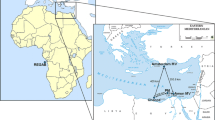
Sampling sites of Serolis paradoxa in the Strait of Magellan near Punta Arenas (PA), the opening to the Atlantic Ocean (AO) and the Falkland Islands (FI).
In this context, the relatively few reports of species endemic to the Falkland Islands are not unexpected [8, 20–22]. On the whole, evidence from marine species supports that the Falkland Islands form a part of the Atlantic Magellan Biogeographic Province and that migration of species between the continental South America and the Falkland Island is occurring repeatedly. However, recent molecular studies have shown that unrecognized cryptic species may be more common than previously assumed [15, 17, 23–33]. They indicate that morphological and ecological similarity may be an unreliable piece of evidence on which to base taxonomic identifications and, by extension, the definition of biogeographic provinces derived from them.
In this study we investigate spatial partitioning of intraspecific molecular polymorphisms in nominal Serolis paradoxa (Fabricius, 1775), a marine benthic shallow-water isopod, using two independent genetic marker systems. S. paradoxa was originally described from the Falkland Islands but is also frequently reported from the Strait of Magellan, the Patagonian coastal shelf (Atlantic and Pacific side), and also from regions further to the equator [5, 20]. For the current taxonomic status and synonyms of S. paradoxa see [3]. The vertical distribution of S. paradoxa ranges from shallow waters (about 5 m, Held pers. observ., Lopaz-Gappa pers. observ.) down to 113 m [34]. Although in the Magellan region S. paradoxa can be locally very abundant [35] it is often encountered at medium densities (about 1 ind/m2[36], Mutschke and Rios pers. comm.). Like almost all isopods, S. paradoxa lacks free-swimming distribution stages and broods its offspring in a ventral brood pouch, the marsupium, and is thus expected to be limited in its dispersal capabilities. No information on the life cycle and the duration of S. paradoxa are known. Based on significantly extended embryonic stages of Antarctic serolid isopods in comparison to non-Antarctic isopods [37] it can be assumed that embryonic development, maturation and brooding of S. paradoxa from the cool-temperate regions, each stage lasts several months. Altogether, S. paradoxa is expected to have very limited dispersal capacity due to these factors.
Direct measurements of dispersal and migration over large geographical distances provide a poor means of assessing effective gene flow. The small number of immigrants needed per generation to appreciably change the genetic structure of a population will not be picked up by realistic sampling schemes. Indirect genetic estimates use tools that interpret the genetic structure of a population as a result of past genetic influx and thus represent an easier and more reliable method [38]. However, in this context historic extrinsic factors that may have exerted a structuring force must be considered when estimating present-day population structure. One extrinsic factor known to have had a major impact on genetic structure and distribution of species are glaciation events [39]. Their influence on the marine fauna is two-fold: large-scale glaciations may directly render entire coastal habitats unavailable during glacial maxima [40–42] and also lead to a decrease in sea level of up to 130 m [41]. The latter may disrupt inshore habitats on either side of an emerging barrier (e.g. appearance of the Panama land bridge [12], or connect shallow water habitats that are disjunct during periods of high sea level (additional file 1).
The focus of the present study is the genetic structure of S. paradoxa from the Falkland Islands and the Patagonian shelf. The present-day situation suggests that the deeper waters on the South American shelf may present an insurmountable barrier to S. paradoxa inhabiting shallow waters around Patagonia and the Falkland Islands. Historically, the lower sea level during glacial maxima may have connected both regions and facilitated migration between habitats thus counteracting independent lineage sorting in the two regions.
By investigating the coherence between gene pools and construction of an approximate timeline, we test whether the disruptive or unifying forces predominated and if the influence of the last glaciations exerted a major influence on the evolutionary history of S. paradoxa. We also test whether the major age difference between marine habitats in the central Strait of Magellan and around the Falkland Islands (< 14 KY BP [43–50] and millions of years [51], respectively) exerted a measurable influence on the genetics of populations living there today. In particular, we test for differences in genetic diversity and patterns of recent population expansions or secondary contact of colonizers from the Atlantic and the Pacific side in central Magellan Strait. We hypothesize that populations in the central Strait of Magellan are genetically less diverse than populations from the coast or the Falkland Islands due to recent range expansion into the Magellan Strait subsequent to the retreat of the glaciers after the last glacial maximum (LGM).
Materials and methods
Taxon sampling
Specimens from the Falkland Islands (FI) were collected by dredging in shallow waters (< 20 m) near Port Stanley from a Zodiac during the ICEFISH 2004 expedition. Specimens from the Strait of Magellan near Punta Arenas (PA) were collected in January 1997 by CH SCUBA diving at two neighbouring stations (500 m apart) and specimens from the Atlantic opening of the Strait of Magellan (AO) were provided from the 2nd Cruce Estrecho in 2003, by Carlos Rios and Erika Mutschke, Universidad de Magallanes, Punta Arenas (see Figure 1 and Table 1). Animals were immediately preserved in 96% ethanol. Microsatellite analyses were performed for 35 specimens from PA, 33 from AO and 23 from FI. A subset of 27 specimens from PA, 22 from AO and 22 from FI were analysed for variation of the 3'-terminus of the mitochondrial 16S rRNA gene.
DNA extraction, PCR, sequencing/genotyping
Total DNA was extracted from muscle tissue using the Qiagen DNeasy Mini Kit according to the standard tissue protocol. Only 100 μl of elution buffer were used to increase DNA concentration.
Microsatellites
Microsatellite markers Spa04, Spa12, Spa34, Spa35, Spa39, Spa42 and Spa43 [32] were applied to assess intraspecific genetic polymorphisms for all specimens from the three sampling sites. Standard 15 μl reactions consisted of 1× PCR HotMaster Buffer, 0.2 mM dNTPs, 0.5–0.75 μM of each primer (one labelled, one unlabelled), 0.03 U/μl HotMaster Taq (Eppendorf, 5-Prime), 0.5 M Betaine (Sigma Aldrich) and 5–20 ng of genomic DNA. Cycling conditions on an epgradient thermocycler (Eppendorf) were 2 min at 94°C followed by 30 to 34 cycles with 20 s at 94°C, 15 s at annealing temperature, 30 s at 65°C. A final extension step of 45 minutes at 65°C was performed to reduce in vitro artefacts due to incomplete adenylation of products [see [32] for details]. PCR products were visualized on 2% TBE agarose gels, diluted 1–15 fold with molgrade water (CARL ROTH) and 1 μl of the diluted product was denatured in a mixture of 14.7 μl HI-DI formamide with 0.3 μl GeneScan ROX 500 size standard (both Applied Biosystems). Allele length scoring was performed using the software GENEMAPPER 4.0 (Applied Biosystems). To minimize genotyping errors [52, 53], up to four independent reactions were performed on a subset of samples to estimate allele calling errors.
