
Abstract
Aim
Complement activation is associated with the pathogenesis of age-related macular degeneration (AMD). We aimed to investigate whether 670-nm light treatment reduces the propagation of complement in a light-induced model of atrophic AMD.
Methods
Sprague–Dawley (SD) rats were pretreated with 9 J/cm2 670-nm light for 3 minutes daily over 5 days; other animals were sham treated. Animals were exposed to white light (1,000 lux) for 24 h, after which animals were kept in dim light (5 lux) for 7 days. Expression of complement genes was assessed by quantitative polymerase chain reaction (qPCR), and immunohistochemistry. Counts were made of C3-expressing monocytes/microglia using in situ hybridization. Photoreceptor death was also assessed using outer nuclear layer (ONL) thickness measurements, and oxidative stress using immunohistochemistry for 4-hydroxynonenal (4-HNE).
Results
Following light damage, retinas pretreated with 670-nm light had reduced immunoreactivity for the oxidative damage maker 4-HNE in the ONL and outer segments, compared to controls. In conjunction, there was significant reduction in retinal expression of complement genes C1s, C2, C3, C4b, C3aR1, and C5r1 following 670 nm treatment. In situ hybridization, coupled with immunoreactivity for the marker ionized calcium binding adaptor molecule 1 (IBA1), revealed that C3 is expressed by infiltrating microglia/monocytes in subretinal space following light damage, which were significantly reduced in number after 670 nm treatment. Additionally, immunohistochemistry for C3 revealed a decrease in C3 deposition in the ONL following 670 nm treatment.
Conclusions
Our data indicate that 670-nm light pretreatment reduces lipid peroxidation and complement propagation in the degenerating retina. These findings have relevance to the cellular events of complement activation underling the pathogenesis of AMD, and highlight the potential of 670-nm light as a non-invasive anti-inflammatory therapy.
Similar content being viewed by others


Introduction
Age-related macular degeneration (AMD) is a global epidemic with an estimated worldwide prevalence of 30 to 50 million [1], and a leading cause of blindness in people aged over 65 (reviewed in [2]). While anti-vascular endothelial growth factor (VEGF) therapy has proved to be of benefit for neovascular or ‘wet’ AMD [3], there are presently no treatments that mitigate progression of photoreceptor loss in the more common atrophic or ‘dry’ form of AMD [4]. Although the pathogenesis of AMD is a complex multifactorial process, the recent discovery of a direct association of complement activation with the incidence of AMD has firmly established inflammation as an important factor mediating its pathogenesis [5, 6].
The complement system is a component of the innate immune response, which provides a rapid host defense against a range of immunological challenges, and aiding in the maintenance of homeostasis [7, 8]. Complement activation initiates a cascade of proteolytic cleavages [9, 10], which augment the ability of the host to initiate humoral defenses against infectious pathogens [11], and promote the removal of potentially noxious substances including extracellular debris [7, 9, 10, 12], immune complexes [8, 13–15], and apoptotic cells [14, 16–19]. When activated and poorly regulated, however, complement may induce the destruction of host tissue (reviewed in [20, 21]). In AMD, a pathogenic role of the complement system has been revealed through a number of gene association studies. These identified a significant association between the Y402H sequence variant in the regulatory gene complement factor H (CFH) with the incidence of AMD [22–25], as well as other susceptibility variants in complement pathway genes C2 [5, 26], CFB [5, 26], and the central component C3 [27–31]. While the precise cellular events that promote complement activity in the degenerating retina are unclear (reviewed in [5]) it is known that oxidative damage to photoreceptors promotes the activation of complement and deposition of C3 protein, as documented in a carboxyethylpyrrole (CEP)-mediated mouse model of AMD [32, 33].
Previous investigations have indicated that oxidative damage may be modulated by exposure to irradiation with 670-nm red light. Exposure to light concentrated in the red to near-infrared light range (630 to 1,000 nm) is known to react with the redox active metal centers of cytochrome c oxidase (a key constituent of the electron transport chain) that results in increased electron transfer and improved mitochondrial respiration, and ATP synthesis [34–36]. Studies have indicated that 670-nm light exposure reduces oxidative damage in models of rotenone-induced neurotoxicity [37], optic nerve transection [38], and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD)-induced toxicity [39]. Moreover, 670-nm light irradiation has been shown to accelerate wound healing [40], and ameliorate tissue damage in models of Parkinson’s disease [41], multiple sclerosis [42], and methanol-induced retinal toxicity [43].
We have previously shown that retinal irradiation with 670-nm light attenuates photoreceptor apoptosis induced by exposure to bright continuous white light (BCL) in rats [44], a model with pathogenic features in common with the atrophic ‘dry’ form of AMD [45–49]. Additionally, we have shown that a suite of complement-related genes are upregulated following BCL exposure, and that C3 is expressed in the retina by infiltrating monocytes/microglia [50]. However, the effect of 670-nm light irradiation on complement propagation in the degenerating retina has not been explored. In the current study we aimed to investigate the expression and localization of complement genes, in correlation with oxidative damage, following 670-nm light treatment and light damage. Our data show that pretreatment with 670-nm light reduces the expression of complement components and receptors including in the retina following BCL exposure. Further, we find a reduction in the recruitment of C3-expressing microglia/macrophages in the retina following 670-nm light, in spatiotemporal coincidence with decreases in the oxidative damage marker 4-hydroxynonenal (4-HNE) [51].
