
Abstract
Autophagy, the major mechanism for degrading long-lived intracellular proteins and organelles, is essential for eukaryotic cell homeostasis. Autophagy also defends the cell against invasion by microorganisms and has important roles in innate and adaptive immunity. Increasingly evident is that HIV-1 replication is dependent on select components of autophagy. Fittingly, HIV-1 proteins are able to modulate autophagy to maximize virus production. At the same time, HIV-1 proteins appear to disrupt autophagy in uninfected cells, thereby contributing to CD4+ cell death and HIV-1 pathogenesis. These observations allow for new approaches for the treatment and possibly the prevention of HIV-1 infection. This review focuses on the relationship between autophagy and HIV-1 infection. Discussed is how autophagy plays dual roles in HIV-1 replication and HIV-1 disease progression.
Similar content being viewed by others
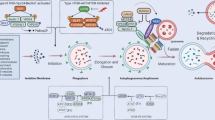


Introduction
Human immunodeficiency virus 1 (HIV-1) establishes a chronic infection that is characterized by persistent virus replication, a systemic decline in CD4+ T cell numbers, accumulating immunologic defects, and the eventual rise of AIDS-defining opportunistic infections and cancers[1]. It is increasingly evident 1 that autophagy, a proteolytic mechanism, plays roles in both HIV-1 replication and disease progression. This review discusses substantial findings from basic research and translational studies of autophagy and HIV-1. Emphasized is the relevance of (macro)autophagy to HIV-1 replication, anti-HIV-1 immune responses, and HIV-1 pathogenesis.
Overview of autophagy
Autophagy, first described many decades ago, has been popularized by recent advances in its cellular and molecular characterization[2]. Although other systems (e.g. microautophagy and chaperone-mediated autophagy) also transport cytoplasmic material to lysosomes, macroautophagy, hereafter referred to as autophagy, is the dominant mechanism for degrading long-lived proteins and organelles[3].
Autophagy can be conceptualized as a three-stage process (Figure1). Stage 1, the initiation of autophagy, is triggered by events that include nutrient starvation, cytokine signaling, and genomic stress. Many of these signals intersect with the mammalian target of rapamycin (mTOR) and act to reverse its inhibitory effects on autophagy[4]. Stage 2, autophagosome synthesis, involves the functions of more than 20 autophagy-related (ATG) genes (Table1), two ubiquitin-like systems (Atg12-Atg5 and LC3-PE), and one lipid kinase signaling complex (PI3K/Beclin-1)[5]. Its end result is the formation of a double-membrane vesicle that contains cytosolic content. Stage 3, the proteolytic stage of autophagy, entails the fusion of mature autophagosomes with lysosomes[6]. Its coordination has recently been attributed to the function of the transcription factor EB (TFEB)[7]. The cytosolic contents are degraded by lysosomal acid hydrolases and then returned to the cytoplasm via channels in the autophagolysosomal membrane. Reactivation of mTOR terminates autophagy[8].

The 3 stages of autophagy. Illustrated are distinct stages in the process of autophagy. i) Autophagy is initiated by pathways that inactivate mTOR. ii) Autophagosome synthesis involves the coupling of LC3-II to the autophagosome membrane and the formation of double membrane vesicles that sequester cytoplasmic material. iii) The final stage of autophagy, proteolysis, entails the fusion of mature autophagosomes with lysosomes and the release of breakdown products into the cytoplasm.
Measuring autophagy
Several methods are commonly used to measure autophagy (Table2)[27]. One approach is to measure, by Western blot procedures, the intracellular levels of two variants of the MAP kinase light chain 3 (LC3) protein: LC3-I and LC3-II. LC3-I is lipidated to form LC3-II and then associates with the autophagosomal membrane upon the induction of autophagy. Thus, measurement of the ratio of LC3-II to LC3-I by Western blot methods is widely used to enumerate autophagic flux; the LC3-II/LC3-I ratio increases upon the induction of autophagy and autophagosome formation. Notably, there are three mammalian isoforms of LC3: LC3A, LC3B, and LC3C. LC3B-II is the only protein known to specifically localize to the autophagosome[28]. Another tactic is to visualize and enumerate autophagosomes by electron microscopy (Figure2). The extraordinary magnification of intracellular constituents enabled by electron microscopy allows for the direct visualization of the double membrane autophagosome structures[29]. The use of LC3-GFP fusion proteins has been helpful for measuring autophagy, allowing for streamlined fluorescent microscopy procedures[30]. Finally, kinetic RT-PCR is regularly used to measure the relative levels of autophagy-related gene transcripts such as BECN1, the gene that encodes Beclin 1 (see Table1).

Example of an autophagosome in an HIV-infected cell. Shown is a transmission electron micrograph of an HIV-1SF33-infected monocyte-derived macrophage (L Ackerman and MS Killian, unpublished data). At higher magnification, the HIV-1 particles (roughly 0.1 μm in diameter) are clearly visible within the autophagosomal structure (inset). Abbreviations: nucleus, N; autophagosome, AP; rough endoplasmic reticulum, RER. Arrows point to the multiple membranes surrounding the autophagosome.
Chemical compounds can also be useful for investigating the different stages of autophagy (see Figure1). Rapamycin inhibits mTOR to induce autophagy[32]. 3-methyladenine (3-MA) inhibits class III phosphatidylinositol 3-kinase (PI3K) and thereby blocks autophagosome formation[33]. Bafilomycin A1 is a specific inhibitor of vacuolar H + ATPase that blocks the fusion between autophagosomes and lysosomes[34]. Thus, 3-MA and Bafilomycin A1 can be used to study the effects of inhibiting the early and late stages of autophagy respectively.
Studies of autophagy must be carefully evaluated with respect to the details of the assays employed and the interpretation of their results[27, 28]. For example, punctate dots in fluorescent microscope images do not necessarily represent autophagosomes, as LC3 can form autophagy-independent aggregates within the cell[35]. In addition, the chemical compounds used to modify autophagic conditions, such as rapamycin, can be toxic to cells at relatively low concentrations[36]. Noteworthy is that prolonged 3-MA treatment in nutrient-rich medium is reported to promote autophagy flux in some cell lines[37]. Therefore, approaches to inhibit autophagy can actually have the opposite effect under certain conditions.
Autophagy and HIV-1 replication
As an obligate intracellular parasite, HIV-1 is dependent on its ability to evade intrinsic cellular defenses including xenophagy - the engulfment and destruction of intracellular microbes by autophagy (Figure3)[38]. Discussed below are studies demonstrating that HIV-1 is able increase virus production by inducing autophagy and evading its proteolytic components.

Autophagy in HIV-1 infection. Left) HIV-1 replication requires early autophagic events for its replication[39], perhaps because the autophagosomal membrane provides a scaffold for virion assembly. Incomplete, or fully formed virions that enter the cell via endocytosis[40], could be degraded by the autophagy (xenophagy) pathway. However, HIV-1 can inhibit the late stage of autophagy to avoid the digestion of virions within autolysosomes. Right) Autophagy is a crucial component of innate and adaptive immune responses to HIV-1 infection. Autophagy is required for the TLR7-mediated signaling of interferon-alpha (IFN-α) production by plasmacytoid dendritic cells (innate immunity) in response to HIV[41]. Autophagy also contributes to proteolytic processing for the presentation of HIV-1 peptides in the context of MHC class II (adaptive immunity).