16S rDNA
The universal primers 16Sar and 16Sbr [54] were used for amplification. Reactions were carried out in 25 μl volumes with 1× HotMaster reaction buffer, 0.2 mM dNTPs, 0.5 μM of each primer, 0.025 U/μl HotMaster Taq (Eppendorf, 5-Prime). Reaction conditions were: Initial denaturation for 2 min at 94°C followed by 36 cycles of 20 s at 94°, 15 s at 46°C and 80 s at 65°C plus a final elongation step of 5 min at 65°C. PCR products were purified using Qiagen QIAquick or Eppendorf Perfectprep Gel cleanup kits. Cycle-sequencing was performed in 10 μl reaction volumes using 1 μM of either 16Sar or 16Sbr primer, 1 μl of the purified template DNA and the BigDye Terminator Kit 3.1 chemistry (Applied Biosystems) according to the recommendations of the manufacturer. Reactions were purified according to the 'modified protocol' of the Qiagen DyeEx Kit. Sequencing was conducted on an ABI 3130xl sequencer.
Data analysis
Microsatellites
Raw data were checked and corrected for genotyping errors using the software MICRO-CHECKER version 2.2.3 [55] and DROPOUT version 1.3 [56]. In addition, MICRO-CHECKER was used to test for the presence of null alleles in populations, i.e. alleles that fail to amplify due to substitutions in the primer binding regions. Corrected genotype tables were converted to specific software formats using the software MSTOOLKIT version 3.1 [57] and CONVERT version 1.3.1 [58]. The program ANIMALFARM version 1.0 [59] was used to test for loci with significantly disproportionate variances that may bias allele-size based distance estimates such as Slatkin's or Rousset's RST estimates [60, 61]. Tests for Hardy Weinberg equilibrium (HWE) and linkage disequilibrium (LD) were performed using GENEPOP version 4.0.6 [62]. Parameter settings: 10,000 dememorization steps, 50 batches, 20,000 MCMC sampling steps. HWE tests aim at testing whether there is a statistically significant deviation of genotype frequencies from those expected according to Mendelian inheritance. Linkage disequilibrium occurs when two genomic loci are not inherited independently, e.g. due to physical linkage or other processes at population level hindering independent recombination of loci.
To assess partitioning of genetic variability within individuals, subpopulations and regions, we performed hierarchical analyses of molecular variance (AMOVA) using ARLEQUIN version 3.11 [63]. Therefore, populations PA and AO were assigned to one group, while FI constituted the other group. In addition, single and multilocus inbreeding coefficients (FIS) and pairwise population coancestry coefficients (FST, similar to Weir and Cockerham's Theta) were estimated as in [64] using GENEPOP. We also calculated pairwise allele-size based differentiation estimates, RST, according to [61] using GENEPOP. Significance was assessed by exact G tests as implemented in GENEPOP. The interpretation of the FST values from multiallelic data is problematic because their maximum values depend on the amount of within-population variation and even in the absence of any shared allele often fail to reach the theoretical maximum of 1 [65–67]. We therefore applied a standardization approach suggested by Hedrick [67] for calculations of GST and derived by Meirmans [68] for Analysis of Variance frameworks (ANOVA).
The main principle of this standardization approach is to correct the maximum possible value for FST as follows: FST(max) is calculated using GENEPOP applying the sampling bias correction suggested by Meirmans [68] using the Software RECODEDATA[68]. F'ST was subsequently calculated by dividing FST by this inferred maximum value.
The standardized F'ST measure calculated range from 0 (populations equifrequent for all alleles) to 1 (populations fixed for different alleles) and therefore makes interpretation of the degree of subdivision much easier and facilitates comparing results among studies.
In addition to these ANOVA based coancestry estimates we performed individual assignment tests using the program STRUCTURE, version 2.2.2 [69] to investigate population subdivision. The advantage of the Bayesian clustering algorithm of STRUCTURE is that no classification of populations has to be done a priori. Assuming HWE and no or only weak LD within subpopulations, STRUCTURE assigns individual genotypes probabilistically to populations and calculates the likelihood of the genotype dataset for a given number of populations (K), i.e. ln Pr (D|K) for K = 1 to K = n, using a Markov Chain Monte Carlo algorithm [69, 70]. For the S. paradoxa data set, the most likely number of populations was inferred without making assumptions on geographic origin of individuals. The number of MCMC steps needed to reach convergence was first estimated by comparing run lengths between 10,000 and 2,000,000 steps. Convergence was generally reached with <5,000 steps. Therefore, for the parameter sets 10 independent runs with a burn-in of 5,000 and subsequent 100,000 MCMC steps were performed with and without assuming recent admixture in the prior model, and considering alleles as correlated and uncorrelated. The number of clusters (K) to infer was defined from K = 1 to K = 4 to allow detection of potential cryptic subpopulations. Alpha was inferred from the data for each population separately. Results from 10 independent runs were analysed in CLUMPP, version 1.1.1 [71] to compute a consensus membership coefficient Q-matrix from all 10 independent Q-matrices. Both the individual Q-matrix and averaged population Q-matrix were visualized using DISTRUCT, version 1.1 [72].
To assess estimates of the present effective population size (N e ), we applied the linkage disequilibrium method proposed by Hill (1981) [73], modified by Waples [74] to account for a bias correction when sample size is much smaller than effective population size. This method is implemented in the program LDNE, version 1.3 [75]. Calculations of N e and the confidence intervals (CI) were estimated considering alleles with a frequency of c ≥ 0.05 and c ≥ 0.02 and ≥ 0.01, respectively.
Tests for historical population bottlenecks were performed using the program BOTTLENECK[76]. Tests implemented in this program are based on the hypothesis that populations that have experienced recent reductions in their effective population size (N e ) show a reduction in both allelic richness and heterozygosity. In populations decreasing in size, the number of alleles (N A ) drops faster than heterozygosity [77] and therefore the observed heterozygosity is larger than the expected heterozygosity (H O > H E ). Conversely, in expanding populations often the number of alleles increases faster than heterozygosity until equilibrium is reached. From the comparison of both parameters, allelic diversity and heterozygosity, it is possible to make inferences regarding historical demography of a population. For each locus and population BOTTLENECK computes distribution of H E expected from the observed N A , given the sample size (n) under the assumption of mutation-drift equilibrium. This distribution is obtained through simulating the coalescent process of n genes under the three possible mutation models, i.e. a) the Infinite Allele Model (IAM), b) the Two-Phase Model (TPM), c) the Stepwise-Mutation Model (SMM). As recommended by Cornuet and Luikart [78] we tested several proportions of the SMM for the TPM (70–90%). Statistical significance of the parameters were inferred applying a Sign-test and a Wilcoxon-rank-test [76, 78, 79].
