
Abstract
Alzheimer’s disease (AD) has been reconceptualized as a dynamic pathophysiological process, where the accumulation of amyloid-beta (Aβ) is thought to trigger a cascade of neurodegenerative events resulting in cognitive impairment and, eventually, dementia. In addition to Aβ pathology, various lines of research have implicated neuroinflammation as an important participant in AD pathophysiology. Currently, neuroinflammation can be measured in vivo using positron emission tomography (PET) with ligands targeting diverse biological processes such as microglial activation, reactive astrocytes and phospholipase A2 activity. In terms of therapeutic strategies, despite a strong rationale and epidemiological studies suggesting that the use of non-steroidal anti-inflammatory drugs (NSAIDs) may reduce the prevalence of AD, clinical trials conducted to date have proven inconclusive. In this respect, it has been hypothesized that NSAIDs may only prove protective if administered early on in the disease course, prior to the accumulation of significant AD pathology. In order to test various hypotheses pertaining to the exact role of neuroinflammation in AD, studies in asymptomatic carriers of mutations deterministic for early-onset familial AD may prove of use. In this respect, PET ligands for neuroinflammation may act as surrogate markers of disease progression, allowing for the development of more integrative models of AD, as well as for the measuring of target engagement in the context of clinical trials using NSAIDs. In this review, we address the biological basis of neuroinflammatory changes in AD, underscore therapeutic strategies using anti-inflammatory compounds, and shed light on the possibility of tracking neuroinflammation in vivo using PET imaging ligands.
Similar content being viewed by others


Background
Research advances over the past decade have led to the reconceptualization of Alzheimer’s disease (AD) as a progressive pathophysiological process in which the accumulation of amyloid-beta (Aβ) is thought to trigger a cascade of neurodegenerative events, including the intracellular accumulation of hyperphosphorylated tau [1, 2]. From a clinical standpoint, AD is viewed as a continuum, comprising a clinically silent phase [3] (characterized by cognitive normality in the presence of AD pathology), a prodromal mild cognitive impairment (MCI) phase [4]—during which individuals exhibit cognitive dysfunction, but of insufficient severity to meet criteria for dementia—and, finally, a dementia phase [5].
In addition to the pathological hallmarks of AD, Aβ and hyperphosphorylated tau, a growing body of literature points to neuroinflammation as an important player in the pathogenesis of AD. Following a set of classic studies implicating the complement factors C1q, C4 and C3 in the formation of amyloid plaques [6, 7], activated microglia and the inflammatory cytokine IL-1 were found to be elevated in AD patients [8, 9]. In addition to its role in the promotion of astrogliosis [10, 11], IL-1 is known to induce marked expression of the amyloid precursor protein (APP) gene [12] and α1-antichymotrypsin [13], both known components of amyloid plaques [14, 15]. Further exploration of complement activation showed that while the opsonizing components were in proximity to amyloid plaques, the terminal components were associated with dystrophic neurites [16, 17]. The importance of the complement system in AD was established shortly after, following the discovery that C1q possessed the ability to bind Aβ and its N-terminal fragments and thus to initiate neuroinflammation via activation of the classical complement pathway [18]. Since this early work, numerous post-mortem immunohistochemical, biochemical, and molecular studies have confirmed the presence of neuroinflammation in the brain of AD subjects (for review, see [19]).
A key issue regarding neuroinflammation in AD is whether this response is beneficial or detrimental in nature. While acute neuroinflammation seems to be an adaptive reaction aiming to restore brain integrity [20], chronic inflammation appears to be an injurious process, resulting in progressive neurodegeneration [21, 22]. Clinical trials using non-steroidal anti-inflammatory drugs (NSAIDs) - initiated on the basis of numerous epidemiological studies suggesting that systemic use of NSAIDs can prevent or delay the onset of AD [23, 24] - have yielded mixed or inconclusive results [25]. However, preliminary results from the Alzheimer's Disease Anti-inflammatory Prevention Trial (ADAPT) may suggest that NSAIDs can be beneficial only if administered early in the disease course, before any symptoms are evident [22]. While this Janus face of neuroinflammation in AD has yet to be fully understood, it is clear that neuroinflammation is an early and continuous process, present from preclinical through late stage AD [26–28].
