
Abstract
Background
Poor response to ovarian stimulation with exogenous gonadotrophins occurs in 9–24% of women undergoing in vitro fertilisation (IVF) treatment, which represents an estimated 4000–10,000 women per year in the UK. Poor responders often have their treatment cycle cancelled because of expected poor outcome.
One treatment strategy that may influence outcome is the choice of pituitary suppression regimen prior to the initiation of ovarian stimulation. The three commonly used pituitary suppression regimens in IVF treatment are:
(1) the GnRH agonist long regimen,
(2) the GnRH agonist short regimen and
(3) the GnRH antagonist regimen.
A systematic review of randomised controlled trials of these pituitary suppression regimens has shown the evidence to be either inconclusive or inconsistent. We therefore designed a three arm randomised trial to evaluate the effectiveness of these regimens in women who had poor ovarian response in a previous IVF treatment cycle.
Methods/design
Consenting, eligible women will be randomised to one of the three regimens using an internet-based trial management programme that ensures allocation concealment and employs block randomisation and minimisation for prognostic variables. The primary outcome is the number of oocytes retrieved. Other outcomes include total dose of follicle stimulating hormone (FSH) used for ovarian stimulation, mature oocytes retrieved, embryos available for transfer, implantation rate and clinical pregnancy rate.
The sample size for this trial has been estimated as 102 participants with 34 participants in each of the three arms. Appropriate interim analysis will be conducted by a Data Monitoring and Ethics Committee (DMEC), and the final analysis will be by intention to treat.
Trial registration
ISRCTN27044628
Similar content being viewed by others


Background
Poor ovarian response, defined as failure of the development of sufficient number of mature follicles to proceed to oocyte retrieval or yielding only a few oocytes following gonadotrophin stimulation in women undergoing in vitro fertilisation (IVF) treatment, occurs in 9–24% of women [1]. Poor ovarian response is likely to be an increasing problem with women delaying childbearing and presenting for treatment later in their reproductive life. In comparison to normal responders these patients have impaired fertilisation rates, lower embryo quality and decreased pregnancy rates [2]. Poor responders often have their treatment cycle cancelled because of expected poor outcome [3]. This causes emotional distress for the couple, as well as a financial burden on the couple or the service provider.
Treatment of poor responders who are undergoing IVF treatment remains a challenge. Various treatment regimens and interventions have been proposed in an effort to improve ovarian response and IVF outcome in this group of patients. These include different regimens for pituitary suppression, controlled ovarian hyperstimulation (COH) as well as adjuvant therapies [4]. Currently, the choice of pituitary suppression regimens proposed for the management of poor responders is mainly based on individual centre's or clinician's preferences, with no single protocol considered to be superior over the other. Of the various available pituitary suppression regimens, the three commonly used ones are:
-
(1)
the GnRH agonist long regimen,
-
(2)
the GnRH agonist short regimen and
-
(3)
the GnRH antagonist regimen.
To address the question of their effectiveness, we performed a systematic review and meta-analysis. Literature searches were conducted in MEDLINE, EMBASE, the Cochrane Database, National Research Register and ISI proceedings to identify randomised trials comparing the effects of the above three regimens. We identified seven randomised controlled trials comparing the regimens with each other.[5–11] There were no trials that compared all three regimens against each other. The quality of these 7 randomised trials was generally poor (only 2/7 trials had adequate allocation concealment reported). These trials were also generally small, leading to imprecision in their findings, even when combined in a meta-analysis (Figure 1). Furthermore, there was substantial clinical and statistical heterogeneity amongst the trials. The findings were inconclusive or inconsistent (Figure 1). A well designed, adequately powered, three arm trial comparing the long GnRH agonist regime versus the short GnRH agonist regime versus the GnRH antagonist regime is therefore needed.

Meta-analysis of the number of oocytes retrieved with different pituitary suppression regimens in poor responders.
Methods/design
Objective
In the proposed trial we will evaluate which of the three commonly used down regulation regimens is the most effective for women who have shown poor response in their previous IVF treatment cycle.
Design
A prospective, allocation concealed, assessor-blind, three-arm randomised-controlled-trial.
Primary endpoints
-
Number of eggs retrieved per IVF treatment cycle started.