16S rDNA
Assembly of forward and reverse strands and editing was performed using the software SEQMAN (Dnastar, Lasergene) and GENEIOUS version 4.0.2 (Biomatters Ltd.). Sequence alignment was performed using MUSCLE version 3.6 [80]. The alignment required no manual correction based on secondary structure information [81]. Sequence variation was analyzed using MEGA 4.0 [82]. Gene diversity and nucleotide diversity according to Nei [83] and Theta based on the number of segregating sites, Theta (S), were calculated with ARLEQUIN version 3.11. Genetic differentiation between populations and between regions [(PA + AO) vs. FI] were assessed using an FST and AMOVA framework as implemented in ARLEQUIN. Assuming neutrality, evidence of a population expansion was tested applying Tajima's D [84] and Fu's F S statistic [85] as implemented in ARLEQUIN applying a coalescent simulation approach generating 10,000 selectively neutral samples for assessment of significance of results. A test for sudden population expansion based on the pairwise mismatch distribution was calculated using ARLEQUIN and significance was assessed by 50,000 pseudo replicates.
A statistical parsimony network with a 95% connection-probability limit was created for the 490-bp alignment using TCS version 1.21 [86]. In addition, two outgroup sequences of the serolid isopods Cuspidoserolis luethjei and Cuspidoserolis johnstoni (GenBank accession numbers AJ269802, AJ269803; see [24]) were aligned to the S. paradoxa sequences using MUSCLE, resulting in a 492-bp alignment. This alignment was used to calculate a neighbor joining tree [87] with bootstrap support (1000 replicates) based on uncorrected p-distances using PAUP* version 4b10 [88].
The coalescent-based MCMC approach implemented in the software BEAST[89] was used to date the splitting event between the different Serolis paradoxa lineages applying both a strict and a relaxed molecular clock model. The sequence model HKY85 was used for modelling sequence evolution [90] together with a predefined mutation rate of 0.37% per million years [91], based on a molecular clock for the serolid isopod Ceratoserolis trilobitoides. Dating times and confidence intervals (CI) were filtered using TRACER version 1.4 [92].
Results
Microsatellites
Seven microsatellite loci were analysed for three populations. All loci were highly polymorphic for all three populations (Table 2). The number of alleles per microsatellite locus ranged from 6 to 23. The observed heterozygosity ranged from 0.0 (Spa39, all specimens homozygous for populations FI) to 0.886. Total heterozygosity was highest in PA, lower in AO and lowest in FI (Table 2). Significant deviations from HWE were detected for loci Spa04 and Spa39 (Table 2). Analyses with MICRO-CHECKER indicate that null alleles may be the cause for inflated homozygosity for these loci. No significant global LD was observed after sequential Bonferroni correction [93]. Based on allele frequencies, the geographically intermediate population AO is clearly more similar to PA than FI (additional file 2). The allele distribution of all microsatellite loci reveal strongly differing frequency spectra with several private and almost fixed different allele patterns between regions (e.g. loci Spa04, Spa12, Spa35, Spa43). Allele length spectra differ between populations but overlap (additional file 2).
Results of ANIMALFARM confirmed that none of the loci contributed disproportionally to distance-based differentiation estimates after Bonferroni or Sidak adjustment of the significance level.
Analyses of the AMOVA indicate that most variation is distributed among individuals (Table 3). ΦIS and the global multilocus inbreeding coefficient FIS are significantly positive for all populations (Table 2, Table 3) indicating further within-population structure. However, a large proportion of variation is distributed among the major geographical regions [(PA + AO) vs. FI] and only a minor but nonetheless significant proportion between populations (Table 3). Strong differences between the two regions were observed for allele frequency patterns at all loci. In particular loci Spa04, Spa12 and Spa43 are nearly fixed for different alleles in populations from the two major regions [(PA, AO) vs. FI] (additional file 2) whereas PA and AO reveal very similar allele frequency patterns. The pronounced differences separating FI from Patagonia are expressed by the high and significant pairwise FST and RST estimates between population PA and FI and populations AO and FI (Table 4). Differentiation estimates were even higher between populations AO and FI. In contrast, FST estimates among populations PA and AO were low albeit significant (P = 0.0005). RST estimates were lower than FST estimates in this study and did not support significance differentiation between PA and AO (Table 4). In general, RST is hypothesized to be larger if an appreciable amount of differentiation between populations is not only caused by drift but by independent mutations in the different, isolated populations according to a stepwise mutation model (SMM). Consequently, RST distance measures are considered a 'memory' of past mutations [60]. FST is superior to RST when populations have diverged mostly by means of random genetic drift and migration m (i.e. mutation rate << migration).
Standardized pairwise FST calculates [67] in this study showed very strong pairwise population differentiation between Patagonia and the Falklands (PA vs FI: 0.86; AO vs FI: 0.91), and much smaller values among the Magellan Strait populations (PA vs AO: 0.063). These values demonstrate that both regions are almost fixed for different alleles at the seven loci investigated. When removing locus Spa39, which is biased due to the presence of null-alleles in population FI, the standardized values did not change, however, the non-standardized FST values were almost twice as high (data not shown).
Inferring the most likely number of populations without making assumptions concerning their delimitation, STRUCTURE identified only two very distinct clusters, which correspond to the two major regions (AP+AO vs. FI; Figure 2). When using the no-admixture model, all individuals are correctly assigned to the two regions with admixture proportions of 1.000 and 0.000, respectively. There was no additional substructure within populations (PA, AO, FI), i.e., when analysing the populations separately Ln Pr (D|K) was highest for K = 1. No significant differentiation between population PA and AO was detected (compare FST estimates, Table 4).

Results of cluster analyses performed with S TRUCTURE (admixture model, allele frequencies correlated) with the highest log likelihood probability. The graphs display the consensus membership coefficients matrices (Q-matrices) for 91 individuals from three populations of Serolis paradoxa using seven microsatellite loci. The genotype of each individual is represented by a single bar, where the proportion of the colour refers to the probability of assignment to a certain cluster.
Estimating the present effective population sizes using the LD approach [74], we consistently received negative Ne estimates with confidence intervals ranging from negative values to infinity, thus indicating very large population sizes [75]. When testing for recent demographic contractions or expansions by looking for deviations from mutation-drift equilibrium under different mutation models using BOTTLENECK we found a significant heterozygosity deficiency under particular mutation models. For population AO there was a significant heterozygosity deficit under both SMM and TPM models (additional file 3), which provides strong evidence for recent population expansion. For FI the evidence for recent population expansion was somewhat weaker: a significant heterozygosity deficit was detected only using the SMM and the Wilcoxon test (P = 0.0195, additional file 3). Thus results of BOTTLENECK do not provide evidence for a similarly drastic decline and subsequent recovery in population size for PA. For population AO under a TPM and a strict SMM, the significant excess of heterozygosity may indicate that this population is expanding presently. Although the evolutionary dynamics of microsatellites are not fully understood [94, 95] it is commonly accepted that the IAM model is not an appropriate descriptor of the mutational dynamics of microsatellite markers and hence that its application often leads to unrealistic conclusions.
In summary, the results from the microsatellite analyses provide evidence for moderate differentiation between the two Patagonian populations and very strong subdivision between populations from Patagonia and the Falkland Islands. Genetic diversity was highest in the center of the Strait of Magellan, lower near its opening towards the Atlantic Ocean and lowest around the Falkland Islands. All populations showed a significant heterozygosity deficit corroborated by high FIS values (Table 2) which may be indicative for inbreeding of local populations.