Recently, positron emission tomography (PET) imaging agents targeting neuroinflammatory processes have been developed and offer the opportunity for non-invasive in vivo tracking of diverse brain inflammatory events (Table 1). Specifically, microglial activation, reactive astrocytosis and increased phospholipase activity are neuroinflammatory events amenable of quantification using PET imaging agents [29–31]. In addition to tracking the progression of AD as a function of neuroinflammatory response, the use of PET imaging agents may help shed light on the interplay between Aβ, hyperphosphorylated tau, and neuroinflammation, possibly leading to improved modeling of AD pathophysiology.
In this review, we describe some of the most important insights provided by PET imaging agents targeting neuroinflammation in AD, revise the evidence provided by preclinical and clinical trials using NSAIDs, and underscore the role that PET biomarkers may play in terms of the development of novel therapeutic strategies, monitoring of disease progression, as well as biomarkers of target engagement.
Imaging microglial activation using PET
Comprising approximately 10% of the cells within the central nervous system [46], microglia constitute the first line of defense against invading pathogens and other harmful agents. Under pathological conditions, microglial cells proliferate and migrate to the site of injury, acquiring phagocytic abilities and releasing various pro-inflammatory mediators [47–50]. In AD, reactive microglia in the vicinity of Aβ plaques have been repeatedly observed in both clinical [51, 52] and experimental studies [53, 54], with experimental models confirming Aβ-mediated release of various neurotoxic molecules by microglia [55–58]. In keeping with the biphasic hypothesis of neuroinflammation, however, additional studies have shown activated microglia to release neuroprotective cytokines such as transforming growth factor-β1, and there may be worsening of AD pathology following microglial inhibition [59].
Currently, PET imaging of microglial activation is possible using molecular agents targeting the 18 kDa translocator protein (TSPO), formerly named the peripheral benzodiazepine receptor (PBR) [60, 61]. Located mainly in parenchymal glial cells, TSPO is present at low concentrations under normal physiological conditions [62], save for the ependyma, choroid plexus, and olfactory nerve layer of the olfactory bulb, which display high densities of TSPO receptors [63, 64]. In response to neuroinflammation, however, TSPO levels undergo a dramatic increase, making it well-suited for assessment of microglial activation [62]. Indeed, numerous studies indicate TSPO to be a sensitive marker of reactive microglia and inflammation secondary to neurodegeneration, including of the AD type (for review see [65, 66]).
Preclinical studies using PET ligands binding TSPO have been performed in transgenic (Tg) rodent models harboring human APP or tau pathogenic mutations. In the case of [11C]PK11195 - the prototypical TSPO ligand - the number of available binding sites (Bmax) was found to be significantly increased in the frontal cortex of AD post-mortem tissue, as compared to controls, while [3H](R)-PK11195 binding correlated significantly with immunohistochemically labeled activated microglia [67]. Likewise, an age-dependent increase in [3H](R)-PK11195 was noted in APP/PS1 Tg mice, in keeping with increased retention of [11C](R)-PK11195 assessed using microPET, which was again correlated with the presence of activated microglia, as determined via histopathological assessment [67]. Similar work conducted using [11C]AC-5216 [33], and [18F]FEDAA1106 [68] - TSPO ligands which are optimized for improved blood-brain barrier permeability, affinity and, in the case of [11C]AC-5216, kinetics - revealed increased TSPO signals in living Tg mice overexpressing human APP (APPE6993Δ) [69]. Importantly, the APPE6993Δ model displays high levels of Aβ in the absence of fibrillary amyloid plaques [70], suggesting that amyloid dysmetabolism per se is sufficient to induce upregulation of TSPO-positive microglia.