Genetic studies
At least 35 genes (see Table1) are involved in autophagy[3, 38]. Several of these autophagy-associated genes have been linked with HIV-1 replication. Using small interfering RNA (siRNA) to knock down host genes in a HeLa-derived (epithelial) cell line, members of protein-conjugation pathways involved in autophagy (ATG7, ATG8, ATG12, and ATG16L2) and lysosomal-associated genes (CLN3, LapTM5) were found to be essential for HIV-1 replication[42]. Similarly, a recent study found that the knockdown of PIK3R4, ATG4A, ATG5, or ATG16 with short hairpin RNA (shRNA) led to the inhibition of HIV-1LAI replication in SupT1 cells (T cell line) without having gross effects on the cell viability[43]. However, in separate studies, treatment of HIV-infected HeLa and H9 cells (a T cell line) with rapamycin to induce autophagy did not increase HIV-1 replication[39]. Thus, additional studies are needed to better determine the relationships between the select autophagy genes and HIV replication.
Studies of T cells
CD4+ T cells are the major targets of HIV-1 infection[1]. Zhou and Spector were the first to report that HIV-1 can down regulate autophagy in productively infected CD4+ T cells[44]. They found that Beclin 1 levels were substantially decreased in primary CD4+ T cells that were infected with HIV-1MN, an X4 strain (i.e., using the CXCR4 co-receptor), in comparison to uninfected cells and to those treated with aldrithiol-2 (AT-2) inactivated HIV-1[44]. In addition, LC3-II levels were reduced in the HIV-infected cells as measured by confocal microscopy and Western blot procedures[44]. The inhibition of autophagy in the HIV-infected cells was found to be reversible by nutrient starvation and rapamycin[44]. This finding suggests that the inhibitory effect of HIV-1 infection on autophagy in CD4+ T cells occurs upstream of mTOR and primarily acts to block the initiation stage of autophagy (see Figure1). Others have observed that HIV-1 infection inhibits autophagy in the MOLT-4 CD4+ T cell line[45]. Infection of MOLT-4 cells with HIV-1NL4-3 (X4) or HIV-1NL4-Ad8, an R5 variant (i.e., using the CCR5 co-receptor), caused reductions in the number of autophagosomes visible by transmission electron microscopy (TEM) and in the levels of LC3-II as measured in Western blots[45]. Notably, the LC3-I level was also reduced in the HIV-infected MOLT-4 cells, possibly reflecting the presence of a broad effect on transcription.
In contrast to the observations above, HIV-1 and HIV-2 infections have been recently reported to induce autophagy in Jurkat cells (a human T cell leukemia cell line) and primary CD4+ T cells[46]. HIV-1MN- and HIV-2Rod-infected Jurkat cells exhibited increased numbers of autophagosomes in electron micrographs and the increased expression of various autophagy-associated genes including ULK1, Atg4D, and BECN1. The inhibition of autophagy with 3-MA (concentration not stated) or through the siRNA-mediated knockdown of BECN1 resulted in decreased levels of HIV-1 RNA in the supernatants of HIV-1MN-infected Jurkat cell cultures. Because 3-MA can be toxic to cells (e.g., when > [0.5 mM]) and the knockdown of BECN1 can slow cell growth[43], potentially reducing HIV replication in an autophagy-independent manner, it is important to note that the cell viability and proliferation was not adequately assessed in these studies. Also reported, primary CD4+ T cells infected with HIV-1 for 3 days were found to exhibit increased Beclin-1 levels in Western blots and increased LC3 immunofluorescence[46]. In light of the findings of decreased autophagy in HIV-infected primary CD4+ T cells by Zhou and Spector[44], the observations of increased autophagy in HIV-infected Jurkat cells[46] could be explained by the presence of inherent differences in autophagy between primary T cells and some immortalized cell-lines. The observations of increased Beclin-1 and LC3 levels in the later studies of HIV-1-infected primary CD4+ T cells[46] are most consistent with the effect of the exposure to HIV (see below) rather than the consequence of HIV infection, particularly as the percent of HIV-infected cells 2 was not established in those studies.
Importantly, HIV-1 also modifies autophagy in uninfected bystander CD4+ T cells. In a seminal paper, Espert et al. reported that the accumulation of autophagosomes and Beclin 1 in umbilical cord blood CD4+ T lymphocytes was similarly induced by i) HEK.293 cells transfected to express HIV-1 Env, ii) rapamycin, and iii) CEM T cells infected with HIV-1NL4-3[47]. Autophagosomes did not accumulate in CD4+ T cells treated with the drugs 3-MA and AMD3100 (a CXCR4 antagonist). Also described in this study was that the Env-induced autophagy preceded apoptotic cell death (see Autophagy and HIV-1 disease progression). In follow-up studies, this group determined that the fusion activity of the HIV-1 envelope glycoprotein gp41 was primarily responsible for this effect[48] and that more than 30 candidate proteins are associated[49]. These proteins were largely involved in degradation processes, redox homeostasis, metabolism and cytoskeleton dynamics, and linked to mitochondrial functions. Espert et al. have more recently reported that R5 Env also induces autophagy and cell death in uninfected CD4+ T cell lines[45]. Thus, the effect of HIV-1 Env on autophagy in CD4+ T cells does not appear to be co-receptor specific. Env triggers a broad decrease in protein synthesis that may act to induce autophagy by reducing levels of the inhibitory protein mTOR[49]. The contrasting observations of inhibited autophagy in HIV-1 infected CD4+ T cells and elevated autophagy in bystander CD4+ T cells suggest that productive infection can reverse the enhancing effect of HIV-1 Env on autophagy in uninfected CD4+ T cells.
Studies of macrophages
Macrophages are another important cellular reservoir of HIV-1[50]. Two groups have reported similar findings regarding autophagy in HIV-infected monocyte-derived macrophages (MDM)[39, 45]. In agreement with studies of HeLa cells[42], the induction of autophagy appears to be necessary for HIV-1 replication in MDM. The infection of MDM, upon exposure to HIV-infected cells, is associated with increased numbers of autophagosomes[45]. Interestingly, HIV was detected in the cells with moderate, but not high, numbers of autophagosomes[45]. HIV-infected MDM, cultured in the presence of 3-MA, exhibited substantial reductions in the production of both R5 and X4 viruses[45]. The siRNA-mediated knockdown of Beclin 1 and Atg7 also diminished virus production in HIV-1SF162-infected MDM and U937 cells (a myelomonocytic cell line)[39]. Conversely, treatment of MDM, THP-1 (a myelomonocytic cell line), and U937 cells with rapamycin to induce autophagy increased HIV-1 production[39]. The late stages of autophagy (i.e., lysosome fusion and proteolysis) reduce HIV-1 production, as evidenced by the effects of bafilomycin A1[39]. While most studies of macrophages indicate that HIV-1 promotes autophagosome formation and inhibits the late proteolytic stage of autophagy, decreased LC3-II levels in HIV-1-infected U937 cells have been reported[44]. Thus, the effects of HIV-1 on autophagy in monocytic cell lines can differ from those found in MDM.
Mechanisms enabling HIV-1 to subvert autophagy in macrophages have been elucidated[39]. Demonstrated is that HIV-1 Gag and Nef interact with the autophagy proteins LC3 and Beclin 1 respectively. The colocalization of Gag with LC3 suggests that autophagy plays a role in the biosynthesis, processing, or assembly of HIV-1 intermediates[39]. Alternatively, the Gag/LC3 colocalization could reflect the targeting of Gag for autophagic degradation. The association between Gag and LC3 appears to be unique to monocytic cells[39]. In binding protein complexes containing Beclin 1, Nef is able to inhibit the proteolytic stages of autophagy and thereby prevent the destruction of HIV-1 intermediates[39]. Thus, HIV-1 Nef acts as an “antiautophagic maturation factor”[39]. The Nef 174DD175 motif that is needed for CD4 downmodulation[51] is required for its interaction with Beclin 1[39]. Recently, Nef has also been shown to interact with immunity-associated GTPase family M (IRGM) to induce autophagy in macrophages[52]. These findings indicate that Nef can have the dual function of initiating autophagy and inhibiting its maturation. Unexplained is the normal replication and cytopathicity exhibited by some Nef-deleted HIV-1 isolates in vitro and in vivo[53, 54].