Secondary endpoints
-
The total dose of follicle stimulating hormone (FSH) used for ovarian stimulation
-
The number of mature eggs retrieved
-
The number of embryos available for transfer
-
The clinical pregnancy rate and
-
The embryo implantation rate
Inclusion Criteria
Any woman undergoing an IVF treatment cycle who fits the criteria to be a "poor responder" is eligible to participate in the trial.
For this study a "poor responder" is defined as a woman who had a previous IVF treatment cycle in which she was stimulated with a daily dose of FSH of 300 IU or more and
-
Produced an inadequate number of mature follicles (three or less follicles measuring > 17 mm) following stimulation for at least nine days; OR
-
Had three or less oocytes retrieved at oocyte retrieval.
Exclusion Criteria
-
Women aged over 40 years.
-
Women with a single ovary.
Randomisation
Third party, distant, internet-based block randomisation with minimisation will be used to ensure randomisation and complete allocation concealment. The aim of minimisation would be to balance for the following prognostic variables (age, body mass index, previous pregnancies and previous live birth). Women may be randomised into the study by internet randomisation [12] or by telephoning a central office.
Blinding
The trial will be a single-blinded study, where the assessors (the doctor performing the egg collection procedure and the embryologist involved in identifying and assessing the eggs) are blinded to the treatment regimen.
Methods
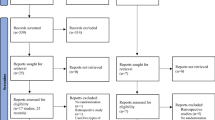
All women who have had poor ovarian response in a previous IVF treatment cycle and who wish to undertake another IVF treatment cycle are invited to participate in the trial. The trial details are explained to the patient by a doctor at their clinic appointment. An information sheet [see Additional file 1] explaining the trial is given which also has a contact number to telephone and speak to the trial investigators. If the woman subsequently agrees to participate in the trial she is requested to sign a consent form [see Additional file 2]. The woman is then randomised to one of the three trial regimens and informed which regimen she has been randomised. Should the woman need more time to decide the clinic doctor will gain verbal permission for the investigators to call and ascertain whether the patient wishes to enrol on the trial. If the patient does wish to enrol then one of the investigators will make an appointment for the patient to sign the consent form. If the woman declines to participate then no further action is required with regard to the trial and the woman will receive standard care. The flowchart in figure 2 describes the participant flow in the trial. Details of all participants and outcomes are recorded on the case report form [see Additional file 3].

Participant flow chart through the PRINT trial.
The treatment regimens
GnRH agonist long regimen (Figure 3)

GnRH agonist long regimen.
Pituitary down-regulation with GnRH agonist, nafarelin (Synarel; Pharmacia, UK) starts on day 21 of the menstrual cycle. Nafarelin nasal spray is taken at a dose of two sniffs, twice a day, where one sniff equals 200 micrograms. The woman will generally have menstruation within two weeks of starting nafarelin at which point she attends the assisted conception unit (ACU) for a transvaginal ultrasound scan (TVS) to confirm down regulation (quiescent ovaries with follicles < 10 mm diameter and endometrium < 5 mm in thickness). On confirmation of down regulation, ovarian stimulation is commenced with FSH, follitropin alfa (Gonal-F; Serono, UK) at 450 IU daily by subcutaneous injection. The dose of the nafarelin nasal spray is halved (one sniff, twice a day) during ovarian stimulation. The woman then attends for a TVS nine days after starting ovarian stimulation and is instructed to administer huaman chorionic gonadotrophin (HCG) subcutaneously when criteria for egg collection are met (see below). FSH and nafarelin are continued until HCG administration.
The approximate duration of this treatment regimen (ie from the start of nafarelin nasal spray, commenced on day 21 of the menstrual cycle, until the pregnancy test is performed) is 6 weeks.
GnRH agonist short regimen (Figure 4)

GnRH agonist short regimen.
Treatment for this protocol starts on day 2 or 3 of the menstrual cycle after a TVS has confirmed the ovaries are quiescent and the endometrium thin (<5 mm). The ovaries are generally quiescent and the endometrium thin at the beginning of a menstrual cycle. On the 2nd or 3rd day of the menstrual cycle the woman commences nafarelin nasal spray (one sniff twice a day, where each sniff equals 200 micrograms). This is continued until administration of the HCG injection (approximately 10 to 15 days from the start of nafarelin nasal spray). On the 3rd or 4th day of the menstrual
cycle the woman commences FSH (450 IU daily by subcutaneous injection). The woman attends for a TVS on day 9 of ovarian stimulation and is instructed to administer HCG when criteria for egg collection are met (see below). FSH and nafarelin are continued until the day of HCG administration.