Clinical studies using [11C](R)-PK11195 in patients with mild-to-moderate AD have shown increased retention in the entorhinal, temporoparietal and posterior cingulate cortices, areas that show decreased glucose use, as measured with [18F]DG-PET [71]. Furthermore, elevated microglial activation, as indexed by high [11C](R)-PK11195 binding within cortical association and striatal regions (see Figure 1), was noted in a group of AD subjects with high Pittsburgh compound B ([11C]PIB) retention, whose mini mental state examination (MMSE) scores were correlated negatively with microglial activation, but not with [11C]PIB binding [29]. Additional studies have provided conflicting results [72]; however, it is possible that [11C](R)-PK11195 sensitivity might be insufficient for detecting microglial activation present in mild-to-moderate AD [73]. In MCI, findings with [11C](R)-PK11195 are inconclusive, with one study showing a small increase in PIB-positive patients relative to controls [74], but others studies reporting no increase, even among patients who subsequently converted to AD [72, 73].

PET biological targets for measuring neuroinflammation in AD. Amyloid-beta (Aβ)1–42 and neurofibrillary tangles (NFTs) - the classic hallmarks of Alzheimer’s disease (AD) - can trigger neuroinflammatory changes, which induces the release of complement factors, cytokines and others inflammatory factors. Positron emission tomography (PET) uses biological surrogates for measuring neuroinflammation. Microglial activation is estimated by the expression of the 18-kDa translocator protein (TSPO), which is mainly found on the outer mitochondrial membrane of the microglial cells under inflammatory conditions. Monoamine oxidase-B (MAO-B), an enzyme usually located on the outer mitochondrial membrane of astrocytes, is proposed as an index of reactive astrocytosis. Radiolabeled arachidonic acid (AA), a phospholipid present in the cell membrane and cleaved by phospholipase A2 (PLA2), can estimate the AA metabolism. AA is the precursor of eicosanoids - prostaglandins and leukotrienes - which are potent mediators of the inflammatory response.
Among second generation TSPO radioligands, increased binding of [11C]DAA1106 has been observed in AD, as compared to controls, though no correlation was found with respect to disease severity [75]. In a follow-up study among patients with MCI, the increased binding was associated with progression to AD over a 5-year follow-up period [39]. In the case of [11C]PBR28, increased binding was noted in AD, but not MCI, despite the latter displaying cerebral amyloidosis and hippocampal atrophy using PIB-PET and magnetic resonance imaging (MRI) volumetry [76]. Furthermore, [11C]PBR28 binding was shown to correlate with clinical severity and gray matter loss, particularly within regions exhibiting the highest density of TSPO [76] (see Figure 2). Finally, [11C]PBR28 binding was found to be higher among patients with early-onset AD (<65 years), particularly within frontal and parietal regions, in keeping with studies showing greater frontoparietal atrophy in patients with early-onset AD [77–80]. Collectively, these findings with [11C]PBR28 suggest that the increased expression of TSPO by activated microglia occurs after progression to AD, and continues as a function of disease progression, in those who develop disease symptoms at an early age.

Illustrative [ 11 C](R)PK11195 PET imaging. Representative [11C](R)PK11195 images in a healthy control (age 65 years) and in a patient with Alzheimer’s disease (AD) dementia (age 68 years). The brain axial view shows increased [11C](R)PK11195 binding in the AD subject (yellow-red spots) in comparison to the healthy control subject. Standardized uptake value (SUV) defined by the ratio of brain to reference region (supervised reference tissue extraction) radioactivity was used for estimating [11C](R)PK11195 binding. Image provided by Dr Paul Edison of the Division of Brain Sciences, Department of Medicine, Imperial College London, UK.