Another distinction between CD4+ lymphocytic and monocytic cells is the effect of HIV-1 Env on autophagy in bystander cells. In contrast to its effect on CD4+ T cells, HIV-1 R5 and X4 Env, when expressed by transfected HEK.293 cells, do not trigger uninfected human monocytic leukemia THP1 cells, MDM, or U937 to undergo autophagy[45]. This distinction could explain the occurrence of CD4+ T cell losses amidst relatively stable monocyte levels in HIV-infected individuals[55].
HIV-1 Tat can have an indirect effect on autophagy in macrophages. In healthy macrophages, autophagy is induced by the pro-inflammatory cytokine interferon-gamma (IFN-γ)[56]. However, pretreatment of monocyte-derived macrophages with HIV-1 Tat, inhibits the stimulatory effect of IFN-γ on autophagy and impairs the antimicrobial functions of the cells[56]. The underlying mechanism involves the ability of Tat to block STAT1 phosphorylation[56]. In other studies, Tat has been found to inhibit autophagy in uninfected macrophages by increasing Akt, Src, and IL-10 production, leading to the silencing of STAT3 and inhibition of autophagy[57].
To summarize, the studies above indicate that HIV-1 proteins disrupt autophagy in HIV-infected and uninfected cells (Table3). The effects of HIV-1 on autophagy are cell-type specific and could be associated with the observed differences in infectivity, virus replication kinetics, and cytopathicity among CD4+ cells of different hematopoietic lineages. In this regard, studies of cell-lines can be misleading with respect to the relationships between HIV and autophagy in primary cells.
Autophagy and anti-HIV-1 immune responses
Distinguishing features of progressive HIV-1 infection include impaired innate and adaptive immune responses and hyper-immune activation[1, 61]. Autophagy is essential for the functionality of innate and adaptive immune responses (see Figure3) and the maintenance of self-tolerance[38, 62]. Thus, autophagy can play important roles in immune cell functions that have direct relevance to HIV-1 infection.
Innate immunity
Innate immune responses provide the earliest host defense against microbial invasion[63]. Cells of the innate immune system use pattern recognition receptors (e.g. Toll-like receptors [TLRs] and nucleotide-binding oligomerization domains [NODs]) to identify highly conserved pathogen-associated molecular patterns (PAMPs, e.g., unmethylated CpG motifs and viral single-stranded RNA)[64]. The cell types of the innate immune system that can exhibit direct anti-HIV-1 activity include plasmacytoid dendritic cells (pDCs), natural killer (NK) cells, and monocytes/macrophages. While pDCs are scarcely present in the blood (< 10 cells per μl), they are the major type-1 interferon (IFN-α) producing cells[65]. pDCs secrete large amounts of IFN-α in response to HIV-infected cells and thereby suppress HIV-1 replication in those cells[66]. The recognition of HIV-infected cells by pDCs appears to be primarily mediated by TLR7, a receptor for single-stranded RNA[67]. Importantly, the production of IFN-α by pDCs in response to TLR7 signaling is dependent on autophagy[68, 69]. Furthermore, pDCs produce IFN-α in response to infectious or AT-2 inactivated HIV-1MN through the induction of autophagy following TLR7 signaling[41]. NK cells are activated by pDCs responding to HIV-1[70] and the ability of NK cells to lyse HIV-infected target cells is enhanced by IFN-α[71]. The activation of macrophages via innate biosensors to secrete anti-HIV cytokines such as the β-chemokines and the macrophage-derived anti-HIV factor (MDAF)[72] can require autophagy[73]. Consequently, autophagy is crucial for suppression of HIV-1 replication by the soluble factors and cytolytic functions of the innate immune system.
Adaptive immunity
In addition to their roles in innate immunity, dendritic cells (DCs) and macrophages promote adaptive immune responses by surveying proteins and secreting cytokines in response to pathogens[74]. In this regard, autophagy can contribute to the processing and presentation of viral peptides in the context of major histocompatibility complex (MHC) class I and II molecules. Autophagy has been observed to enhance the presentation of HSV-1 antigens on the MHC-I molecules of macrophages[75]. In contrast, the inhibition of autophagy in DCs by 3-MA prevents the presentation of HIV-1 antigens on MHC-II, but not cross-presentation on MHC-I to CD8+ T cells[76]. Also, treatment of DCs with 3-MA to inhibit autophagy results in reduced MHC-II expression and impairs antigen presentation of respiratory syncytial virus (RSV)[77]. In other studies, most Influenza A virus antigens were presented to CD4+ T cells by MHC-II on DCs without a requirement for autophagy[78]. These heterogeneous observations suggest that the contribution of autophagy to antigen presentation via MHC-I and II molecules could be pathogen and cell-type specific.
The cytokines produced by DCs and macrophages function to regulate autophagy in other immune cells by intersecting with pathways upstream of mTOR (see Figure1)[62]. In general, Th1 cytokines (e.g. IFN-γ) upregulate autophagy, while Th2 cytokines (e.g. IL-4 and IL-13) abrogate this process[79, 80]. Notably, an intense cytokine storm occurs during acute HIV-1 infection and large amounts of IFN-γ, TNF-α, and IL-10 are produced by T cells throughout the course of disease progression[81]. Thus, the cytokine response to HIV-1 infection can influence autophagy in distal cells and tissues and thereby have pathogenic consequences (see below).
Self-tolerance
The tight regulation of antiviral immune responses is necessary to prevent autoimmunity. The autophagic mechanisms used by DCs to direct adaptive immune responses (see above) are also used to promote self-tolerance. This function of autophagy is exemplified in murine models. Athymic mice implanted with Atg5−/− thymus tissue exhibit inflammation of the gut (colitis) and other organs, indicating that autophagy is crucial for thymic selection and self- tolerance[82]. Implicating a role for autophagy in human autoimmune disorders are findings that 1) polymorphisms in NOD2 and ATG16L1 are associated with susceptibility to the inflammatory gut disorder Crohn's disease[83], and 2) the interaction between the two proteins encoded by these genes is essential for normal autophagy in DCs[84, 85]. The dysregulation of autophagy by viral proteins, perhaps due to Env-mediated effects on bystander CD4+ T regulatory cells, could contribute to aspects of autoimmunity observed in HIV-1 infection[86].
Autophagy and HIV-1 disease progression
As a general homeostasis mechanism, autophagy functions to maintain the health of cells and tissues. Aberrant autophagy has been implicated in a variety of neurodegenerative disorders, cancers, and autoimmune diseases[87]. Hence, the dysregulation of autophagy by HIV, as discussed below, could play a role in the broad pathology of HIV-1 infection (Table4).
Persistent virus replication and CD4+ T cell loss
HIV-1 establishes a chronic viral infection with persistent virus replication that is the underlying cause of HIV-1 disease progression[1]. As discussed (see Autophagy and HIV-1 replication), HIV-1 subverts autophagy to promote virus replication in the infected cell. Also, the importance of autophagy in antiviral immunity was reviewed (see Autophagy and anti-HIV-1 immune responses). Because the primary targets of HIV-1 infection are CD4+ cells that play key roles in antiviral immunity, the dysregulation of autophagy in these cells further promotes the chronicity of HIV-1 infection. As described above, HIV-1 Env promotes hyper-autophagy in bystander CD4+ T lymphocytes. This effect is associated with increased apoptotic cell death and is proposed to be a major mechanism for CD4+ T cell loss[47]. During acute HIV-1-infection, massive losses of CD4+ T cells occur in gastrointestinal tissues[92, 93], perhaps due to the increased sensitivity of these cells to Env-mediated autophagic cell death. Conceivably, elite controllers of HIV-1 infection, who exhibit undetectable viral loads and maintain stable CD4+ T cell counts[94], differ in their regulation of autophagy. In this regard, the restriction of virus replication by xenophagy could be an important contributor to HIV-1 latency.