The approximate duration of this treatment regimen (ie from the start of nafarelin nasal spray, commenced on day 2 or 3 of the menstrual cycle, until the pregnancy test is performed) is 4 weeks.
GnRH antagonist regimen (Figure 5)

GnRH antagonist regimen.
Treatment with this regimen starts on day 2 or 3 of the menstrual cycle. The woman will attend the ACU for a TVS and if the ovaries are quiescent and the endometrium thin (<5 mm) she will commence ovarian stimulation with a daily dose of 450 IU of FSH subcutaneously. Following this a TVS is performed on day 6 of ovarian stimulation to identify the leading follicle. Scans are continued until the leading follicle has a diameter of 14 mm which is usually between days 6 to day 8 of ovarian stimulation. When the leading follicle has reached a diameter of 14 mm, GnRH antagonist, cetrorelix (Cetrotide; Serono, UK) 0.25 mg daily is administered subcutaneously. The next TVS is performed on day 9 of ovarian stimulation, when the woman is instructed to administer HCG if criteria are met (see below). FSH and cetrorelix are continued until HCG administration.
The approximate duration of this treatment regimen (ie from the start of FSH injections, commenced on day 2 or 3 of the menstrual cycle, until the pregnancy test is performed) is 4 weeks.
The following steps of the treatment are the same for all the three treatment regimens.
Criteria for HCG administration
When ≥ 3 follicles attain a mean diameter of ≥ 17 mm, HCG, choriogonadotrophin alfa injection (Ovitrelle; Serono, UK) 6,500 IU is administered subcutaneously.
Egg collection (oocyte retrieval)
Egg collection is performed 34–38 hours after the HCG injection. Egg collection is routinely done under heavy sedation.
Embryo transfer (replacement of embryos)
Embryo Transfer is performed two, three or five days following the day of egg collection, based on the number and quality of embryos available.
Luteal support
All women undergoing IVF treatment are advised to self administer progesterone pessaries (Cyclogest; Alpharma, UK) 400 mg daily starting on the day of egg collection until the day of the pregnancy test, then until 8 weeks of pregnancy if the treatment is successful.
Pregnancy test
All women are requested to do a home urinary pregnancy test 14 days from the day of egg collection.
Early pregnancy scans
All women having IVF treatment have two early pregnancy scans at 6 and 8 weeks gestation.
Trial statistics
Number of participants
Sample size calculation was based on the observed differences in eggs collected from existing literature as well as on the judgement on what constitutes as a clinically Minimally Important Difference (MID), which was judged to be increasing the number of oocytes retrieved from 3 to 5. For this difference of 2 oocytes retrieved, with a standard deviation of 2.5 (as observed in the existing literature), for a power of 90% and an alpha of 5%, 102 women in total will need to be recruited (34 each of the three arms of the trial).
Statistical analysis
The analysis will be by intention to treat, and will be carried out in the following four steps:
Step 1: Summarising trial data
Baseline data and outcome data will be summarised separately. For continuous variables (eg age and FSH level), we will examine the distribution of the observations, and if normally distributed we will then summarise them as means with standard deviations (SDs). If they are non-normally distributed, then medians and inter-quartile ranges (IRQs) will be reported. For dichotomous data (eg clinical pregnancy), we will provide proportions (or percentages).
For our main outcome measure (the number of oocytes collected), as the number of oocytes is not a dichotomous or continuous variable but is a count, and as it is more likely not to be normally distributed, a model for count such as the negative binomial model will be used for analysis.
In addition to the baseline and outcome data, we will also summarise the recruitment numbers, those participants lost to follow-up, protocol violations and other relevant data.
Step 2: Inter-group comparisons
A test for overall comparison (eg ANOVA, or if assumption for ANOVA are not met, a non-parametric equivalence such as Kruskal-Wallis) will be employed for each outcome across all three interventions (A, B, and C), and if this is found to be significant (at a p-value of ≤ 0.05), then we will proceed to pair-wise comparisons (i.e., A vs B, B vs C, A vs C). We appreciate that multitudes of pair-wise comparisons can suffer from Type I (false-positive) error, and will therefore adjust for multiplicity of comparisons (by using steps such as Bonferroni and Tukey's procedure).