Methods
Animals and light exposure
All experiments conducted were in accordance with the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research. Adult Sprague–Dawley (SD) rats were born and reared in dim cyclic light conditions (12 h light, 12 h dark) with an ambient light level of approximately 5 lux.
Prior to BCL exposure, some animals were preconditioned with 670-nm light using a WARP75 670 nm LED array (QBMI Photomedicine, Barneveld, WI, USA) while others were sham treated. Animals in the treatment group were exposed to 9 J/cm2 of 670-nm light for 3 minutes daily over a period of 5 days, according to our protocol described previously [44]. All animals were then dark adapted for a minimum of 15 h then transferred to individual cages designed to allow light to enter unimpeded. BCL exposure commenced consistently at 9:00 am, and was achieved using a cold-white fluorescent light source positioned above the cages (18 W, Cool White; TFC, Taipei, Taiwan), at an intensity of approximately 1,000 lux at the cage floor. BCL exposure was maintained for 24 h, after which animals were immediately returned to dim cyclic conditions for a post-exposure period of 7 days. This timepoint was chosen for analysis since maximal upregulation of C3, recruitment of C3-expressing monocytes/microglia, and formation of the lesion occurs at this time [50]. Age-matched dim-reared animals, either treated or sham treated with 670 nm, served as controls. Animals were then killed and retinal tissue obtained for analysis.
Tissue collection and processing
Animals were killed with an overdose of barbiturate administered by an intraperitoneal injection (60 mg/kg bodyweight, Valabarb; Virbac, Milperra, Australia). The eye from some animals was marked at the superior surface for orientation then enucleated and processed for cryosectioning, while the retina from others was excised through an incision in the cornea and prepared for RNA extraction.
Eyes for cryosectioning were immediately immersion fixed in 4% paraformaldehyde in 0.1 M phosphate-buffered saline (PBS) (pH 7.3) for 3 h at room temperature, then processed as previously described [46], and cryosectioned at 16 μm. Retinas for RNA extraction were immediately immersed in chilled RNAlater solution (cat. no. 7024; Ambion, Austin, TX, USA), then stored in according to the manufacturer’s instructions. RNA was then extracted from each sample and analyzed following previously established methodology [52, 53].
Quantitative real-time polymerase chain reaction (qPCR)
First-strand cDNA synthesis was performed using a protocol described previously [53]. Gene amplification was measured using commercially available TaqMan® hydrolysis probes (Applied Biosystems, Foster City, CA, USA), the details of which are provided in Table 1. The hydrolysis probes were applied following a previously established qPCR protocol [53]. The fold change was determined using the ΔΔCq method. Expression of the target gene was normalized relative to the expression of the reference gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which has shown no appreciable change following BCL exposure previously [50].
In situ hybridization
To investigate the localization of C3 mRNA transcripts in the retina following BCL, a riboprobe to C3 was generated for in situ hybridization on cyrosections of retinal tissue, as described in previous studies conducted by our group [50, 54]. The C3 riboprobe was hybridized overnight at 57°C, and then washed in saline sodium citrate (pH 7.4) at 60°C.
Outer nuclear layer (ONL) thickness measurements
Thickness of the ONL in each age group was measured in increments of 1 mm along the full length of retinal cyrosections cut in the parasaggital plane (superioinferior), which were in close proximity to the vertical meridian. The DNA-specific dye bisbenzamide (Calbiochem, La Jolla, CA, USA) was used to visualize the cellular layers. ONL thickness was calculated as the ratio of ONL thickness to the distance between the outer and inner limiting membranes (OLM-ILM), to take into account any obliquely cut sections. The total ONL ratio from each retina is the average of two retina sections at comparable locations.
Immunohistochemistry
Cryosections from each group were used for immunohistochemistry with an antibody against complement C3 (1:50, cat. no. ab11887; Abcam, Cambridge, MA, USA), 4-HNE (1:200, cat. no. HNE11-S; Alpha Diagnostic, San Antonio, TX, USA), and ionized calcium binding adaptor molecule 1 (IBA1) (1:1,000, cat. no. 019–19741; Wako, Osaka, Japan). Immunohistochemistry was performed using methodology previously described [53]. Immunofluorescence was viewed using a Zeiss laser scanning microscope (Carl Zeiss, Jena, Germany), and acquired using PASCAL software (Zeiss, v4.0). Images were prepared for publication using Adobe Photoshop software.
Quantification of C3-expressing nuclei
Counts of C3-expressing nuclei were performed on retinal cryosections stained for C3 using in situ hybridization (as described above); identification of these C3-expressing nuclei as monocytes/microglia was confirmed in a previous investigation by our group [50]. Counts of C3-expressing nuclei were carried out along the full length of retinal sections cut in the parasaggital plane (superoinferior) close to the vertical meridian, in adjacent fields measuring 1 mm across.
Statistical analysis
Statistical analysis was performed using one-way analysis of variance (ANOVA) with Tukey’s multiple comparison post test. For each analysis, differences with a P value <0.05 were considered statistically significant.
Results
Quantification of ONL thickness following BCL exposure and 670-nm light treatment
The effect of 670-nm light pretreatment on photoreceptor survival following BCL was assessed using ONL thickness measurements across the retina (Figure 1). The average thickness ratio of the ONL decreased by 27% following exposure to BCL, compared to dim-reared animals (P <0.05). In contrast, animals treated with 670-nm light prior to BCL exposure showed substantial preservation of ONL thickness compared to those exposed to BCL alone (P <0.05), which was comparable to ONL thickness of dim-reared animals (P >0.05). No change was observed in ONL thickness in dim-reared animals treated with 670-nm light, compared to untreated controls (P >0.05).