Opportunistic infections and cancers
The impairment of autophagy in cells of the innate and adaptive immune systems could facilitate the rise of opportunistic infections. Irregularities in the dendritic cell compartment become prevalent with progression to AIDS[1], including a decline in pDC number and function[79, 95]. Thus, disruption of autophagy in dendritic cells as some studies suggest, could be an important contributor to HIV-1 disease progression. Monocyte-derived dendritic cells exposed to HIV-1 exhibit reduced LC3-II expression, decreased TLR responses, and impaired antigen presentation functions[76]. These inhibitory effects of HIV-1 on autophagy in DCs were mediated by Env-induced mTOR signaling[76]. Also, the interaction between HIV-1 Nef and Beclin-1 (discussed above) could inhibit autophagic pathways that protect against tumorigenesis[96]. Thus, by inhibiting autophagy-dependent mechanisms in DCs, HIV-1 could allow for opportunistic infections and cancers to evade innate and adaptive immune responses.
Neurodegenerative disease
The clearance of misfolded and aggregate proteins via autophagy plays a protective role against neurodegenerative disorders such as Huntington’s and Parkinson’s disease[97, 98]. Defects in this housekeeping function of autophagy are linked with HIV-associated neurologic diseases. Autophagic markers are moderately increased in brain tissues of persons having HIV-encephalitis (HIVE) and HIV-associated dementia (HAD) in comparison to HIV-infected brains having no impairment[58, 88]. HAD is associated with increased levels of a byproduct of CXCL12 (i.e., SDF-1), a chemokine that blocks neuronal autophagy[89, 99]. Soluble factors in cultures of simian immunodeficiency virus (SIV)-infected microglia also inhibit neuronal autophagy[100]. In other studies, the exposure of neuronal cells to HIV-1 gp120 resulted in increased Beclin 1, LC3-II and ATG5 levels[58]. This bystander effect of HIV-1 Env that increases autophagy in neuronal cells is similar to the one described above for CD4+ T cells (see Autophagy and HIV-1 replication).
In addition to Env, the HIV-1 transactivator protein, Tat, can also disrupt autophagy in the neurological system. Tat is detectable in the blood and cerebrospinal fluid of HIV-infected individuals[101] and has been shown to be cytotoxic to human brain microvascular endothelial cells[102]. Human umbilical vein endothelial cells (HUVEC), when exposed to cell culture medium conditioned by HeLa-Tat cells, exhibit increased levels of Nox4-dependent H2O2 production, endoplasmic reticulum (ER) stress, and autophagy[60]. Thus, circulating Tat may contribute to HIV-1 neuropathogenesis through an autophagy-dependent mechanism.
Cardiovascular disease and frailty
Receiving increasing attention is evidence that HIV-1-infected individuals age at a faster rate than others[103]. In this regard, autophagy may contribute to the increased rates of cardiovascular disease (CVD) and frailty observed in HIV-infected individuals. Hyper-autophagy has an important role in several types of cardiomyopathy by functioning as a death pathway[104]. CVD, particularly congestive heart failure, is strongly associated with a frailty phenotype (e.g., increased weakness, slowness, exhaustion, anergy, and weight loss) that becomes increasingly prevalent among older adults[105, 106]. Independent of age, a strong inverse correlation exists between frailty and CD4+ T cell counts among HIV-infected individuals[107]. Thus, increased autophagic CD4+ T cell death due to Env-mediated effects[47] could potentially contribute to increased frailty. Also, autophagy influences longevity in eukaryotic organisms[108] and therefore aberrant autophagy could be an important frailty factor in the context of HIV-1 infection.
Implications for autophagy therapeutics
Irregular autophagy (e.g. Env-mediated hyper-autophagy), as a contributor to HIV-1 disease progression, could be therapeutically managed using a variety of pharmacologic agents (Table5). Autophagy modifiers are being clinically evaluated for the treatment of Huntington's disease[109], renal cell carcinoma[110], aging[111], and other autophagy-related disorders[112]. Autophagy enhancing candidates include clonidine, minoxidil, verapamil, and STF-62247[109, 110]. Also, the protease inhibitors nelfinavir and saquinavir are under evaluation for their autophagy-enhancing activities[90, 113]. In this regard, autophagy needs further study in HIV-infected subjects receiving protease inhibitors. Of note, very low concentrations of rapamycin (e.g., < 1 nM) can have anti-HIV-1 activity in vitro[114, 115]. This finding makes rapamycin an attractive candidate for evaluation in the treatment of HIV-1 infection[116]. Moreover, recent evidence suggests that the autophagy promoting effects of vitamin D could be of therapeutic benefit to HIV-infected individuals[36]. Because HIV-1 can require autophagy for virus replication and this process becomes induced upon exposure to HIV, drugs that inhibit autophagy could potentially be used to lower HIV replication and to reduce hyper-autophagy levels. Candidate drugs include wortmannin and other PI3K inhibitors that are being evaluated for their potential clinical use as autophagy blockers[117].
In light of the contributions of autophagy to classical antigen presentation and innate pathogen sensory mechanisms, autophagy modifiers could also be helpful for an HIV-1 vaccine strategy. Indeed, the observation that rapamycin-enhanced autophagy increases antigen presentation by DCs and macrophages opens novel approaches for boosting adaptive immunity in immunocompromised individuals or in the context of vaccination[130]. Furthermore, the potential effects of exposure to HIV-1 proteins, such as gp120, on autophagy (see Table3) should be evaluated in the context of vaccine studies.
Conclusions
Many questions remain pertaining to the relationship between autophagy and HIV-1 infection. Yet unknown is whether or not the ability of HIV-1 to promote autophagy in bystander CD4+ T cells renders those cells more susceptible to HIV-1 infection. Also unexplained are the differential effects of HIV-1 on autophagy in macrophages and CD4+ T cells. In addition to the need for further basic research, translational studies are needed to establish the magnitude of the effects of HIV-1 on autophagy in HIV-1 infected individuals. Moreover, few studies have evaluated autophagy in animal models of HIV infection.
In summary, the importance of autophagy in HIV-1 infection is becoming increasingly clear. Direct effects of HIV-1 on autophagy include the subversion of autophagy in HIV-infected cells and the induction of hyper-autophagy in bystander CD4+ T cells. Because HIV-1 targets key cytokine-producing and immunoregulatory cells, its disruption of autophagy in these cells can have broad pathogenic consequences. Indeed, autophagy appears to play a dual role in HIV-1 infection and disease progression.
Endnotes
1 An April 2012 search ofhttp://www.pubmed.gov using the terms “HIV AND autophagy” returned 55 entries in the database (Killian MS, independent observation).
2 With typical procedures and HIV-1 stock concentrations (e.g. 1 μg/ml p24 equivalents), the frequency of productively HIV-infected primary CD4+ T cell blasts remains relatively low at day 3 post-infection e.g., generally less than 25% of the cells exhibit intracellular p24 or CD4 down-modulation (Killian MS, independent observation).
References
Levy JA: HIV and the pathogenesis of AIDS. 2007, ASM Press, Washington, D.C
Yang Z, Klionsky DJ: Eaten alive: a history of macroautophagy. Nat Cell Biol. 2010, 12: 814-822. 10.1038/ncb0910-814
Tanida I: Autophagy basics. Microbiol Immunol. 2011, 55: 1-11. 10.1111/j.1348-0421.2010.00271.x
Jung CH, Ro SH, Cao J, Otto NM, Kim DH: mTOR regulation of autophagy. FEBS Lett. 2010, 584: 1287-1295. 10.1016/j.febslet.2010.01.017
Xie Z, Klionsky DJ: Autophagosome formation: core machinery and adaptations. NatCell Biol. 2007, 9: 1102-1109.