Second-generation 18-kDa translocator protein ligands and the rs6971 polymorphism
In contrast to PET studies using [11C]PK11195 - which binds with similar affinity across subjects [81] - interpretation of studies using second-generation ligands for the TSPO has been rendered difficult by substantial inter-individual variability in binding affinity, ranging from 4- to 50-fold [82]. Recently, a common single-nucleotide polymorphism (rs6971) in exon 4 of the TSPO gene has been identified as the key determinant of TSPO ligand affinity [83, 84]. Based on the rs6971 polymorphism, subjects are currently divided in three groups: high-affinity binders (HAb), mixed-affinity binders (MAb) and low-affinity binders (LAb), with binding class determined on the basis of the number of high- versus low-affinity sites [81, 82]. While the binding variation between HAb and MAb is around 30% [85], a difference approaching 80% has been observed between between HAb and LAb [76]. Adjusting for the rs6971 polymorphism - either via genotyping or by leucocyte binding assay - has been shown to result in more accurate quantitation of TSPO availability [76], as well as offering potential benefits of increased statistical power and smaller required sample size in the case of clinical studies (see Figure 3).

Illustrative [ 11 C]PBR28 PET imaging. Representative [11C]PBR28 images in a healthy control (age 61 years) and in a patient with Alzheimer’s disease (AD) dementia (age 57 years). The brain axial view show increased [11C]PBR28 binding in the AD subject (yellow-red spots) in comparison to the healthy control subject. Of note, both subjects are high-affinity binders. Distribution volume corrected for free fraction of the radioligand in plasma (V T /f P) was used for estimating [11C]PBR28 binding values. Image provided by Drs William Kreisl and Robert Innis of the Molecular Imaging Branch, National Institute of Mental Health-NIMH, USA.
Cannabinoid receptor type 2: a potential target for imaging microglial activation using PET
Under physiological conditions, the cannabinoid receptor type 2 (CB2) is expressed in very low concentrations in the brain [86, 87]. Recent studies, however, have demonstrated microglial overexpression of CB2 in AD following Aβ deposition [88, 89]. An attractive alternative to TSPO ligands, CB2 radiopharmaceuticals, such as [11C]Sch225336 [44] and [11C]A-836339 [90], are potential imaging biomarkers for estimating activated microglia in the brain. Indeed, work using [11C]A-836339 has provided the first in vivo evidence of CB2 upregulation in APPswe/PS1dE9 mice [90], an animal model presenting Aβ deposition similar to that seen in AD. Moreover, preliminary findings highlight the CB2 receptor as a potential therapeutic target, with use of selective CB2 agonist shown to reduce microgliosis, promote Aβ clearance, and improve cognitive performance in both Tg APP 2576 mice [91] and in rats with cognitive impairment following bilateral microinjections of Aβ at the level of the hippocampus [92]. In addition, Tg amyloid mice lacking the CB2 receptor have been shown to exhibit significantly increased levels of soluble Aβ1–42 and plaque deposition [93]. Though the use CB2 imaging agents may play a role in monitoring the effectiveness of CB2-specific interventions, more studies are needed. The link between TSPO and CB2 expression, however, remains elusive.
Imaging reactive astrocytes using PET
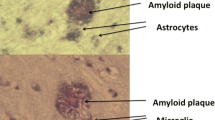
Astrocytes are the most prevalent cells in the central nervous system, outnumbering neurons by at least five-fold [94]. These specialized glial cells dynamically interact with neurons modulating diverse signaling pathways as well as synapse formation [95–97]. Similar to microglia, astrocytes become reactive in response to a variety of detrimental stimuli [98]. In AD, increased expression of glial fibrillary acidic protein (GFAP) is typically observed in immunohistochemical studies of post-mortem brain tissue, indicating an increased number of reactive astrocytes [99], with GFAP-positive astrocytes noted at the margins of amyloid deposits [100]. Though the astrocytic network is thought to exert a neuroprotective role via the sequestration/degradation of Aβ [101–105], its involvement is likewise believed to extend in deleterious directions, including the amplification of cortical amyloid deposition via propagation of intercellular calcium waves [106].