Measurements of outer nuclear layer (ONL) thickness following 670-nm light treatment and bright continuous white light (BCL) exposure. The thickness of the ONL decreased significantly following exposure to BCL, compared to both dim-reared and dim-reared + 670 nm groups (P <0.05). In comparison, the ONL was significantly thicker in animals treated with 670-nm light prior to BCL exposure (P <0.05). Dim reared n = 3, dim reared + 670 nm n = 3, light damage n = 3, light damage + 670-nm light n = 3; error bars represent SEM. *Significant change using analysis of variance (ANOVA) with Tukey’s post test where P <0.05.
Immunoreactivity for 4-HNE in the retina
Immunoreactivity (IR) for 4-HNE was detected in the inner (IS) and outer (OS) segments at low levels in both 670-nm light and untreated control retinas (Figure 2A,B, arrow). Following exposure to BCL, IR for 4-HNE was more intense (Figure 2C,D) and formed multiple deposits throughout the IS and OS region area (arrow). In animals treated with 670-nm light prior to BCL however, levels of 4-HNE IR were lower the IS and OS region area (Figure 2E), and more comparable to dim reared.

Immunoreactivity (IR) for 4-hydroxynonenal (4-HNE; red) in the retina following 670-nm light treatment and exposure to bright continuous white light (BCL). (A,B) IR for 4-HNE was faintly detected in the inner (IS) and outer segments (OS) of both untreated (A) and 670-nm light-treated (B) dim-reared animals (arrows). (C,D) IR for 4-HNE was more intense in the IS and OS (arrow) following exposure to BCL. (E) Treatment with 670-nm light prior to BCL resulted in a marked reduction in IR for 4-HNE (arrow), compared to light damage alone (C). (F) Negative controls showed no specific staining for 4-HNE following BCL exposure. ONL = outer nuclear layer; OS = outer segments.
Modulation of complement-related gene expression with 670-nm light pretreatment
We examined the expression of six complement-related genes using qPCR (Figure 3) that were identified following BCL in microarray analysis conducted by us previously [50]; these include complement components C1s, C2, C3, and C4b, as well as anaphylatoxin receptors C3aR1, and C5r1. Following exposure to BCL, the expression of components C1s, C2, C3, C4b increased significantly compared to dim reared (P <0.05, Figure 3A). Differential expression of C3, and C4b peaked at 1,009% and 1,204% following BCL, while C1s and C2 showed more modest increases (389% and 348% respectively). C3aR1 and C5r1 also showed robust increases in expression following BCL exposure (359% and 278% respectively, P <0.05, Figure 3B). In animals treated with 670-nm light prior to BCL, the expression of all complement components (Figure 3A) and receptors (Figure 3B) assessed was substantially lower than those subjected to BCL alone (P <0.05). The differential expression of C2 in particular was reduced to only 21% in the 670-nm light-treated BCL group, and was statistically indistinguishable from dim-reared animals (P >0.05). There was no significant change in the expression of complement genes in dim-reared animals treated with 670-nm light, compared to untreated controls (P <0.05).

Expression of complement-related genes in the retina by quantitative polymerase chain reaction (qPCR) following 670-nm light treatment and bright continuous white light (BCL) exposure. (A,B) The expression of complement components C1s, C2, C3, C4b (A) and receptors C3aR1, C5r1 (B) increased significantly following BCL relative to dim-reared animals (P <0.05). In animals treated with 670-nm light prior to BCL exposure, the expression of all complement-related genes assessed was substantially reduced, compared to those exposed to BCL alone (P <0.05). Dim-reared animals exposed 670-nm light showed no significant modulation of complement gene expression relative to controls (P >0.05). Dim reared n = 3, dim reared + 670 nm n = 3, light damage n = 3, light damage + 670-nm light n = 3; error bars represent SEM. *Significant change using analysis of variance (ANOVA) with Tukey’s post test where P <0.05.
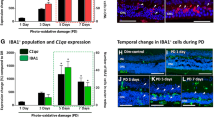
Localization of C3 mRNA and protein in the retina following BCL and 670-nm light treatment
Because of its central role in the activation and propagation of complement, we selected C3 for further investigation in relation to 670-nm light treatment. The localization of C3 expression in the retina was assessed with in situ hybridization (Figure 4). C3 was expressed in the retina by C3-expressing nuclei IR for the monocyte/microglia marker IBA1 (Figure 4C-F), consistent with our previous investigation [50]. C3-expressing nuclei were sparsely distributed in the retinal vasculature in dim-reared animals, at near zero per retina on average (Figure 4A, histogram). Following exposure to BCL, we detected a substantial increase (P <0.05) in the number of C3-positive cells to 26.7 per retina (Figure 4, histogram), which were mostly associated with the degenerative remains of photoreceptors in the ONL (Figure 4C,D, arrows). In contrast, pretreatment with 670-nm light resulted in a dramatic reduction in the number of C3-expressing nuclei to near zero following BCL (P <0.05), consistent with dim-reared animals.