Tong J, Yan X, Yu L: The late stage of autophagy: cellular events and molecular regulation. Protein Cell. 2010, 1: 907-915. 10.1007/s13238-010-0121-z
Settembre C, Di Malta C, Polito VA, Garcia Arencibia M, Vetrini F, Erdin S, Erdin SU, Huynh T, Medina D, Colella P: TFEB links autophagy to lysosomal biogenesis. Science. 2011, 332: 1429-1433. 10.1126/science.1204592
Yu L, McPhee CK, Zheng L, Mardones GA, Rong Y, Peng J, Mi N, Zhao Y, Liu Z, Wan F: Termination of autophagy and reformation of lysosomes regulated by mTOR. Nature. 2010, 465: 942-946. 10.1038/nature09076
Lee JW, Park S, Takahashi Y, Wang HG: The association of AMPK with ULK1 regulates autophagy. PLoS One. 2010, 5: e15394- 10.1371/journal.pone.0015394
Hosokawa N, Hara T, Kaizuka T, Kishi C, Takamura A, Miura Y, Iemura S, Natsume T, Takehana K, Yamada N: Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009, 20: 1981-1991. 10.1091/mbc.E08-12-1248
Hosokawa N, Sasaki T, Iemura S, Natsume T, Hara T, Mizushima N: Atg101, a novel mammalian autophagy protein interacting with Atg13. Autophagy. 2009, 5: 973-979. 10.4161/auto.5.7.9296
Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, Mizushima N: FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol. 2008, 181: 497-510. 10.1083/jcb.200712064
Webber JL, Tooze SA: New insights into the function of Atg9. FEBS Lett. 2010, 584: 1319-1326. 10.1016/j.febslet.2010.01.020
Proikas-Cezanne T, Waddell S, Gaugel A, Frickey T, Lupas A, Nordheim A: WIPI-1alpha (WIPI49), a member of the novel 7-bladed WIPI protein family, is aberrantly expressed in human cancer and is linked to starvation-induced autophagy. Oncogene. 2004, 23: 9314-9325. 10.1038/sj.onc.1208331
Itakura E, Kishi C, Inoue K, Mizushima N: Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell. 2008, 19: 5360-5372. 10.1091/mbc.E08-01-0080
Weidberg H, Shvets E, Elazar Z: Biogenesis and cargo selectivity of autophagosomes. Annu Rev Biochem. 2011, 80: 125-156. 10.1146/annurev-biochem-052709-094552
Velikkakath AK, Nishimura T, Oita E, Ishihara N, Mizushima N: Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol Biol Cell. 2012, 5: 896-909.
Mizushima N, Yamamoto A, Hatano M, Kobayashi Y, Kabeya Y, Suzuki K, Tokuhisa T, Ohsumi Y, Yoshimori T: Dissection of autophagosome formation using Apg5-deficient mouse embryonic stem cells. J Cell Biol. 2001, 152: 657-668. 10.1083/jcb.152.4.657
Geng J, Klionsky DJ: The Atg8 and Atg12 ubiquitin-like conjugation systems inmacroautophagy. 'Protein modifications: beyond the usual suspects' reviewseries. EMBO Rep. 2008, 9: 859-864. 10.1038/embor.2008.163
Itoh T, Fujita N, Kanno E, Yamamoto A, Yoshimori T, Fukuda M: Golgi-resident small GTPase Rab33B interacts with Atg16L and modulates autophagosome formation. Mol Biol Cell. 2008, 19: 2916-2925. 10.1091/mbc.E07-12-1231
Yuan W, Stromhaug PE, Dunn WA: Glucose-induced autophagy of peroxisomes in Pichia pastoris requires a unique E1-like protein. Mol Biol Cell. 1999, 10: 1353-1366.
Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z: LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J. 2010, 29: 1792-1802. 10.1038/emboj.2010.74
Tanida I, Tanida-Miyake E, Komatsu M, Ueno T, Kominami E: Human Apg3p/Aut1p homologue is an authentic E2 enzyme for multiple substrates, GATE-16, GABARAP, and MAP-LC3, and facilitates the conjugation of hApg12p to hApg5p. J Biol Chem. 2002, 277: 13739-13744. 10.1074/jbc.M200385200
Hemelaar J, Lelyveld VS, Kessler BM, Ploegh HL: A single protease, Apg4B, is specific for the autophagy-related ubiquitin-like proteins GATE-16, MAP1-LC3, GABARAP, and Apg8L. J Biol Chem. 2003, 278: 51841-51850. 10.1074/jbc.M308762200
Ganley IG, Wong PM, Gammoh N, Jiang X: Distinct autophagosomal-lysosomal fusion mechanism revealed by thapsigargin-induced autophagy arrest. Mol Cell. 2011, 42: 731-743. 10.1016/j.molcel.2011.04.024
Yang Z, Klionsky DJ: An overview of the molecular mechanism of autophagy. CurrTopMicrobiolImmunol. 2009, 335: 1-32.
Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A: Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008, 4: 151-175.
Barth S, Glick D, Macleod KF: Autophagy: assays and artifacts. J Pathol. 2010, 221: 117-124. 10.1002/path.2694
Yla-Anttila P, Vihinen H, Jokitalo E, Eskelinen EL: Monitoring autophagy by electron microscopy in Mammalian cells. Methods Enzymol. 2009, 452: 143-164.
Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y: In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004, 15: 1101-1111.
Mizushima N, Yoshimori T, Levine B: Methods in mammalian autophagy research. Cell. 2010, 140: 313-326. 10.1016/j.cell.2010.01.028
Raught B, Gingras AC, Sonenberg N: The target of rapamycin (TOR) proteins. Proc Natl Acad Sci U S A. 2001, 98: 7037-7044. 10.1073/pnas.121145898
Seglen PO, Gordon PB: 3-Methyladenine: specific inhibitor of autophagic/lysosomal protein degradation in isolated rat hepatocytes. ProcNatlAcadSciUSA. 1982, 79: 1889-1892.
Klionsky DJ, Elazar Z, Seglen PO, Rubinsztein DC: Does bafilomycin A1 block the fusion of autophagosomes with lysosomes?. Autophagy. 2008, 4: 849-950.
Kuma A, Matsui M, Mizushima N: LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy. 2007, 3: 323-328.
Campbell GR, Spector SA: Hormonally active vitamin D3 (1alpha, 25- dihydroxycholecalciferol) triggers autophagy in human macrophages that inhibits HIV-1 infection. J Biol Chem. 2011, 286: 18890-18902. 10.1074/jbc.M110.206110
Wu YT, Tan HL, Shui G, Bauvy C, Huang Q, Wenk MR, Ong CN, Codogno P, Shen HM: Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. 2010, 285: 10850-10861. 10.1074/jbc.M109.080796
Levine B, Mizushima N, Virgin HW: Autophagy in immunity and inflammation. Nature. 2011, 469: 323-335. 10.1038/nature09782
Kyei GB, Dinkins C, Davis AS, Roberts E, Singh SB, Dong C, Wu L, Kominami E, Ueno T, Yamamoto A: Autophagy pathway intersects with HIV-1 biosynthesis and regulates viral yields in macrophages. JCell Biol. 2009, 186: 255-268.