16S rDNA
We sequenced a 490 bp fragment of the 16S rRNA gene for a subset of 27 specimens from population PA, 22 from AO and 22 from FI (Table 1) to test whether the strong pattern of differentiation inferred using fast evolving microsatellites is also traceable with a slower evolving gene. The amplified fragment was AT-rich as typical for this gene (A 39.3%, C 13.1%, G 14.0%, T 33.6%; [54]). Of the seventeen polymorphic positions, ten were parsimony informative and seven represented singletons. Substitutions were located only in loop regions of the rRNA gene fragment (folding model: Drosophila melanogaster 16S rRNA, [81]). Eleven haplotypes were characterized (HT1-HT11, Table 5). The statistical parsimony network constructed is characterized by two shallow subnetworks (≤ 4 segregating sites) representing the Patagonian vs. the Falkland Islands populations, which are connected by a long internal branch (8 segregating sites, Figure 3). None of the haplotypes was shared among specimens from different regions. Phylogenetic analyses revealed that specimens from both regions form two reciprocally monophyletic clades each supported by high bootstrap values (Figure 4). The average uncorrected pairwise distance between both groups was 2.1% (1.7% for transitions; 0.4% for transversions). Variation within groups was an order of magnitude lower (0.3% among FI and 0.4% among PA and AO; Figure 4). These values are amongst the lower values observed between reproductively isolated species of serolid isopods and other crustaceans [24, 26–28, 31]. This dominant partitioning of genetic diversity by regions was supported by high and highly significant (P < 0.001) values for pairwise population differentiation (FST) between populations from both regions: PA:FI FST = 0.95; AO:FI, FST = 0.97). The results of the AMOVA indicate that 90.48% of the total variance is partitioned between the two regions [(PA and AO) vs. (FI)], corresponding to a high value of ΦCT = 0.905 (Table 6). However, a lower but highly significant proportion of total variance was also distributed between populations AO and PA (4.70%), corresponding to the value of ΦSC of -0.494. Pairwise differentiation estimates support that gene flow is also restricted within the Strait of Magellan (PA:AO FST = 0.45; P < 0.001).

Statistical parsimony network of 16S rDNA haplotypes of Serolis paradoxa from the Strait of Magellan (white and grey) and the Falkland Islands (black). Branches in subnetwork represent one substitution except for the branch connecting HT4 and HT7, which differ by eight mutations.

Neighbor joining tree based on uncorrected p-distances of 3'-terminus of the mitochondrial 16S rRNA gene for the 11 haplotypes from S. paradoxa sampled from Patagonia (HT1-HT6, HT11) and the Falkland Islands (HT7-10). Sequences of Cuspidoserolis luethjei, AJ269802 and C. johnstoni, AJ269803 [15] were used as outgroup. Numbers on branches represent bootstrap support (1000 replicates).
Nucleotide diversity and gene diversity were highest for population PA, lower for AO and lowest for FI (Table 7). Estimates of Theta (S) were also higher for PA than for AO and FI (Table 7). Tajima's D and Fu's Fs were negative and significantly different from zero for FI, and for Fu's Fs only for population AO (Table 7). Recent population expansions are frequently associated with negative values of D and Fs because under these circumstances mutation generates more and closely related haplotypes than are eliminated by genetic drift. It should be considered that according to Fu [85] a significance level of 5% corresponds to P = 0.02, thus Fu's Fs for population AO is not significant. The mismatch analyses could not the reject the assumptions of sudden population expansion for all populations (additional file 4). In summary, there is a stronger signature for population expansion in AO and FI than in PA based on the 16S rDNA data.
Dating of the time to the most recent common ancestor (tMRCA) between both S. paradoxa lineages using a strict molecular clock for the mutation rate differed for the two monophyletic lineages. For the Patagonian taxa as ingroup the tMRCA inferred was 0.948 MY (5% CI 0.344 MY, 95% CI 1.658 MY) and for the Falkland Islands lineage 0.643 MY (5% CI 0.136 MY, 95% CI 1.207 MY). Thus, from both inferences, evidence for a splitting event in the mid-Pleistocene is supported.
Discussion
The genetic variability within the nominal species Serolis paradoxa turned out to have extensive spatial structure. The differences in mutation rates and coalescent dynamics of the two marker systems help describe present-day population structure and reconstruct historical demographic processes.
Two genetically distinct lineages
There is strong evidence of divergence between populations from Patagonia and the Falkland Islands, supported by microsatellite and mitochondrial data. The dominant feature of the intraspecific variability of mitochondrial DNA data for the Patagonian populations (PA and AO) and the Falkland Island population is that populations form two shallow subnetworks, corresponding to the two geographic regions. The nuclear microsatellite markers support the geographic partitioning of variation with high and significant FST, RST differentiation values and strong support from Bayesian cluster analyses (Figure 2).
The geographic positions of our sampling locations along an East-West axis might suggest testing for isolation by distance effects (IBD). However, in the context of this study, the IBD is an inappropriate method and it is unlikely that this would become more meaningful even if more intermediate sampling locations were available. This is because the central Strait of Magellan became available for (re)colonization only very recently, approximately 9–14 KY BP [46, 48]. This rapid range expansion is typically accompanied by loss of alleles and an excess of homozygosity [96] which violates a mutation-drift equilibrium assumed by the IBD model. Investigating distance effects on the distribution of intraspecific variance inside the Magellan Strait offers a means to trace the recolonization of this young habitat and would be appropriate for IBD but this requires more fine-scaled sampling and is outside the scope of this paper.
Absence of effective gene flow between the Falkland Islands and Patagonia is strongly suggested by nearly fixed population specific differences in fast evolving microsatellites and the perfect congruence of haplotype identity and geography for the 16S rDNA data. The long branch connecting the two groups of haplotypes (Figure 3) and their reciprocal monophyly (Figure 4) indicates complete lineage sorting in both groups. The magnitude of genetic differentiation between 16S genotypes is on the order of magnitude known for reproductively isolated species [26–28, 97]. Speciation ultimately involves the irreversible disruption of a once contiguous gene pool into two [98]. The recognition of species thus centers around direct or indirect evidence for gene flow between them. Our data from two independent molecular markers are in line with the expectations of two independently evolving lineages. The patterns and magnitude of the remaining differences do not suggest the presence of additional cryptic species inside (PA and AO) vs. (FI), they indicate, however, that gene flow is restricted even within the Strait of Magellan. The congruence between both marker systems supports that the 16S rRNA gene tree reflects the species tree rather than being a result of shared ancestral polymorphisms [99] or other processes affecting mitochondrial genes (see [100] for review).