In situ hybridization for C3 mRNA in the retina following 670-nm light and bright continuous white light (BCL) exposure. (A-E) Representative images from the superior mid-periphery show in situ hybridization for C3 mRNA in the retina. Retinas from dim-reared (A) and 670-nm light-treated dim-reared (B) animals showed no staining for C3 mRNA in the retina except for infrequent C3-expressing nuclei associated with the retina vasculature (data not shown). In BCL-exposed animals (C-D) C3-expressing nuclei were more numerous in the outer nuclear layer (ONL) and outer segments within the lesion area, while none were observed in those treated with 670-nm light prior to BCL exposure (E). (F,G) C3 expression (dark grey) in sections counterimmunolabelled with anti-IBA1 (green), showing immunoreactivity in C3-expressing nuclei within the degenerating ONL (arrows) following BCL. Histogram: Quantification of C3-expressing nuclei per retina showed a dramatic increase from near zero in dim-reared animals to 26.7 following BCL exposure (P <0.05). In contrast, the number of C3-expressing nuclei was reduced to near zero in animals pretreated with 670-nm light following BCL (P <0.05). Dim reared n = 3, dim reared + 670-nm light n = 3, light damage n = 3, light damage + 670-nm light n = 3; error bars represent SEM. *Significant change using analysis of variance (ANOVA) with Tukey’s post test where P <0.05.
IR for C3 protein was performed using an antibody against C3 (Figure 5A-J). C3 was detected faintly within the retinal vasculature (Figure 5A, arrows), and the choroid (data not shown) of dim-reared animals. Following BCL exposure we detected strong C3-IR in multiple deposits throughout the ONL (Figure 5C,D, arrows) and the layer of outer segments (Figure 5E,F, arrows) at the site of the developing lesion. Using coimmunolabelling for 4-HNE, we observed colocalization between C3-IR deposits in the outer segment layer and aggregations of 4-HNE IR (Figure 5I-L, arrows) in animals exposed to BCL alone. In animals treated with 670-nm light prior to BCL however, C3-IR was nearly absent from in the ONL and outer segments (Figure 5G), with a distribution similar to dim-reared controls (Figure 5G, arrows).

Immunoreactivity (IR) for C3 (red) in the retina following 670-nm light treatment and exposure to bright continuous white light (BCL). (A-F) Representative images of C3 immunoreactivity taken from the superior mid-periphery. (A,B) IR for C3 was faintly detected in retinal vasculature (arrows) of dim-reared retinas (A), as well as those treated with 670-nm light (B), at comparable levels of IR. (C-F) Following BCL exposure, IR for C3 was observed in multiple deposits throughout the outer nuclear layer (ONL) in the lesion area (C-D, arrows) and the layer of outer segments at the edge of the lesion (E-F, arrows). (G) C3-IR was vastly reduced in the ONL and outer segments of animals treated with 670-nm light prior to BCL, and appeared similar to dim-reared controls (arrows). (H) Negative controls showed no specific staining for C3 following BCL exposure. (I-L) C3-IR (red) in the outer segments of sections counterimmunolabelled with anti-4-hydroxynonenal (4-HNE) (green), shows colocalization for aggregations of 4-HNE immunoreactivity (arrows) following BCL. C = choroid; ONL = outer nuclear layer; OS = outer segment layer.
Discussion
The results of this study demonstrate the efficacy of irradiation with 670-nm light in suppressing lipid peroxidation and complement propagation following BCL exposure through several novel findings. First, we show that 670-nm light irradiation reduces immunoreactivity for 4-HNE in the inner and outer segment region following BCL exposure. Second, using qPCR we show that 670-nm light pretreatment inhibits the expression of a suite of complement genes following BCL, including components from the classical pathway (C1s, C2, C3, C4b), and anaphylatoxin receptors (C3aR1, C5r1). Third, we show a decrease in C3-expressing monocytes/microglia to the ONL following 670-nm light pretreatment, which coincides with reduced deposition of C3 protein in the ONL and photoreceptor outer segments.
Previous investigations, including our own, have shown that irradiation of the retina with various 670-nm light paradigms reduces photoreceptor degeneration following exposure to BCL [44, 55]. The current study, however, is the first to show that retinal pretreatment with 670-nm light reduces the expression of complement-related genes following BCL exposure. These include classical components C1s, C2, and C4b, which mediate assembly of the C3 convertase [9], as well as macrophage receptor genes C3aR1 and C5r1 that recognize cleavage products of C3 and C5 respectively [20]. Furthermore, we show that irradiation with 670-nm light reduces the recruitment of microglial cells that synthesize and deposit C3 protein in the outer retina following BCL exposure. Our findings are also consistent with a previous study demonstrating a reduction IBA1 positive microglia in aging mice following irradiation with 670-nm light [36]. While complement activation has beneficial properties such as aiding debris clearance by recruited phagocytes (reviewed in [10]), other experimental evidence suggests that robust complement propagation may be detrimental following injury. Indeed, a study using mice deficient in the regulatory gene complement factor D (CFD) indicates that complement propagation exacerbates photoreceptor death following light damage [56].
The present findings suggest that 670-nm light pretreatment attenuates oxidative damage to photoreceptors and reduces inflammation, which may in turn reduce stimulation of the complement cascade. These effects of 670-nm light may be due to an interaction with mitochondria, since compromised mitochondrial function may initiate proinflammatory signaling pathways, and generation of reactive oxygen species (reviewed in [57]). This is supported by the findings of Kokkinpoulos and colleagues, who found that 670 nm irradiation increases mitochondrial membrane polarization (and consequently and ATP synthesis) in vitro, and reduces deposition of C3 in aged mice [36].