Uchil PD, Mothes W: HIV Entry Revisited. Cell. 2009, 137: 402-404. 10.1016/j.cell.2009.04.033
Zhou D, Kang KH, Spector SA: Production of Interferon alpha by Human Immunodeficiency Virus Type 1 in Human Plasmacytoid Dendritic Cells Is Dependent on Induction of Autophagy. J Infect Dis. 2012, 205: 1258-1267. 10.1093/infdis/jis187
Brass AL, Dykxhoorn DM, Benita Y, Yan N, Engelman A, Xavier RJ, Lieberman J, Elledge SJ: Identification of host proteins required for HIV infection through a functional genomic screen. Science. 2008, 319: 921-926. 10.1126/science.1152725
Eekels JJ, Sagnier S, Geerts D, Jeeninga RE, Biard-Piechaczyk M, Berkhout B: Inhibition of HIV-1 replication with stable RNAi-mediated knockdown of autophagy factors. Virol J. 2012, 9: 69- 10.1186/1743-422X-9-69
Zhou D, Spector SA: Human immunodeficiency virus type-1 infection inhibits autophagy. AIDS. 2008, 22: 695-699. 10.1097/QAD.0b013e3282f4a836
Espert L, Varbanov M, Robert-Hebmann V, Sagnier S, Robbins I, Sanchez F, Lafont V, Biard-Piechaczyk M: Differential role of autophagy in CD4 T cells and macrophages during X4 and R5 HIV-1 infection. PLoSONE. 2009, 4: e5787-
Wang X, Gao Y, Tan J, Devadas K, Ragupathy V, Takeda K, Zhao J, Hewlett I: HIV-1 and HIV-2 infections induce autophagy in Jurkat and CD4(+) T cells. Cell Signal. 2012, 24: 1414-1419. 10.1016/j.cellsig.2012.02.016
Espert L, Denizot M, Grimaldi M, Robert-Hebmann V, Gay B, Varbanov M, Codogno P, Biard-Piechaczyk M: Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. JClinInvest. 2006, 116: 2161-2172.
Denizot M, Varbanov M, Espert L, Robert-Hebmann V, Sagnier S, Garcia E, Curriu M, Mamoun R, Blanco J, Biard-Piechaczyk M: HIV-1 gp41 fusogenic function triggers autophagy in uninfected cells. Autophagy. 2008, 4: 998-1008.
Molina L, Grimaldi M, Robert-Hebmann V, Espert L, Varbanov M, Devaux C, Granier C, Biard-Piechaczyk M: Proteomic analysis of the cellular responses induced in uninfected immune cells by cell-expressed X4 HIV-1 envelope. Proteomics. 2007, 7: 3116-3130. 10.1002/pmic.200700306
Carter CA, Ehrlich LS: Cell biology of HIV-1 infection of macrophages. AnnuRevMicrobiol. 2008, 62: 425-443.
Roeth JF, Collins KL: Human immunodeficiency virus type 1 Nef: adapting to intracellular trafficking pathways. MicrobiolMolBiolRev. 2006, 70: 548-563.
Gregoire IP, Richetta C, Meyniel-Schicklin L, Borel S, Pradezynski F, Diaz O, Deloire A, Azocar O, Baguet J, Le Breton M: IRGM Is a Common Target of RNA Viruses that Subvert the Autophagy Network. PLoS Pathog. 2011, 7: e1002422- 10.1371/journal.ppat.1002422
Gorry PR, McPhee DA, Verity E, Dyer WB, Wesselingh SL, Learmont J, Sullivan JS, Roche M, Zaunders JJ, Gabuzda D: Pathogenicity and immunogenicity of attenuated, nef-deleted HIV-1 strains in vivo. Retrovirology. 2007, 4: 66- 10.1186/1742-4690-4-66
Terwilliger EF, Langhoff E, Gabuzda D, Zazopoulos E, Haseltine WA: Allelic variation in the effects of the nef gene on replication of human immunodeficiency virus type 1. Proc Natl Acad Sci U S A. 1991, 88: 10971-10975. 10.1073/pnas.88.23.10971
Abel PM, McSharry C, Galloway E, Ross C, Severn A, Toner G, Gruer L, Wilkinson PC: Heterogeneity of peripheral blood monocyte populations in human immunodeficiency virus-1 seropositive patients. FEMS Microbiol Immunol. 1992, 5: 317-323.
Li JC, Au KY, Fang JW, Yim HC, Chow KH, Ho PL, Lau AS: HIV-1 trans-activator protein dysregulates IFN-gamma signaling and contributes to the suppression of autophagy induction. AIDS. 2011, 25: 15-25. 10.1097/QAD.0b013e328340fd61
Van Grol J, Subauste C, Andrade RM, Fujinaga K, Nelson J, Subauste CS: HIV-1 inhibits autophagy in bystander macrophage/monocytic cells through Src-Akt and STAT3. PLoS One. 2010, 5: e11733- 10.1371/journal.pone.0011733
Spector SA, Zhou D: Autophagy: an overlooked mechanism of HIV-1 pathogenesis and neuroAIDS?. Autophagy. 2008, 4: 704-706.
Au K, Li J, Yim H, Fang J, Lau A: PP1-095 HIV Tat modulation of IFN-g-induced expression of autophagy-associated genes in primary human blood macrophages. Cytokine. 2009, 48: 46-90.
Wu RF, Ma Z, Liu Z, Terada LS: Nox4-derived H2O2 mediates endoplasmic reticulum signaling through local Ras activation. Mol Cell Biol. 2010, 30: 3553-3568. 10.1128/MCB.01445-09
Killian MS, Levy JA: HIV/AIDS: 30 years of progress and future challenges. Eur J Immunol. 2011, 41: 3401-3411. 10.1002/eji.201142082
Munz C: Enhancing immunity through autophagy. Annu Rev Immunol. 2009, 27: 423-449. 10.1146/annurev.immunol.021908.132537
Borrow P: Innate immunity in acute HIV-1 infection. Curr Opin HIV AIDS. 2011, 6: 353-363. 10.1097/COH.0b013e3283495996
Delgado M, Singh S, de Haro S, Master S, Ponpuak M, Dinkins C, Ornatowski W, Vergne I, Deretic V: Autophagy and pattern recognition receptors in innate immunity. ImmunolRev. 2009, 227: 189-202.
Siegal FP, Kadowaki N, Shodell M, Fitzgerald-Bocarsly PA, Shah K, Ho S, Antonenko S, Liu YJ: The nature of the principal type 1 interferon-producing cells in human blood. Science. 1999, 284: 1835-1837. 10.1126/science.284.5421.1835
Schmidt B, Ashlock BM, Foster H, Fujimura SH, Levy JA: HIV-infected cells are major inducers of plasmacytoid dendritic cell interferon production, maturation, and migration. Virology. 2005, 343: 256-266. 10.1016/j.virol.2005.09.059
Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S: Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004, 303: 1526-1529. 10.1126/science.1093620
Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A: Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science. 2007, 315: 1398-1401. 10.1126/science.1136880
Guiducci C, Ghirelli C, Marloie-Provost MA, Matray T, Coffman RL, Liu YJ, Barrat FJ, Soumelis V: PI3K is critical for the nuclear translocation of IRF-7 and type I IFN production by human plasmacytoid predendritic cells in response to TLR activation. Journal of Experimental Medicine. 2008, 205: 315-322. 10.1084/jem.20070763
Alter G, Suscovich TJ, Teigen N, Meier A, Streeck H, Brander C, Altfeld M: Single- stranded RNA derived from HIV-1 serves as a potent activator of NK cells. J Immunol. 2007, 178: 7658-7666.
Hervas-Stubbs S, Perez-Gracia JL, Rouzaut A, Sanmamed MF, Le Bon A, Melero I: Direct effects of type I interferons on cells of the immune system. Clin Cancer Res. 2011, 17: 2619-2627. 10.1158/1078-0432.CCR-10-1114
Mikulak J, Gianolini M, Versmisse P, Pancino G, Lusso P, Verani A: Biological and physical characterization of the X4 HIV-1 suppressive factor secreted by LPS stimulated human macrophages. Virology. 2009, 390: 37-44. 10.1016/j.virol.2009.04.005
Sanjuan MA, Dillon CP, Tait SW, Moshiach S, Dorsey F, Connell S, Komatsu M, Tanaka K, Cleveland JL, Withoff S, Green DR: Toll-like receptor signaling in macrophages links the autophagy pathway to phagocytosis. Nature. 2007, 450: 1253-1257. 10.1038/nature06421
Rossi M, Young JW: Human dendritic cells: potent antigen-presenting cells at the crossroads of innate and adaptive immunity. J Immunol. 2005, 175: 1373-1381.