Evolutionary history of nominal Serolis paradoxa
Southern Hemisphere glaciations differently affected both regions: The Falkland Islands were little affected by glacial advances [51], thus S. paradoxa was able to survive by following the rising and falling sea levels. In Patagonia, however, major parts of today's distribution of nominal S. paradoxa became unavailable due to ice coverage and/or low sea levels. Western Patagonia was covered by a contiguous ice shield similar to the Antarctic Peninsula today and the central Strait of Magellan was inundated only after the LGM, approximately 14-9 KY BP [46]. Contrary to the situation around the Falkland Islands where S. paradoxa was presumably continuously present over evolutionary times, this species was forced to immigrate into the Strait of Magellan only recently after the retreat of the glaciers. Surprisingly, genetic diversity estimates for population PA from central Magellan Strait (Table 7) indicate that the population has the highest genetic diversity and shows almost no signs for recent population expansion (Table 7, additional file 3, 4), although colonization of a new habitat is often accompanied by a loss of genetic diversity (founder event). In comparison, population FI is less diverse for the 16S rDNA with one dominant haplotype only (HT7) and reveals strong evidence for recent population expansion. Diversity estimates decline from population PA in the west to population FI in the east, which seems counterintuitive as the effects of past glaciations are likely to have been much more severe for PA than for AO and FI. This apparent contradiction may, however, be explained by the fact that the Magellan Strait was recolonized after the LGM not only from the Atlantic but also the Pacific side, thus receiving allelic diversity from different source populations. In the contact zone in the central Magellan Strait, this scenario explains the inflated genetic diversity estimates for PA.
In summary, our data are in agreement with the following scenario: Populations of an ancestral species were separated geographically and evolved in allopatry (Falkland Islands vs. Pacific and Atlantic side of Patagonia). Applying a rate for the accumulation of substitutions in 16S rDNA estimated by Held [91] for the serolid isopod Ceratoserolis trilobitoides (Eights, 1833) with a rate of 0.37% per MY for transitions and transversions, the time of divergence was estimated to have occurred several hundreds of thousands of years before present. Thus the initial separation of lineages predates the last glaciations and took place in mid-Pleistocene (average estimates 0.643 – 0.948 BP). In view of the strong genomic signatures of differentiation between Patagonia and the Falkland Islands we must therefore reject the hypothesis that low sea levels during glacial periods led to significantly elevated levels of gene flow between populations of S. paradoxa due to greater proximity of shallow-water habitats. A similar argument applies to potential migration between Patagonia and the Falkland Islands via passive rafting on drifting substrates. Although there are major directional ocean currents that frequently transport substrates suitable for transportation of even rather immobile species [101–103] this apparently played no role in the recent evolutionary history of S. paradoxa. This species exclusively inhabits soft-bottom shallow waters and is frequently half-buried in the sediment (Held pers. observ.). Its capability to colonize new island habitats and maintain genetic continuity across barriers to dispersal and over evolutionary times is therefore small. Further sampling effort should focus on sampling specimens from the West Falkland Islands. It cannot be excluded that members of both lineages live in sympatry today.
Reliability and systematic bias in differentiation estimates
The equilibrium FST estimate for totally isolated populations based on microsatellites can reach the maximum value of FST = 1 only theoretically. Due to the high mutation rate of microsatellites [94, 104, 105] and often a restricted allelic spectrum ([106], but see [107]), the intrapopulation variability is generally very high in particular after a long time of independent evolution of large populations. Applying Meirman's standardization approach for pairwise FST, differentiation between PA and FI is 0.86, between AO and FI 0.91 and among the Magellan Strait populations PA and AO 0.063 and thus about three times larger than without this correction. These values underpin that populations from Patagonia and the Falkland Islands are almost fixed for different alleles at the seven loci investigated. The results point out the importance of the recently introduced standardization approach [67, 68] in order to allow for easier comparison and interpretation of the data. Differentiation estimates of the 16S rDNA yield comparable results. Differentiation was significant between all three populations. Although populations AO and PA shared the most common haplotypes, FST estimates between PA and AO revealed much higher differentiation than inferred using microsatellite data. The most plausible explanation is that the fourfold smaller effective population size of mitochondrial DNA [108] lead to much stronger effects of genetic drift, resulting in higher differentiation estimates.
In principle, differentiation estimates can also be biased due to comparing samples obtained in different years (PA: 1997, AO: 2003, FI: 2004). However, as only few years, corresponding to even fewer generations of S. paradoxa, separate the samples and no major disturbances in the regions were reported for the time in-between the samplings. Thus, we regard this a negligible issue.
Concerning the dating of the split between the two lineages, it must be stated that genetic distances between two lineages increase much faster than predicted by molecular clocks if populations experience population bottlenecks [65]. Thus, the realistic tMRCA between the two lineages might be shorter than the estimated mean using the molecular clock. In addition, it is not entirely certain if the molecular clock can be applied to S. paradoxa. The time estimates are based on 16S rRNA substitution rates commonly used for other Crustacea [97].
Taxonomic and conservation status of the newly delimited species
The genetic data strongly suggest that nominal Serolis paradoxa (Fabricius, 1775) consists of two reproductively isolated species one of which occurs in Patagonia while the other is presumably confined to shallow waters around the Falkland Islands. As the type was originally described by Fabricius as Oniscus paradoxum Fabricius, 1775 from the Falkland Island the species from Patagonia is in need of formal description and a scientific name.
The occurrence of cryptic species has important implications for the conservation of biodiversity in general [109]. If a cryptic species is not recognized, unique and endangered local faunas cannot be efficiently protected. However, the estimates of effective population size for both species contained inside nominal Serolis paradoxa imply that both are highly abundant and neither needs to be considered endangered.
Conclusion
In summary, our data prove low but significant differentiation among populations within the Strait of Magellan and the absence of effective gene flow among populations from the Strait of Magellan and the Falkland Islands. In fact, specimens from both regions belong to two cryptic lineages that probably diverged in the mid-Pleistocene and may already represent reproductively isolated species. The 16S rDNA data supports a genetically rich central Strait of Magellan population, an intermediate population near the Atlantic opening of the Strait of Magellan and a genetically depauperate Falkland Island population. The results are in line with the expectations of colonization of the central Strait of Magellan from both sides of Patagonia after the last glacial maximum approximately 9-14 KY after deglaciation of the habitat and rise of sea levels.
While the fauna of the Falkland Islands has often been accepted to share most of their faunal inventory with Patagonia our results indicate that shallow water species with low mobility may in fact be strongly differentiated populations of one species or even reproductively isolated species.
References
Avise JC: Phylogeography. The history and formation of species. 2000, Cambridge: Harvard University Press
Brandt A: Origin of Antarctic Isopoda (Crustacea, Malacostraca). Marine Biology. 1992, 113: 415-423.
Wägele J: Notes on Antarctic and South American Serolidae (Crustacea, Isopoda) with remarks on the phylogenetic biogeography and a description of new genera. Zoologische Jahrbücher Abteilung für Systematik Ökologie und Geographie der Tiere. 1994, 121: 3-69.