Irradiation with 670-nm light has previously been shown to reduce markers of oxidative damage, such as manganese superoxide dismutase (MnSOD), in other degenerative models [37–39], and our current findings indicate that 670-nm light therapy decreases production of 4-HNE, a byproduct of lipid peroxidation [58], in photoreceptors following BCL exposure. We also find deposition of C3 occurs in spatiotemporal coincidence with increases in 4-HNE following BCL exposure. This is consistent with the investigations by Hollyfield and colleagues, in which mice immunized with the oxidative damage byproduct CEP develop AMD-like retinal degeneration and show increased deposition of complement C3 in the outer retina [32, 33]. Several investigations also show that exposing RPE cultures to photo-oxidative stress, or to oxidized photoreceptor outer segments, reduces their ability to express the complement regulatory gene CFH [59, 60], thus promoting complement activation.
The mechanism by 670 nm modulates the recruitment of C3-expressing microglia is unclear, though it may be related to reduction of oxidative stress and/or reduced expression of proinflammatory factors. Irradiation with 670-nm light reduces expression of the proinflammatory cytokine tumor necrosis factor α (TNFα) in the retina of aged mice [36], and the chemokine Ccl2 (a chemoattractant for monocytes/microglia [61, 62]) following light damage [63]. Furthermore, accumulation of lipofuscin constituents, such as the bisretinoid A2E, may promote activation and deposition of complement by microglia in vitro, by simultaneously reducing synthesis of the inhibitor CFH while increasing synthesis of CFB, a promoter of the alternative pathway [64]. Increased generation of C3 activation products has also been observed in RPE cells that have accumulated photo-oxidative products of A2E [65].
Relevance to complement propagation in human retinal degeneration
Our findings, in conjunction with the results from other investigations, have previously shown that light damage in rats shares pathological features in common with ‘dry’ AMD [45–49]. Like the widely used laser-induced model of neovascular AMD, this model employs an acute damaging stimulus to evoke long-term, site-specific changes in the retina. Although the rat retina lacks a macula and its key specialization the fovea centralis, it includes a feature that is homologous to the foveal region: the area centralis, in the superiotemporal portion of the retina [66–68]. Following light damage this region develops focal degeneration of photoreceptors and RPE cells, and associated changes to the blood-retinal barrier, which mimics many histopathological aspects of advanced ‘dry’ AMD [45–48].
Complement activation is well established in the literature as a key factor in the pathogenesis of AMD. Key among these are gene association studies which show an association of polymorphisms in a range of complement-related genes with the pathogenesis of AMD (reviewed in [5]), including a string of investigations which have identified a strong association with C2 [5, 26] and C3 [27–31]. Additionally, histological analyses of post-mortem AMD eyes indicate that complement components and regulatory proteins are present in drusen deposits (reviewed in [5]), particularly activation products of C3 such as C3d and C3b [69–71]. We show in our investigation that irradiation of the retina with 670-nm light substantially reduces the expression C2 and C3, as well as the deposition of C3 protein in the outer retina, following light-induced degeneration. Moreover, mitochondrial dysfunction and oxidative damage are thought to be involved in the pathogenesis of AMD [72–74], and several recent investigations have shown levels of oxidative biomarkers are elevated in patients with AMD, compared to controls [75, 76]. As such, 670-nm light irradiation may have potential as a non-invasive intervention to reduce activation of complement in neural degenerations including atrophic AMD.
Conclusions
Our findings suggest that retinal irradiation with 670-nm light substantially reduces the propagation of complement in the retina following light-induced degeneration, and is associated with a concomitant reduction in oxidative damage to the photoreceptors. Consequently, these findings further clarify the role of complement and oxidative damage in retinal degeneration, and suggest that 670-nm light irradiation may be a useful strategy to control detrimental propagation of inflammatory responses in retinal degenerations including atrophic AMD.
References
Ambati J: Age-related macular degeneration and the other double helix. The Cogan Lecture. Invest Ophthalmol Vis Sci 2011, 52:2165–2169.
Ambati J, Ambati BK, Yoo SH, Ianchulev S, Adamis AP: Age-related macular degeneration: etiology, pathogenesis, and therapeutic strategies. Surv Ophthalmol 2003, 48:257–293.
Grisanti S, Tatar O: The role of vascular endothelial growth factor and other endogenous interplayers in age-related macular degeneration. Prog Retin Eye Res 2008, 27:372–390.
Wong WT, Kam W, Cunningham D, Harrington M, Hammel K, Meyerle CB, Cukras C, Chew EY, Sadda SR, Ferris FL: Treatment of geographic atrophy by the topical administration of OT-551: results of a phase II clinical trial. Invest Ophthalmol Vis Sci 2010, 51:6131–6139.
Anderson DH, Radeke MJ, Gallo NB, Chapin EA, Johnson PT, Curletti CR, Hancox LS, Hu J, Ebright JN, Malek G, Hauser MA, Rickman CB, Bok D, Hageman GS, Johnson LV: The pivotal role of the complement system in aging and age-related macular degeneration: hypothesis re-visited. Prog Retin Eye Res 2010, 29:95–112.
Donoso LA, Kim D, Frost A, Callahan A, Hageman G: The role of inflammation in the pathogenesis of age-related macular degeneration. Surv Ophthalmol 2006, 51:137–152.
Paidassi H, Tacnet-Delorme P, Garlatti V, Darnault C, Ghebrehiwet B, Gaboriaud C, Arlaud GJ, Frachet P: C1q binds phosphatidylserine and likely acts as a multiligand-bridging molecule in apoptotic cell recognition. J Immunol 2008, 180:2329–2338.
Walport MJ: Complement. Second of two parts. N Engl J Med 2001, 344:1140–1144.
Gasque P: Complement: a unique innate immune sensor for danger signals. Mol Immunol 2004, 41:1089–1098.