In PET imaging, the enzyme monoamine oxidase B (MAO-B) has been proposed as a biomarker for in vivo quantification of astrocytosis in AD [107]. Located on the outer mitochondrial membrane, MAO-B occurs predominantly in astrocytes [108, 109], and can be imaged using 11C-deuterium-L-deprenyl ([11C]-DED), a radiopharmaceutical exhibiting high affinity and specificity for MAO-B [110, 111]. Catalyzing the oxidative deamination of catecholamines, MAO-B density has been shown to be highly expressed in astrocytes surrounding amyloid plaques [112] and seems to contribute to neurodegeneration by disrupting oxidative homeostasis [113].
Clinical studies using [11C]DED have demonstrated increased tracer retention in MCI and AD, with binding highest among PIB + MCI individuals [30]. These findings suggest that reactive astrocytosis may be present early on in the course of AD, in keeping with previous hypotheses [99, 114], as well as with findings of increased [11C]DED binding in the earliest Braak stages of AD post-mortem tissue [115]. Several important factors remain unanswered with respect to this study, however. First, [11C]DED binding may be underestimated since it is highly dependent on cerebral blood flow [111, 116, 117], a parameter known to be reduced in AD [118–121]. Moreover, it is unclear the degree to which the [11C]DED signal represents reactive astrocytosis, since MAO-B is also found within serotonergic neurons and non-reactive astrocytes [108, 109]. Another confounder that needs to be addressed is the fact that MAO-B seems to increase with age in almost all brain regions (with the exception of the cingulate gyrus) in healthy human subjects [111].
Imaging phospholipase A2 using PET
Microglia derived inflammatory cytokines are capable of binding astrocytic cytokine receptors that are coupled to cytosolic phospholipase A2 (cPLA2), and secretory phospholipase A2 (sPLA2) [122]. Activation of these Ca2+-dependent enzymes results in the hydrolysis of membrane phospholipids, liberating arachidonic acid (AA) [123, 124], itself a precursor of pro-inflammatory eicosanoids including prostaglandins and leukotrienes [125, 126]. Moreover, nitric oxide, released as part of this reaction, can likewise promote AA hydrolysis via cPLA2 via postsynaptic ionotropic N-methyl-D-aspartate glutamate receptors [127, 128], as can Aβ [129, 130]. Such mechanisms have been noted in AD, including increased expression of cPLA2 and sPLA2, elevated cytokine levels, increased glutamatergic markers and different forms of accumulated Aβ [129–131], as well as increased cerebrospinal fluid (CSF) levels of AA metabolites [132]. On the basis of these markers, AA metabolism has been hypothesized to be elevated in AD [31].
Preliminary results obtained using radiolabeled AA (1-[11C]-AA) support this hypothesis, with elevated incorporation coefficients noted in neocortical areas shown to have high densities of neuritic plaques with activated microglia [31]. To the extent that the elevated binding of 1-[11C]-AA represents the upregulation of AA metabolism, PET with 1-[11C]-AA may prove of use in the assessment of investigations in patients with AD.
Neuroinflammation as a therapeutic target in AD
Following an initial report of unexpectedly low prevalence of AD among patients with rheumatoid arthritis [133], numerous epidemiological studies have indicated a reduced incidence of AD among users of NSAIDs (for a review, see [134]). A systematic review suggests that duration of NSAID use of at least 2 years is required to reduce risk estimates [135], while the apparent protective effects of NSAIDs is diminished among older individuals, even to the point of disappearance [136, 137] or, in one study, reversal [138]. The apparent protective effect may also be more pronounced among carriers of the apolipoprotein E (APOE) ϵ4 allele [24, 137, 139, 140]. In general, non-aspirin compounds have been associated with greater protective effects, relative to aspirin compounds [134], and no protective effects have been suggested among users of acetaminophen [141].