Knox GA, Lowry JK: A comparison between the benthos of the Southern Ocean and the North Polar Ocean with special reference of the Amphipoda and the Polychaeta. Polar Oceans. Edited by: Dunbar MJ. 1977, Calgary: Arctic Institute of North America, 423-462.
Lancellotti DA, Vasquez JA: Zoogeografía de macroinvertebrados bentónicos de la costa de Chile: contribución para la conservación marina. Revista Chilena de Historia Natural. 2002, 73: 99-129.
McDowall RM: Falkland Islands biogeography: converging trajectories in the Southern Ocean. Journal of Biogeography. 2005, 32: 49-62.
Longhurst AR: Ecological geography of the sea. 2007, Burlington, MA, USA: Academic Press
Montiel San Martin A: Biodiversity, zoogeography and ecology of polychaetes from the Magellan region and adjacent areas. Berichte zur Polarforschung. 2005, 505: 1-112.
Adie RJ: The position of the Falkland Islands in a reconstruction of Gondwanaland. Geological Magazine. 1952, 89: 401-410.
Mitchell C, Taylor GK, Cox KG, Shaw J: Are the Falkland Islands a rotated microplate?. Nature. 1986, 319: 131-134.
Marshall JEA: The Falkland Islands: A key element in Gondwana palaeogeography. Tectonics. 1994, 13: 499-514.
Knowlton N: Sibling species in the sea. Annual Review of Ecology and Systematics. 1993, 24: 189-216.
Palumbi SR: Genetic divergence, reproductive isolation and marine speciation. Annual Review of Ecology and Systematics. 1994, 25: 547-572.
Bohonak AJ: Dispersal, gene flow, and population structure. Quarterly Review of Biology. 1999, 74: 21-43.
Knowlton N: Molecular genetic analyses of species boundaries in the sea. Hydrobiologia. 2000, 420: 73-90.
Gallardo MH, Penaloza L, Clasing E: Gene flow and allozymic population structure in the clam Venus antiqua (King of Broderip), (Bivalvia, Veneriidae) from Southern Chile. Journal of Experimental Marine Biology and Ecology. 1998, 230: 193-205.
Hunter RL, Halanych KM: Evaluating connectivity in the brooding brittle star Astrotoma agassizii across the drake passage in the Southern Ocean. Journal of Heredity. 2008, 99: 137-48.
Pérez-Barros P, D'Amato M, Guzmán NV, Lovrich GA: Taxonomic status of two South American sympatric squat lobsters, Munida gregaria and Munida subrugosa (Crustacea: Decapoda: Galatheidae), challenged by DNA sequence information. Biological Journal of the Linnean Society. 2008, 94: 421-434.
Thornhill DJ, Mahon AR, Norenburg JL, Halanych KM: Open-ocean barriers to dispersal: a test case with the Antarctic Polar Front and the ribbon worm Parborlasia corrugatus (Nemertea: Lineidae). Molecular Ecology. 2008, 17: 5104-5117.
Brandt A: Colonization of the Antarctic shelf by the Isopoda (Crustacea, Malacostraca). Berichte zur Polarforschung. 1991, 98: 1-240.
Brandt A, Linse K, Mühlenhardt-Siegel U: Biogeography of Crustacea and Mollusca of the Subantarctic and Antarctic regions. Scientia Marina. 1999, 63: 383-389.
Linse K: New records of shelled marine molluscs at Bouvet Island and preliminary assessment of their biogeographic affinities. Polar Biology. 2006, 29: 120-127.
Allcock AL, Brierley AS, Thorpe JP, Rodhouse PG: Restricted gene flow and evolutionary divergence between geographically separated populations of the Antarctic octopus Paraledone turqueti. Marine Biology. 1997, 129: 97-102.
Held C: Phylogeny and biogeography of serolid isopods (Crustacea, Isopoda, Serolidae) and the use of ribosomal expansion segments in molecular systematics. Mol Phylogenet Evol. 2000, 15 (2): 165-178.
Page TJ, Linse K: More evidence of speciation and dispersal across the Antarctic Polar Front through molecular systematics of Southern Ocean Limatula (Bivalvia: Limidae). Polar Biology. 2002, 25: 818-826.
Held C: Molecular evidence for cryptic speciation within the widespread Antarctic crustacean Ceratoserolis trilobitoides (Crustacea, Isopoda). Antarctic biology in a global context. Edited by: Huiskes AH, Gieskes WW, Rozema J, Schorno RM, van der Vies SM, Wolff WJ. 2003, Leiden: Backhuys Publishers, 135-139.
Held C, Wägele J-W: Cryptic speciation in the giant Antarctic isopod Glyptonotus antarcticus (Isopoda: Valvifera: Chaetiliidae). Scientia Marina. 2005, 69: 175-181.
Raupach MJ, Wägele JW: Distinguishing cryptic species in Antarctic Asellota (Crustacea: Isopoda) – a preliminary study of mitochondrial DNA in Acanthaspidia drygalskii. Antarctic Science. 2006, 18: 191-198.
Linse K, Cope T, Lörz A-N, Sands C: Is the Scotia Sea a centre of Antarctic marine diversification? Some evidence of cryptic speciation in the circum-Antarctic bivalve Lissarca notorcadensis (Arcoidea: Philobryidae). Polar Biology. 2007, 30: 1059-1068.
Wares JP, Daley S, Wetzer R, Toonen RJ: An evaluation of cryptic lineages of Idotea balthica (Isopoda: Idoteidae): Morphology and microsatellites. Journal of Crustacean Biology. 2007, 27: 643-648.
Wilson NG, Hunter RL, Lockhart SJ, Halanych KM: Multiple lineages and absence of panmixia in the "circumpolar" crinoid Promachocrinus kerguelensis from the Atlantic sector of Antarctica. Marine Biology. 2007, 152: 895-904.
Leese F, Kop A, Agrawal S, Held C: Isolation and characterization of microsatellite markers from the marine isopods Serolis paradoxa and Septemserolis septemcarinata (Crustacea: Peracarida). Molecular Ecology Resources. 2008, 8: 818-821.
Mahon AR, Arango CP, Halanych KM: Genetic diversity of Nymphon (Arthropoda: Pycnogonida: Nymphonidae) along the Antarctic Peninsula with a focuson Nymphon australe Hodgson 1902. Marine Biology. 2008, 155: 315-323.
Bastida R, Torti MR: Los isópodos Serolidae de la Argentina. Clave para su reconocimiento. Physis Sección A. 1973, 32: 19-46.
Gappa L, Sueiro M: The subtidal macrobenthic assemblages of Bahia San Sebastian (Tierra del Fuego, Argentina). Polar Biology. 2007, 30: 679-687.
Rios C, Mutschke E, Morrison E: Biodiversidad bentónica sublitoral en el estrecho de Magallanes, Chile. Revista de Biologia Marina y Oceanografia. 2003, 38: 1-12.
Wägele J: On the reproductive biology of Ceratoserolis trilobitoides (Crustacea: Isopoda): Latitudinal variation of fecundity and embryonic development. Polar Biology. 1987, 7: 11-24.