The statistical procedures for pair-wise comparisons will depend on the nature of the data: for example, for dichotomous outcomes, we will use Fishers Exact Test or chi-square as appropriate, and for continuous outcomes we will use t-test if the observations in each trial arm are normally distributed; if non-normally distributed, then Mann-Whitney-U test will be employed. Although p-values will be reported, the focus will be on providing 95% confidence intervals around point estimates as these are more useful in interpreting the findings of the trial.
Step 3: Sub-group analysis
We will give emphasis to planned (a priori) sub-groups in our analyses. However, we are aware sub-groups analysis can suffer from false positive (due to multiplicity of comparisons) and false negative (due to reduced sample sizes) results, and will place limited importance in subgroup findings in relation to the overall (global) findings. We will use post-hoc subgroup analysis only for the purpose of hypothesis generation.
Step 4: Adjustments and sensitivity analyses
If randomisation fails to achieve balanced groups, then we will perform secondary analyses in which we will adjust for unbalanced prognostic factors using procedures such as logistic regression. If the primary unadjusted analysis and secondary adjusted analysis are at discordance, then we will give greater weighting to the primary analysis in the interpretation of trial findings.
For issues such losses to follow-up, missing data, and protocol violations, we will attempt sensitivity analyses to explore the effect of these factors on the trial findings. As a secondary analysis, we will adjust for missing data using imputation techniques to explore the effects of such imputations on the trial findings.
Interim analysis and data and safety monitoring
The trial will be monitored by a Data Monitoring and Ethics Committee (DMEC). The DMEC has members with clinical and statistical background, who have no conflict of interest relating to the three trial protocols and have no involvement in running of any part of the trial. The DMEC is responsible for Data and Safety Monitoring. During the trial the DMEC will perform interim analysis and review unblinded outcome data for safety and efficacy. The first interim analysis is scheduled to take place after primary outcome data are available for 40% of the trial participants, and subsequently at four monthly intervals.
Ethics and confidentiality
This trial will be conducted according to the Principles of Good Clinical Practice as defined in the Medicines for Human Use (Clinical Trials) Amended Regulations 2006, the Research Governance Framework for Health & Social Care 2005 and the Data Protection Act. The trial has already received a favourable ethical opinion from a Regional Ethics Committee (REC).
Patient notes containing their personal and treatment details will be kept within the ACU according to the statutory requirements of the HFE Act 1990 and the strict confidentiality that it requires. Notes from patients who have achieved a pregnancy will be kept or archived for 50 years. Patients will need to give their written consent before any of their treatment details or personal information is passed to their General Practitioners or any other persons who are not covered by an HFEA licence.
Abbreviations
- ACU:
-
Assisted Conception Unit
- ANOVA:
-
Analysis of variance
- COH:
-
Controlled ovarian hyperstimulation
- DMEC:
-
Data Monitoring and Ethics Committee
- FSH:
-
Follicle stimulating hormone
- GnRH:
-
Gonadotrophin releasing hormone
- HFEA:
-
Human Fertilisation and Embryology Act
- HCG:
-
Human chorionic gonadotrophin
- IRQ:
-
Inter-quartile range
- ISRCTN:
-
International Standardised Randomised Controlled Trial Number
- IU:
-
International Unit
- IVF:
-
In vitro fertilisation
- MID:
-
Minimally Important Difference
- PRINT:
-
Poor responders intervention
- REC:
-
Regional Ethics Committee
- SD:
-
Standard deviation
- TVS:
-
Transvaginal scan
- UK:
-
United Kingdom
References
Keay SD, Liversedge NH, Mathur RS, Jenkins JM: Assisted conception following poor ovarian reponse to gonadotrophin stimulation. Br J Obstet Gynaecol. 1997, 104 (5): 521-527.
Mahutte NG, Arici A: Poor responders: does the protocol make a difference?. Current Opinion in Obstetrics and Gynaecology. 2002, 14: 275-281. 10.1097/00001703-200206000-00005.
Lashen H, Ledger W, Lopez-Bernal A, Barlow D: Poor responders to ovulation induction: is proceeding to in-vitro fertlization worthwhile?. Human Reproduction. 1999, 14: 964-969. 10.1093/humrep/14.4.964.