Gasque P, Dean YD, McGreal EP, VanBeek J, Morgan BP: Complement components of the innate immune system in health and disease in the CNS. Immunopharmacology 2000, 49:171–186.
Frank MM, Fries LF: The role of complement in inflammation and phagocytosis. Immunol Today 1991, 12:322–326.
Sim RB, Kishore U, Villiers CL, Marche PN, Mitchell DA: C1q binding and complement activation by prions and amyloids. Immunobiology 2007, 212:355–362.
Davies KA, Schifferli JA, Walport MJ: Complement deficiency and immune complex disease. Springer Semin Immunopathol 1994, 15:397–416.
Botto M: C1q knock-out mice for the study of complement deficiency in autoimmune disease. Exp Clin Immunogenet 1998, 15:231–234.
Navratil JS, Korb LC, Ahearn JM: Systemic lupus erythematosus and complement deficiency: clues to a novel role for the classical complement pathway in the maintenance of immune tolerance. Immunopharmacology 1999, 42:47–52.
Korb LC, Ahearn JM: C1q binds directly and specifically to surface blebs of apoptotic human keratinocytes: complement deficiency and systemic lupus erythematosus revisited. J Immunol 1997, 158:4525–4528.
Taylor PR, Carugati A, Fadok VA, Cook HT, Andrews M, Carroll MC, Savill JS, Henson PM, Botto M, Walport MJ: A hierarchical role for classical pathway complement proteins in the clearance of apoptotic cells in vivo. J Exp Med 2000, 192:359–366.
Trouw LA, Blom AM, Gasque P: Role of complement and complement regulators in the removal of apoptotic cells. Mol Immunol 2008, 45:1199–1207.
Gullstrand B, Martensson U, Sturfelt G, Bengtsson AA, Truedsson L: Complement classical pathway components are all important in clearance of apoptotic and secondary necrotic cells. Clin Exp Immunol 2009, 156:303–311.
Gasque P, Neal JW, Singhrao SK, McGreal EP, Dean YD, Van BJ, Morgan BP: Roles of the complement system in human neurodegenerative disorders: pro-inflammatory and tissue remodeling activities. Mol Neurobiol 2002, 25:1–17.
van Beek J, Elward K, Gasque P: Activation of complement in the central nervous system: roles in neurodegeneration and neuroprotection. Ann N Y Acad Sci 2003, 992:56–71.
Edwards AO, Ritter R 3rd, Abel KJ, Manning A, Panhuysen C, Farrer LA: Complement factor H polymorphism and age-related macular degeneration. Science 2005, 308:421–424.
Klein RJ, Zeiss C, Chew EY, Tsai JY, Sackler RS, Haynes C, Henning AK, SanGiovanni JP, Mane SM, Mayne ST, Bracken MB, Ferris FL, Ott J, Barnstable C, Hoh J: Complement factor H polymorphism in age-related macular degeneration. Science 2005, 308:385–389.
Hageman GS, Anderson DH, Johnson LV, Hancox LS, Taiber AJ, Hardisty LI, Hageman JL, Stockman HA, Borchardt JD, Gehrs KM, Smith RJ, Silvestri G, Russell SR, Klaver CC, Barbazetto I, Chang S, Yannuzzi LA, Barile GR, Merriam JC, Smith RT, Olsh AK, Bergeron J, Zernant J, Merriam JE, Gold B, Dean M, Allikmets R: A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc Natl Acad Sci U S A 2005, 102:7227–7232.
Haines JL, Hauser MA, Schmidt S, Scott WK, Olson LM, Gallins P, Spencer KL, Kwan SY, Noureddine M, Gilbert JR, Schnetz-Boutaud N, Agarwal A, Postel EA, Pericak-Vance MA: Complement factor H variant increases the risk of age-related macular degeneration. Science 2005, 308:419–421.
Jakobsdottir J, Conley YP, Weeks DE, Ferrell RE, Gorin MB: C2 and CFB genes in age-related maculopathy and joint action with CFH and LOC387715 genes. PLoS One 2008, 3:e2199.
Spencer KL, Olson LM, Anderson BM, Schnetz-Boutaud N, Scott WK, Gallins P, Agarwal A, Postel EA, Pericak-Vance MA, Haines JL: C3 R102G polymorphism increases risk of age-related macular degeneration. Hum Mol Genet 2008, 17:1821–1824.
Park KH, Fridley BL, Ryu E, Tosakulwong N, Edwards AO: Complement component 3 (C3) haplotypes and risk of advanced age-related macular degeneration. Invest Ophthalmol Vis Sci 2009, 50:3386–3393.
Maller JB, Fagerness JA, Reynolds RC, Neale BM, Daly MJ, Seddon JM: Variation in complement factor 3 is associated with risk of age-related macular degeneration. Nat Genet 2007, 39:1200–1201.
Yates JR, Sepp T, Matharu BK, Khan JC, Thurlby DA, Shahid H, Clayton DG, Hayward C, Morgan J, Wright AF, Armbrecht AM, Dhillon B, Deary IJ, Redmond E, Bird AC, Moore AT, Genetic Factors in AMD Study Group: Complement C3 variant and the risk of age-related macular degeneration. N Engl J Med 2007, 357:553–561.