Neigel JE: Population genetics and demography of marine species. Marine biodiversity: Patterns and processes. Edited by: Ormond RF, Gage JD, Angel MV. 1997, Cambridge: Cambridge University Press, 274-292.
Hewitt GM: The genetic legacy of the quaternary ice ages. Nature. 2000, 405: 907-913.
Anderson JB, Shipp SS, Lowe AL, Weller JS, Mosola AB: The Antarctic ice sheet during the last glacial maximum and its subsequent retreat history: a review. Quaternary Science Reviews. 2002, 21: 49-70.
Huybrechts P: Sea-level changes at the LGM from ice-dynamic reconstructions of the Greenland and Antarctic ice sheets during the glacial cycles. Quaternary Science Reviews. 2002, 21: 203-231.
Ingolfsson O: Quaternary glacial and climate history of Antarctica. Quaternary Glaciations – Extent and Chronology, Part III. Edited by: Ehlers J, Gibbard PL. 2004, Elsevier, 3-43.
Polanski J: The maximum glaciation in the Argentine Cordillera. Geological Society of America, Inc. Special Paper. 1965, 84: 453-472.
Flint RF, Fidalgo F: Glacial drift in the eastern Argentine Andes between latitude 41° 10'S and latitude 43° 10'S. Geological Society of America Bulletin. 1969, 80: 1043-1052.
Hulton N, Sugden D, Payne A, Clapperton C: Glacier modeling and the climate of Patagonia during the last glacial maximum. Quaternary Research. 1994, 42: 1-19.
Clapperton CM, Sugden DE, Kaufman DS, McCulloch RD: The last glaciation in central Magellan Strait, southernost Chile. Quaternary Research. 1995, 44: 133-148.
Benn DI, Clapperton CM: Glacial Sediment-Landform Associations and Paleoclimate during the Last Glaciation, Strait of Magellan, Chile. Quaternary Research. 2000, 54: 13-23.
McCulloch RD, Bentley MJ, Purves RS, Hulton NRJ, Sugden DE, Clapperton CM: Climatic inferences from glacial and palaeoecological evidence at the last glacial termination, southern South America. Journal of Quaternary Sciences. 2000, 15: 409-417.
Rostami K, Peltier WR, Mangini A: Quaternary marine terraces, sea-level changes and uplift history of Patagonia, Argentina: comparisons with predictions of the ICE-4G (VM2) model of the global process of glacial isostatic adjustment. Quaternary Science Reviews. 2000, 19: 1495-1525.
Hulton NRJ, Purves RS, McCulloch RD, Sugden DE, Bentley MJ: The Last Glacial Maximum and deglaciation in southern South America. Quaternary Science Reviews. 2002, 21: 233-241.
Clapperton CM: Quaternary glaciations in the Southern Ocean and the Antarctic Peninsula area. Quaterary Science Reviews. 1990, 9: 229-252.
Pompanon F, Bonin A, Bellemain E, Taberlet P: Genotyping errors: causes, consequences and solutions. Nature Reviews Genetics. 2005, 6: 847-859.
Hoffman JI, Amos W: Microsatellite genotyping errors: detection approaches, common sources and consequences for paternal exclusion. Molecular Ecology. 2005, 14: 599-612.
Simon C, Frati F, Beckenbach A, Crespi B, Liu H, Flook P: Evolution, Weighting, and Phylogenetic Utility of Mitochondrial Gene-Sequences and A Compilation of Conserved Polymerase Chain-Reaction Primers. Annals of the Entomological Society of America. 1994, 87: 651-701.
van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P: MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Molecular Ecology Notes. 2004, 4: 535-538.
McKelvey KS, Schwartz MS: DROPOUT: A program to identify problem loci and samples for noninvasive genetic samples in a capture-mark-recapture framework. Molecular Ecology Notes. 2005, 5: 716-718.
Park SDE: Trypanotolerance in west African cattle and the population genetic effects of selection. 2001, University of Dublin
Glaubitz JC: CONVERT: A user-friendly program to reformat diploid genotypc data for commonly used population genetic software packages. Molecular Ecology Notes. 2004, 4: 309-310.
Landry PA, Koskinen MT, Primmer CR: Deriving evolutionary relationships among populations using microsatellites and (deltamu)(2): all loci are equal, but some are more equal than others. Genetics. 2002, 161: 1339-47.
Slatkin M: A measure of population subdivision based on microsatellite allele frequencies. Genetics. 1995, 139: 457-462.
Rousset F: Equilibrium values of measures of population subdivision for stepwise mutation processes. Genetics. 1996, 142: 1357-1362.
Rousset F: GENEPOP '007: a complete reimplementation of the GENEPOP software for Windows and Linux. Molecular Ecology Notes. 2008, 8: 103-106.
Excoffier L, Laval G, Schneider S: Arlequin ver. 3.0: an integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online. 2005, 1: 47-50.
Weir BS, Cockerham CC: Estimating F-statistics for the analysis of population structure. Evolution. 1984, 38: 1358-1370.
Hedrick P: Highly variable loci and their interpretation in evolution and conservation. Evolution. 1999, 53: 313-318.
Whitlock MC, McCauley DE: Indirect measures of gene flow and migration: FST not equal to 1/(4Nm + 1). Heredity. 1999, 82 (Pt 2): 117-25.
Hedrick P: A standardized genetic differentiation measure. Evolution. 2005, 59: 1633-1638.
Meirmans PG: Using the AMOVA framework to estimate a standardized genetic differentiation measure. Evolution. 2006, 60: 2399-402.
Pritchard JK, Stephens M, Donnelly P: Inference of population structure using multilocus genotype data. Genetics. 2000, 155: 945-959.
Falush D, Stephens M, Pritchard JK: Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics. 2003, 164: 1567-1587.
Jakobsson M, Rosenberg NA: CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007, 23: 1801-1806.
Rosenberg NA: Distruct: a program for the graphical display of population structure. Molecular Ecology Notes. 2004, 4: 137-138.
Hill WG: Estimation of effective population size from data on linkage disequilibrium. Genetical Research. 1981, 38: 209-216.
Waples RS: A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. Conservation Genetics. 2006, 7: 167-184.
Waples RS, Do C: LDNe: a program for estimating effective population size from data on linkage disequilibrium. Molecular Ecology Resources. 2008, 8: 753-756.
Piry S, Luikart G, Cornuet JM: BOTTLENECK: A computer program for detecting recent reductions in the effective population size using allele frequency data. Journal of Heredity. 1999, 90: 502-503.
Nei M, Maruyama T, Chakraborty R: The bottleneck effect and genetic variability in populations. Evolution. 1975, 29: 1-10.
Cornuet JM, Luikart G: Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics. 1996, 144: 2001-14.
Luikart G, Allendorf FW, Cornuet JM, Sherwin WB: Distortion of allele frequency distributions provides a test for recent population bottlenecks. Journal of Heredity. 1998, 89: 238-247.
Edgar RC: MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004, 32: 1792-1797.