Shanbhag S, Aucott L, Bhattacharya S, Hamilton MA, Mc Tavish AR: Interventions for 'poor responders' to controlled ovarian hyperstimulation (COH) in in-vitro fertilisation (IVF). Cochrane Database of Systematic Reviews. 2007, CD004379-DOI:10.1002/14651858.CD004379.pub 2., 1
Akman MA, Erden HF, Tosun SB, Bayazit N, Aksoy E, Bahceci M: Comparison of agonistic flare-up-protocol and antagonistic multiple dose protocol in ovarian stimulation of poor responders: results of a prospective randomized trial. Human Reproduction. 2001, 16: 868-870. 10.1093/humrep/16.5.868.
Weissman A, Farhi J, Royburt M, Nahum H, Glezerman M, Levran D: Prospective evaluation of two stimulation protocols for low responders who were undergoing in vitro fertilization-embryo transfer. Fertility & Sterility. 2003, 79: 886-892. 10.1016/S0015-0282(02)04928-2.
Cheung LP, Lam PM, Lok IH, Chiu TT, Yeung SY, Tjer CC, Haines CJ: GnRH antagonist versus long GnRH agonist protocol in poor responders undergoing IVF: a randomized controlled trial. Human Reproduction. 2005, 20: 616-621. 10.1093/humrep/deh668.
Marci R, Caserta D, Dolo V, Tatone C, Pavan A, Moscarini M: GnRH antagonist in IVF poor-responder patients: results of a randomized trial. Reproductive Biomedicine Online. 2005, 11: 189-193.
Malmusi S, La Marca A, Giulini S, Xella S, Tagliasacchi D, Marsella T, Volpe A: Comparison of a gonadotropin-releasing hormone (GnRH) antagonist and GnRH agonist flare-up regimen in poor responders undergoing ovarian stimulation. Fertility & Sterility. 2005, 84: 402-406. 10.1016/j.fertnstert.2005.01.139.
Schmidt DW, Bremner T, Orris JJ, Maier DB, Benadiva CA, Nulsen JC: A randomized prospective study of microdose leuprolide versus ganirelix in in vitro fertilization cycles for poor responders. Fertility & Sterility. 2005, 83: 1568-1571. 10.1016/j.fertnstert.2004.10.053.
De Placido G, Mollo A, Clarizia R, Strina I, Conforti S, Alviggi C: Gonadotropin-releasing hormone (GnRH) antagonist plus recombinant luteinizing hormone vs. A standard GnRH agonist short protocol in patients at risk for poor ovarian response. Fertility & Sterility. 2006, 85: 247-250. 10.1016/j.fertnstert.2005.07.1280.
Poor Responders INtervention Trial. [http://www.medscinet.com/print]
Acknowledgements
Source of funding for the study: Assisted Conception Unit, Guy's and St Thomas' NHS Foundation Trust.
Source of funding for the authors and for the manuscript preparation: none.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
SKS participated in the design of the study and was involved in drafting the manuscript.
AC participated in the design and development including the statistical analysis plan.
YK participated in drafting the manuscript and revising it critically for important intellectual content.
PB conceived of the study and participated in its design.
Electronic supplementary material
12978_2007_40_MOESM1_ESM.doc
Additional file 1: Poor Responders Intervention Trial – Participant information sheet. The data provided represents the information that is given to all women who are eligible to participate in the study. (DOC 96 KB)
12978_2007_40_MOESM2_ESM.doc
Additional file 2: Poor Responders Intervention Trial – Consent form. The data provided represents the consent form that is signed by eligible women wishing to participate in the trial. (DOC 78 KB)
12978_2007_40_MOESM3_ESM.doc
Additional file 3: Trial case report form. The data provided represents the form in which data from all participating women are recorded. (DOC 205 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Sunkara, S.K., Coomarasamy, A., Khalaf, Y. et al. A three-arm randomised controlled trial comparing Gonadotrophin Releasing Hormone (GnRH) agonist long regimen versus GnRH agonist short regimen versus GnRH antagonist regimen in women with a history of poor ovarian response undergoing in vitro fertilisation (IVF) treatment: Poor responders intervention trial (PRINT). Reprod Health 4, 12 (2007). https://doi.org/10.1186/1742-4755-4-12
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-4755-4-12