Despriet DD, van Duijn CM, Oostra BA, Uitterlinden AG, Hofman A, Wright AF, ten Brink JB, Bakker A, de Jong PT, Vingerling JR, Bergen AA, Klaver CC: Complement component C3 and risk of age-related macular degeneration. Ophthalmology 2009, 116:474–480. e472
Hollyfield JG, Bonilha VL, Rayborn ME, Yang X, Shadrach KG, Lu L, Ufret RL, Salomon RG, Perez VL: Oxidative damage-induced inflammation initiates age-related macular degeneration. Nat Med 2008, 14:194–198.
Hollyfield JG: Age-related macular degeneration: the molecular link between oxidative damage, tissue-specific inflammation and outer retinal disease: the Proctor lecture. Invest Ophthalmol Vis Sci 2010, 51:1275–1281.
Karu TI, Pyatibrat LV, Kalendo GS: Photobiological modulation of cell attachment via cytochrome c oxidase. Photochem Photobiol Sci 2004, 3:211–216.
Wong-Riley MT, Bai X, Buchmann E, Whelan HT: Light-emitting diode treatment reverses the effect of TTX on cytochrome oxidase in neurons. Neuroreport 2001, 12:3033–3037.
Kokkinopoulos I, Colman A, Hogg C, Heckenlively J, Jeffery G: Age-related retinal inflammation is reduced by 670 nm light via increased mitochondrial membrane potential. Neurobiol Aging in press
Liang HL, Whelan HT, Eells JT, Wong-Riley MT: Near-infrared light via light-emitting diode treatment is therapeutic against rotenone- and 1-methyl-4-phenylpyridinium ion-induced neurotoxicity. Neuroscience 2008, 153:963–974.
Fitzgerald M, Bartlett CA, Payne SC, Hart NS, Rodger J, Harvey AR, Dunlop SA: Near infrared light reduces oxidative stress and preserves function in CNS tissue vulnerable to secondary degeneration following partial transection of the optic nerve. J Neurotrauma 2010, 27:2107–2119.
Lim J, Sanders RA, Yeager RL, Millsap DS, Watkins JB 3rd, Eells JT, Henshel DS: Attenuation of TCDD-induced oxidative stress by 670 nm photobiomodulation in developmental chicken kidney. J Biochem Mol Toxicol 2008, 22:230–239.
Whelan HT, Smits RL Jr, Buchman EV, Whelan NT, Turner SG, Margolis DA, Cevenini V, Stinson H, Ignatius R, Martin T, Cwiklinski J, Philippi AF, Graf WR, Hodgson B, Gould L, Kane M, Chen G, Caviness J: Effect of NASA light-emitting diode irradiation on wound healing. J Clin Laser Med Surg 2001, 19:305–314.
Quirk BJ, Desmet KD, Henry M, Buchmann E, Wong-Riley M, Eells JT, Whelan HT: Therapeutic effect of near infrared (NIR) light on Parkinson’s disease models. Front Biosci (Elite Ed) 2012, 4:818–823.
Muili KA, Gopalakrishnan S, Meyer SL, Eells JT, Lyons JA: Amelioration of experimental autoimmune encephalomyelitis in C57BL/6 mice by photobiomodulation induced by 670 nm light. PLoS One 2012, 7:e30655.
Eells JT, Henry MM, Summerfelt P, Wong-Riley MT, Buchmann EV, Kane M, Whelan NT, Whelan HT: Therapeutic photobiomodulation for methanol-induced retinal toxicity. Proc Natl Acad Sci U S A 2003, 100:3439–3444.
Albarracin R, Eells J, Valter K: Photobiomodulation protects the retina from light-induced photoreceptor degeneration. Invest Ophthalmol Vis Sci 2011, 52:3582–3592.
Marc RE, Jones BW, Watt CB, Vazquez-Chona F, Vaughan DK, Organisciak DT: Extreme retinal remodeling triggered by light damage: implications for age related macular degeneration. Mol Vis 2008, 14:782–806.
Rutar M, Provis JM, Valter K: Brief exposure to damaging light causes focal recruitment of macrophages, and long-term destabilization of photoreceptors in the albino rat retina. Curr Eye Res 2010, 35:631–643.
Sullivan R, Penfold P, Pow DV: Neuronal migration and glial remodeling in degenerating retinas of aged rats and in nonneovascular AMD. Invest Ophthalmol Vis Sci 2003, 44:856–865.
Marco-Gomariz MA, Hurtado-Montalban N, Vidal-Sanz M, Lund RD, Villegas-Perez MP: Phototoxic-induced photoreceptor degeneration causes retinal ganglion cell degeneration in pigmented rats. J Comp Neurol 2006, 498:163–179.
Wielgus AR, Collier RJ, Martin E, Lih FB, Tomer KB, Chignell CF, Roberts JE: Blue light induced A2E oxidation in rat eyes - experimental animal model of dry AMD. Photochem Photobiol Sci 2010, 9:1505–1512.
Rutar M, Natoli R, Valter K, Provis JM: Analysis of complement expression in light-induced retinal degeneration: Synthesis and deposition of C3 by microglia/macrophages is associated with focal photoreceptor degeneration. Invest Ophthalmol Vis Sci 2011,52(8):5347–5358.
Esterbauer H, Schaur RJ, Zollner H: Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 1991, 11:81–128.
Natoli R, Provis J, Valter K, Stone J: Gene regulation induced in the C57BL/6J mouse retina by hyperoxia: a temporal microarray study. Mol Vis 2008, 14:1983–1994.
Rutar M, Natoli R, Valter K, Provis JM: Early focal expression of the chemokine Ccl2 by Müller cells during exposure to damage-inducing bright continuous light. Invest Ophthalmol Vis Sci 2011, 52:2379–2388.