Gutell RR: Collection of small subunit (16S- and 16S-like) ribosomal RNA structures. Nucleic Acids Research. 1993, 21: 3051-3054.
Tamura K, Dudley J, Nei M, Kumar S: MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol. 2007, 24 (8): 1596-1599.
Nei M: Molecular evolutionary genetics. 1987, New York: Columbia University Press
Tajima F: Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics. 1989, 123: 585-595.
Fu Y-X: Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics. 1997, 147: 915-923.
Clement M, Posada D, Crandall KA: TCS: a computer program to estimate gene genealogies. Molecular Ecology. 2000, 9: 1657-1659.
Saitou N, Nei M: The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol. 1987, 4 (4): 406-425.
Swofford DL: PAUP*. Phylogenetic analysis using parsimony (* and other methods). 1998
Drummond AJ, Rambaut A: BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology. 2007, 7: 214-
Hasegawa M, Kishino H, Yano T: Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. Journal of Molecular Evolution. 1985, 22: 160-174.
Held C: No evidence for slow-down of molecular substitution rates at subzero temperatures in Antarctic serolid isopods (Crustacea, Isopoda, Serolidae). Polar Biology. 2001, 24: 497-501.
Rambaut A, Drummond AJ: Tracer v1.4. 2007, [http://beast.bio.ed.ac.uk/Tracer]
Rice WR: Analyzing tables of statistical tests. Evolution. 1989, 43: 223-225.
Ellegren H: Microsatellites: Simple sequences with complex evolution. Nature Reviews Genetics. 2004, 5: 435-445.
Buschiazzo E, Gemmell NJ: The rise, fall and renaissance of microsatellites in eukaryotic genomes. Bioessays. 2006, 28: 1040-50.
Ibrahim KM, Nichols RA, Hewitt GM: Spatial patterns of genetic variation generated by different forms of dispersal during range expansion. Heredity. 1996, 77: 282-291.
Schubart CD, Diesel R, Hedges SB: Rapid evolution to terrestrial life in Jamaican crabs. Nature. 1998, 393: 363-365.
Coyne JA, Orr HA: Speciation. 2004, Sinauer Associates, Inc
Moore WS: Inferring phylogenies from mtDNA variation: Mitochondrial-gene trees versus nuclear-gene trees. Evolution. 1995, 49: 718-726.
Ballard JW, Whitlock MC: The incomplete natural history of mitochondria. Molecular Ecology. 2004, 13: 729-744.
Helmuth B, Veit RR, Holberton R: Long-distance dispersal of a subantarctic brooding bivalve (Gaimardia trapesina) by kelp-rafting. Marine Biology. 1994, 120: 421-426.
Thiel M, Gutow L: The ecology of rafting in the marine environment – I – The floating substrata. Oceanography And Marine Biology: An Annual Review. 2005, 42: 181-263.
Leese FL, Agrawal SA, Held CH: An exception to the rule? Long-distance dispersal of the brooding benthic isopods Septemserolis septemcarinata from remote Southen Ocean islands. Journal of Biogeography. 2008, under review
Weber JL, Wong C: Mutation of human short tandem repeats. Human Molecular Genetics. 1993, 2: 1123-1128.
Schug MD, Hutter CM, Wetterstrand KA, Gaudette MS, Mackay TFC, Aquadro CF: The mutation rates of di-, tri- and tetranucleotide repeats in Drosophila melanogaster. Molecular Biology and Evolution. 1998, 15: 1751-1760.
Nauta MJ, Weissing FJ: Constraints on allele size at microsatellite loci: implications for genetic differentiation. Genetics. 1996, 143: 1021-1032.
Primmer CR, Ellegren H, Saino N, Moller AP: Directional evolution in germline microsatellite mutations. Nature Genetics. 1996, 13: 391-393.
Birky CW, Maruyama T, Fuerst P: An approach to population and evolutionary genetic theory for genes in mitochondria and chloroplasts and some results. Genetics. 1983, 103: 513-527.
Bickford D, Lohman DJ, Sohdi NS, Ng PK, Meier R, Winker K, Ingram KK, Das I: Cryptic species as a window on diversity and conservation. Trends Ecol Evol. 2007, 22 (3): 148-155.
Acknowledgements
We thank Erika Mutschke and Carlos Rios (Universidad de Magallanes, Punta Arenas) for providing us with material from the 2nd Cruce estrecho and helpful information, 2003. We also thank Andrea Eschbach and Shobhit Agrawal (AWI Bremerhaven) for technical assistance in this study. This work was supported by a DFG grant HE-3391/3 to CH, NSF grant OPP-0132032 to H.W. Detrich III, and a DAAD scholarship to FL and AK. This is publication number 22 from the ICEFISH Cruise of 2004.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
CH and FL designed the study and sampled the specimens. FL and AK carried out the molecular genetic analyses. FL performed the computational analyses, was responsible for the interpretation of population genetic data and drafted the manuscript. CH served as PI for the project and made substantial contributions to the interpretation of the data and together with FL wrote the final manuscript. JWW participated in the coordination of the study and contributed to revising the manuscript. AK helped draft the manuscript. All authors read and approved the final version of the manuscript.
Electronic supplementary material
12983_2008_87_MOESM1_ESM.tiff
Additional file 1: Terrestrial (black) and marine habitats (grey, 50 m bathymetry isolines) available at last glacial maximum (approximately 21 KY BP, left panel) and at the present interglacial (right panel). During last glacial maximum the sea level was considerably lower thus bringing shallow water habitats of the Patagonian Coast and the Falkland Islands in close proximity. Adapted from [49]. (TIFF 686 KB)
12983_2008_87_MOESM2_ESM.pdf
Additional file 2: Allele frequencies at seven microsatellite loci for three Serolis paradoxapopulations (PA = Punta Arenas, AO = Atlantic opening of the Strait of Magellan, FI = Falkland Islands). Private alleles are shown in bold. (PDF 64 KB)
12983_2008_87_MOESM3_ESM.pdf
Additional file 3: Statistical tests for significant heterozygosity (H) excess or deficiency in three populations of Serolis paradoxa(PA, AO, FI) assuming three different mutation models (IAM, TPM, SMM). P-values of the Sign Test and Standardized Differences Test and one-tailed probability for heterozygosity deficiency are based on a 1000 permutations. Significant P-values are printed in bold. The TPM was adjusted to allow for 80% mutations according to a SMM and 20% to an IAM model. (PDF 55 KB)
12983_2008_87_MOESM4_ESM.pdf
Additional file 4: Mismatch analysis based on the 16S rDNA data sets. Confidence intervals (CI give the 5% and 95% values, respectively for the parameters estimated by 50,000 bootstrap replicates. (PDF 43 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Leese, F., Kop, A., Wägele, JW. et al. Cryptic speciation in a benthic isopod from Patagonian and Falkland Island waters and the impact of glaciations on its population structure. Front Zool 5, 19 (2008). https://doi.org/10.1186/1742-9994-5-19
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-9994-5-19