English L, Chemali M, Duron J, Rondeau C, Laplante A, Gingras D, Alexander D, Leib D, Norbury C, Lippe R, Desjardins M: Autophagy enhances the presentation of endogenous viral antigens on MHC class I molecules during HSV-1 infection. NatImmunol. 2009, 10: 480-487.
Blanchet FP, Moris A, Nikolic DS, Lehmann M, Cardinaud S, Stalder R, Garcia E, Dinkins C, Leuba F, Wu L: Human immunodeficiency virus-1 inhibition of immunoamphisomes in dendritic cells impairs early innate and adaptive immune responses. Immunity. 2010, 32: 654-669. 10.1016/j.immuni.2010.04.011
Morris S, Swanson MS, Lieberman A, Reed M, Yue Z, Lindell DM, Lukacs NW: Autophagy-Mediated Dendritic Cell Activation Is Essential for Innate Cytokine Production and APC Function with Respiratory Syncytial Virus Responses. J Immunol. 2011, 187: 3953-3961. 10.4049/jimmunol.1100524
Comber JD, Robinson TM, Siciliano NA, Snook AE, Eisenlohr LC: Functional macroautophagy induction by influenza A virus without a contribution to major histocompatibility complex class II-restricted presentation. J Virol. 2011, 85: 6453-6463. 10.1128/JVI.02122-10
Martinson JA, Roman-Gonzalez A, Tenorio AR, Montoya CJ, Gichinga CN, Rugeles MT, Tomai M, Krieg AM, Ghanekar S, Baum LL, Landay AL: Dendritic cells from HIV-1 infected individuals are less responsive to toll-like receptor (TLR) ligands. Cell Immunol. 2007, 250: 75-84. 10.1016/j.cellimm.2008.01.007
Harris J, Master SS, De Haro SA, Delgado M, Roberts EA, Hope JC, Keane J, Deretic V: Th1-Th2 polarisation and autophagy in the control of intracellular mycobacteria by macrophages. Vet Immunol Immunopathol. 2009, 128: 37-43. 10.1016/j.vetimm.2008.10.293
Norris PJ, Pappalardo BL, Custer B, Spotts G, Hecht FM, Busch MP: Elevations in IL-10, TNF-alpha, and IFN-gamma from the earliest point of HIV Type 1 infection. AIDS Research and Human Retroviruses. 2006, 22: 757-762. 10.1089/aid.2006.22.757
Nedjic J, Aichinger M, Emmerich J, Mizushima N, Klein L: Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature. 2008, 455: 396-400. 10.1038/nature07208
Massey DC, Parkes M: Genome-wide association scanning highlights two autophagy genes, ATG16L1 and IRGM, as being significantly associated with Crohn's disease. Autophagy. 2007, 3: 649-651.
Cooney R, Baker J, Brain O, Danis B, Pichulik T, Allan P, Ferguson DJ, Campbell BJ, Jewell D, Simmons A: NOD2 stimulation induces autophagy in dendritic cells influencing bacterial handling and antigen presentation. Nature Medicine. 2010, 16: 90-97. 10.1038/nm.2069
Travassos LH, Carneiro LA, Ramjeet M, Hussey S, Kim YG, Magalhaes JG, Yuan L, Soares F, Chea E, Le Bourhis L: Nod1 and Nod2 direct autophagy by recruiting ATG16L1 to the plasma membrane at the site of bacterial entry. Nat Immunol. 2010, 11: 55-62. 10.1038/ni.1823
Stratton R, Slapak G, Mahungu T: Kinloch-de Loes S: Autoimmunity and HIV. CurrOpinInfectDis. 2009, 22: 49-56.
Mijaljica D, Prescott M, Devenish RJ: Autophagy in disease. Methods Mol Biol. 2010, 648: 79-92. 10.1007/978-1-60761-756-3_5
Zhou D, Masliah E, Spector SA: Autophagy is increased in postmortem brains of persons with HIV-1-associated encephalitis. J Infect Dis. 2011, 203: 1647-1657. 10.1093/infdis/jir163
Zhu Y, Vergote D, Pardo C, Noorbakhsh F, McArthur JC, Hollenberg MD, Overall CM, Power C: CXCR3 activation by lentivirus infection suppresses neuronal autophagy: neuroprotective effects of antiretroviral therapy. FASEB J. 2009, 9: 2928-2941.
McLean K, VanDeVen NA, Sorenson DR, Daudi S, Liu JR: The HIV protease inhibitor saquinavir induces endoplasmic reticulum stress, autophagy, and apoptosis in ovarian cancer cells. GynecolOncol. 2009, 112: 623-630.
Gills JJ, Lopiccolo J, Dennis PA: Nelfinavir, a new anti-cancer drug with pleiotropic effects and many paths to autophagy. Autophagy. 2008, 4: 107-109.
Veazey RS, DeMaria M, Chalifoux LV, Shvetz DE, Pauley DR, Knight HL, Rosenzweig M, Johnson RP, Desrosiers RC, Lackner AA: Gastrointestinal tract as a major site ofCD4+ T cell depletion and viral replication in SIV infection. Science. 1998, 280: 427-431. 10.1126/science.280.5362.427
Brenchley JM, Schacker TW, Ruff LE, Price DA, Taylor JH, Beilman GJ, Nguyen PL, Khoruts A, Larson M, Haase AT, Douek DC: CD4+ T cell depletion during all stages of HIV disease occurs predominantly in the gastrointestinal tract. Journal of Experimental Medicine. 2004, 200: 749-759. 10.1084/jem.20040874
Deeks SG, Walker BD: Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity. 2007, 27: 406-416. 10.1016/j.immuni.2007.08.010
Killian MS, Fujimura SH, Hecht FM, Levy JA: Similar changes in plasmacytoid dendritic cell and CD4 T-cell counts during primary HIV-1 infection and treatment. AIDS. 2006, 20: 1247-1252. 10.1097/01.aids.0000232231.34253.bd
Edinger AL, Thompson CB: Defective autophagy leads to cancer. Cancer Cell. 2003, 4: 422-424. 10.1016/S1535-6108(03)00306-4
Garcia-Arencibia M, Hochfeld WE, Toh PP, Rubinsztein DC: Autophagy, a guardian against neurodegeneration. Semin Cell Dev Biol. 2010, 21: 691-698. 10.1016/j.semcdb.2010.02.008
Winslow AR, Rubinsztein DC: The Parkinson disease protein alpha-synuclein inhibits autophagy. Autophagy. 2011, 7: 429-431. 10.4161/auto.7.4.14393
Lipinski MM, Hoffman G, Ng A, Zhou W, Py BF, Hsu E, Liu X, Eisenberg J, Liu J, Blenis J: A genome-wide siRNA screen reveals multiple mTORC1 independent signaling pathways regulating autophagy under normal nutritional conditions. Dev Cell. 2010, 18: 1041-1052. 10.1016/j.devcel.2010.05.005
Alirezaei M, Kemball CC, Whitton JL: Autophagy, inflammation and neurodegenerative disease. Eur J Neurosci. 2011, 33: 197-204. 10.1111/j.1460-9568.2010.07500.x
Xiao H, Neuveut C, Tiffany HL, Benkirane M, Rich EA, Murphy PM, Jeang KT: Selective CXCR4 antagonism by Tat: implications for in vivo expansion of coreceptor use by HIV-1. Proc Natl Acad Sci U S A. 2000, 97: 11466-11471. 10.1073/pnas.97.21.11466
Khan NA, Di Cello F, Nath A, Kim KS: Human immunodeficiency virus type 1 tat-mediated cytotoxicity of human brain microvascular endothelial cells. J Neurovirol. 2003, 9: 584-593.