Cornish EE, Madigan MC, Natoli R, Hales A, Hendrickson AE, Provis JM: Gradients of cone differentiation and FGF expression during development of the foveal depression in macaque retina. Vis Neurosci 2005, 22:447–459.
Qu C, Cao W, Fan Y, Lin Y: Near-infrared light protect the photoreceptor from light-induced damage in rats. Adv Exp Med Biol 2010, 664:365–374.
Rohrer B, Guo Y, Kunchithapautham K, Gilkeson GS: Eliminating complement factor D reduces photoreceptor susceptibility to light-induced damage. Invest Ophthalmol Vis Sci 2007, 48:5282–5289.
Green DR, Galluzzi L, Kroemer G: Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333:1109–1112.
Tanito M, Elliott MH, Kotake Y, Anderson RE: Protein modifications by 4-hydroxynonenal and 4-hydroxyhexenal in light-exposed rat retina. Invest Ophthalmol Vis Sci 2005, 46:3859–3868.
Chen M, Forrester JV, Xu H: Synthesis of complement factor H by retinal pigment epithelial cells is down-regulated by oxidized photoreceptor outer segments. Exp Eye Res 2007, 84:635–645.
Lau LI, Chiou SH, Liu CJ, Yen MY, Wei YH: The effect of photo-oxidative stress and inflammatory cytokine on complement factor H expression in retinal pigment epithelial cells. Invest Ophthalmol Vis Sci 2011, 52:6832–6841.
Nakazawa T, Hisatomi T, Nakazawa C, Noda K, Maruyama K, She H, Matsubara A, Miyahara S, Nakao S, Yin Y, Benowitz L, Hafezi-Moghadam A, Miller JW: Monocyte chemoattractant protein 1 mediates retinal detachment-induced photoreceptor apoptosis. Proc Natl Acad Sci U S A 2007, 104:2425–2430.
Yoshimura T, Leonard EJ: Identification of high affinity receptors for human monocyte chemoattractant protein-1 on human monocytes. J Immunol 1990, 145:292–297.
Natoli R, Zhu Y, Valter K, Bisti S, Eells J, Stone J: Gene and noncoding RNA regulation underlying photoreceptor protection: microarray study of dietary antioxidant saffron and photobiomodulation in rat retina. Mol Vis 2010, 16:1801–1822.
Ma W, Coon S, Zhao L, Fariss RN, Wong WT: A2E accumulation influences retinal microglial activation and complement regulation. Neurobiol Aging in press
Zhou J, Jang YP, Kim SR, Sparrow JR: Complement activation by photooxidation products of A2E, a lipofuscin constituent of the retinal pigment epithelium. Proc Natl Acad Sci U S A 2006, 103:16182–16187.
Rapaport DH, Stone J: The area centralis of the retina in the cat and other mammals: focal point for function and development of the visual system. Neuroscience 1984, 11:289–301.
Fukuda Y: A three-group classification of rat retinal ganglion cells: histological and physiological studies. Brain Res 1977, 119:327–344.
Rowe MH, Dreher B: Functional morphology of beta cells in the area centralis of the cat’s retina: a model for the evolution of central retinal specializations. Brain Behav Evol 1982, 21:1–23.
Johnson LV, Leitner WP, Staples MK, Anderson DH: Complement activation and inflammatory processes in Drusen formation and age related macular degeneration. Exp Eye Res 2001, 73:887–896.
Anderson DH, Mullins RF, Hageman GS, Johnson LV: A role for local inflammation in the formation of drusen in the aging eye. Am J Ophthalmol 2002, 134:411–431.
Crabb JW, Miyagi M, Gu X, Shadrach K, West KA, Sakaguchi H, Kamei M, Hasan A, Yan L, Rayborn ME, Salomon RG, Hollyfield JG: Drusen proteome analysis: an approach to the etiology of age-related macular degeneration. Proc Natl Acad Sci U S A 2002, 99:14682–14687.
Winkler BS, Boulton ME, Gottsch JD, Sternberg P: Oxidative damage and age-related macular degeneration. Mol Vis 1999, 5:32.
Beatty S, Koh H, Phil M, Henson D, Boulton M: The role of oxidative stress in the pathogenesis of age-related macular degeneration. Surv Ophthalmol 2000, 45:115–134.
Jarrett SG, Lin H, Godley BF, Boulton ME: Mitochondrial DNA damage and its potential role in retinal degeneration. Prog Retin Eye Res 2008, 27:596–607.
Totan Y, Yagci R, Bardak Y, Ozyurt H, Kendir F, Yilmaz G, Sahin S, Sahin Tig U: Oxidative macromolecular damage in age-related macular degeneration. Curr Eye Res 2009, 34:1089–1093.
Lau LI, Liu CJ, Wei YH: Increase of 8-hydroxy-2′-deoxyguanosine in aqueous humor of patients with exudative age-related macular degeneration. Invest Ophthalmol Vis Sci 2010, 51:5486–5490.
Acknowledgements
This work was funded by the Australian Research Council Centres of Excellence Program Grant (CE0561903).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
MVR designed and conducted the experiments, conducted the analysis, and wrote the paper; RCN designed the experiments; RA conducted the experiments; KV designed the experiments; and JMP designed the experiments, and wrote the paper. All authors read and approved the final manuscript.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License ( http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Rutar, M., Natoli, R., Albarracin, R. et al. 670-nm light treatment reduces complement propagation following retinal degeneration. J Neuroinflammation 9, 257 (2012). https://doi.org/10.1186/1742-2094-9-257
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-2094-9-257