Effros RB, Fletcher CV, Gebo K, Halter JB, Hazzard WR, Horne FM, Huebner RE, Janoff EN, Justice AC, Kuritzkes D: Aging and infectious diseases: workshop on HIV infection and aging: what is known and future research directions. Clin Infect Dis. 2008, 47: 542-553. 10.1086/590150
Martinet W, Knaapen MW, Kockx MM, De Meyer GR: Autophagy in cardiovascular disease. Trends MolMed. 2007, 13: 482-491. 10.1016/j.molmed.2007.08.004.
Newman AB, Gottdiener JS, McBurnie MA, Hirsch CH, Kop WJ, Tracy R, Walston JD, Fried LP: Associations of subclinical cardiovascular disease with frailty. J Gerontol A Biol Sci Med Sci. 2001, 56: M158-M166. 10.1093/gerona/56.3.M158
Fried LP, Tangen CM, Walston J, Newman AB, Hirsch C, Gottdiener J, Seeman T, Tracy R, Kop WJ, Burke G, McBurnie MA: Frailty in older adults: evidence for a phenotype. J Gerontol A Biol Sci Med Sci. 2001, 56: M146-M156. 10.1093/gerona/56.3.M146
Desquilbet L, Margolick JB, Fried LP, Phair JP, Jamieson BD, Holloway M, Jacobson LP: Relationship between a frailty-related phenotype and progressive deterioration of the immune system in HIV-infected men. J Acquir Immune Defic Syndr. 2009, 50: 299-306. 10.1097/QAI.0b013e3181945eb0
Vellai T, Takacs-Vellai K, Sass M, Klionsky DJ: The regulation of aging: does autophagy underlie longevity?. Trends Cell Biol. 2009, 19: 487-494. 10.1016/j.tcb.2009.07.007
Williams A, Sarkar S, Cuddon P, Ttofi EK, Saiki S, Siddiqi FH, Jahreiss L, Fleming A, Pask D, Goldsmith P: Novel targets for Huntington's disease in an mTORindependent autophagy pathway. Nat Chem Biol. 2008, 4: 295-305. 10.1038/nchembio.79
Turcotte S, Chan DA, Sutphin PD, Hay MP, Denny WA, Giaccia AJ: A molecule targeting VHL-deficient renal cell carcinoma that induces autophagy. Cancer Cell. 2008, 14: 90-102. 10.1016/j.ccr.2008.06.004
Bergamini E: Autophagy: a cell repair mechanism that retards ageing and age-associated diseases and can be intensified pharmacologically. Mol Aspects Med. 2006, 27: 403-410. 10.1016/j.mam.2006.08.001
Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ: Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007, 6: 304-312. 10.1038/nrd2272
Gills JJ, Lopiccolo J, Tsurutani J, Shoemaker RH, Best CJ, Abu-Asab MS, Borojerdi J, Warfel NA, Gardner ER, Danish M: Nelfinavir, A lead HIV protease inhibitor, is a broad-spectrum, anticancer agent that induces endoplasmic reticulum stress, autophagy, and apoptosis in vitro and in vivo. Clin Cancer Res. 2007, 13: 5183-5194. 10.1158/1078-0432.CCR-07-0161
Roy J, Paquette JS, Fortin JF, Tremblay MJ: The immunosuppressant rapamycin represses human immunodeficiency virus type 1 replication. Antimicrob Agents Chemother. 2002, 46 (11): 3447-55. 2002, 46:3447–3455 10.1128/AAC.46.11.3447-3455.2002
Heredia A, Amoroso A, Davis C, Le N, Reardon E, Dominique JK, Klingebiel E, Gallo RC, Redfield RR: Rapamycin causes down-regulation of CCR5 and accumulation of anti-HIV beta-chemokines: an approach to suppress R5 strains of HIV-1. Proc Natl Acad Sci USA. 2003, 100: 10411-10416. 10.1073/pnas.1834278100
Donia M, McCubrey JA, Bendtzen K, Nicoletti F: Potential use of rapamycin in HIV infection. Br J Clin Pharmacol. 2010, 70: 784-793. 10.1111/j.1365-2125.2010.03735.x
Ng SS, Tsao MS, Nicklee T, Hedley DW: Wortmannin inhibits pkb/akt phosphorylation and promotes gemcitabine antitumor activity in orthotopic human pancreatic cancer xenografts in immunodeficient mice. ClinCancer Res. 2001, 7: 3269-3275.
Hidvegi T, Ewing M, Hale P, Dippold C, Beckett C, Kemp C, Maurice N, Mukherjee A, Goldbach C, Watkins S: An autophagy-enhancing drug promotes degradation of mutant alpha1-antitrypsin Z and reduces hepatic fibrosis. Science. 2010, 329: 229-232. 10.1126/science.1190354
Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC: Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol. 2005, 170: 1101-1111. 10.1083/jcb.200504035
Hundeshagen P, Hamacher-Brady A, Eils R, Brady NR: Concurrent detection of autolysosome formation and lysosomal degradation by flow cytometry in a highcontent screen for inducers of autophagy. BMC Biol. 2011, 9: 38- 10.1186/1741-7007-9-38
Underwood BR, Imarisio S, Fleming A, Rose C, Krishna G, Heard P, Quick M, Korolchuk VI, Renna M, Sarkar S: Antioxidants can inhibit basal autophagy and enhance neurodegeneration in models of polyglutamine disease. Hum Mol Genet. 2010, 19: 3413-3429. 10.1093/hmg/ddq253
Sarkar S, Davies JE, Huang Z, Tunnacliffe A, Rubinsztein DC: Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. JBiolChem. 2007, 282: 5641-5652.
Scarlatti F, Bauvy C, Ventruti A, Sala G, Cluzeaud F, Vandewalle A, Ghidoni R, Codogno P: Ceramide-mediated macroautophagy involves inhibition of protein kinase B and up-regulation of beclin 1. J Biol Chem. 2004, 279: 18384-18391. 10.1074/jbc.M313561200
Balgi AD, Fonseca BD, Donohue E, Tsang TC, Lajoie P, Proud CG, Nabi IR, Roberge M: Screen for chemical modulators of autophagy reveals novel therapeutic inhibitors of mTORC1 signaling. PLoS One. 2009, 4: e7124- 10.1371/journal.pone.0007124
Shintani T, Klionsky DJ: Autophagy in health and disease: a double-edged sword. Science. 2004, 306: 990-995. 10.1126/science.1099993
Donohue E, Tovey A, Vogl AW, Arns S, Sternberg E, Young RN, Roberge M: Inhibition of autophagosome formation by the benzoporphyrin derivative verteporfin. J Biol Chem. 2011, 286: 7290-7300. 10.1074/jbc.M110.139915
Blommaart EF, Krause U, Schellens JP, Vreeling-Sindelarova H, Meijer AJ: The phosphatidylinositol 3-kinase inhibitors wortmannin and LY294002 inhibit autophagy in isolated rat hepatocytes. EurJBiochem. 1997, 243: 240-246.
Haspel J, Shaik RS, Ifedigbo E, Nakahira K, Dolinay T, Englert JA, Choi AM: Characterization of macroautophagic flux in vivo using a leupeptin-based assay. Autophagy. 2011, 7: 629-642. 10.4161/auto.7.6.15100
Hoyvik H, Gordon PB, Berg TO, Stromhaug PE, Seglen PO: Inhibition of autophagic-lysosomal delivery and autophagic lactolysis by asparagine. J Cell Biol. 1991, 113: 1305-1312. 10.1083/jcb.113.6.1305
Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL, Eissa NT: Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nature Medicine. 2009, 15: 267-276. 10.1038/nm.1928
Acknowledgements
This publication was made possible with support from the National Institutes of Health (P30 AI027763) and from the California HIV/AIDS Research Program (ID09-SF-058). I thank Dr. Jay Levy for his critical review of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interest
The author declares to have no competing interests.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Killian, M.S. Dual role of autophagy in HIV-1 replication and pathogenesis. AIDS Res Ther 9, 16 (2012). https://doi.org/10.1186/1742-6405-9-16
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-6405-9-16