Several mechanisms have been proposed to explain the possible protective effects of NSAIDs in AD, including the reduction of brain inflammation by inhibition of cyclooxygenase (COX)-mediated synthesis of pro-inflammatory prostaglandins [101]. Indeed, the beneficial effects of NSAIDs on memory performance in transgenic mouse models of AD have been proposed to relate directly to their blockade of COX activity [102–105, 142, 143], and not to their ability to lower levels of inflammatory cytokines, TNF-α or IL-1β [144]. A second proposed mechanistic hypothesis pertains to the ability of NSAIDs to inhibit processing of the APP or the production or aggregation of Aβ. A subset of NSAIDs (including ibuprofen and indomethacin) have been shown in vitro and in APP-Tg mouse models to preferentially lower levels of amyloidogenic Aβ1–42, independently of their COX-inhibiting activity [145]. These NSAIDs are thought to stimulate non-amyloidogenic processing of APP via enhancement of α-secretase activity [146], to decrease secretion of Aβ in cell lines stimulated with pro-inflammatory cytokines [147], and to decrease the expression of α1-antichymotrypsin, an acute phase protein known to accelerate the development of amyloid pathology in APP-Tg mice [148]. It is noteworthy, however, that a large meta-analysis of NSAID use in human samples has failed to show a distinction between those NSAIDs that modulate γ-secretase activity in vitro and others, and between ibuprofen and naproxen in particular [149]. Finally, NSAIDs that are known to inhibit the multimerization of Aβ in vitro[150] may also inhibit the aggregation of Aβ via direct interaction [151], although human neuropathological studies have failed to show this [152, 153].
The hypothesis that neuroinflammation plays a role in the pathogenesis of AD - and that its suppression via the use of anti-inflammatory compounds may prevent or delay the onset of AD - provided the rationale for a series of clinical trials utilizing various anti-inflammatory drugs [154]. While initial pilot studies using indomethacin (a COX-1 preferential inhibitor) and diclofenac (a non-selective COX inhibitor) combined with the gastroprotective agent misoprostol, suggested benefits in mild-to-moderate AD [155, 156], gastrointestinal problems (a side effect commonly associated with inhibition of COX1) resulted in a high drop-out rate. Follow-up studies with nimesulide [157], celecoxib [158] or rofecoxib [159, 160] - selective COX-2 inhibitors - showed no therapeutic effect [157–160]. Likewise, despite encouraging results in animal models, a 1-year clinical trial using the non-selective COX inhibitor ibuprofen showed no significant overall effects on cognitive and clinical outcomes in patients with mild-to-moderate AD (although positive results were seen in APOE ϵ4 carriers, with the opposite pattern observed in non-carriers) [161]. A primary prevention trial (ADAPT) using naproxen (mixed COX-1/COX-2 inhibitor) and celecoxib (selective COX-2 inhibitor) in individuals at risk for AD was terminated early due to perceived cardiovascular side effects [162], but with results that suggested that these NSAIDs may in fact accelerate disease progression if initiated in individuals already displaying MCI or substantial AD pathophysiology without symptoms. Their use among individuals without AD pathophysiology may prove beneficial, however; a hypothesis supported by CSF biomarker values obtained at 21 to 42 months follow-up [134].
Though the failure of clinical trials using NSAIDs has been ascribed to timing of intervention, duration of treatment, dosage, and drug class, the underlying problem remains the lack of consensus surrounding whether neuroinflammation causes neurodegeneration in AD, or is simply a protective response to primary pathological processes. At present, these findings offer only limited data making it difficult to evaluate the therapeutic utility of NSAIDs in AD. However, piecing together findings from preclinical work in AD-like Tg models, the finding of protective effects in APOE ϵ4 carriers and the overall results of the ADAPT trial, it seems likely that NSAIDs may exert a protective effect but only if administered early on in the disease course. Further studies are required to provide support for this idea.
Potential role of PET in monitoring responsivity to anti-inflammatory therapies in AD
The literature suggests that neuroinflammation occurs early in the course of AD, likely as a response to Aβ and pathologically phosphorylated forms of tau, and that early use of NSAIDs may prove effective in individuals with minimal AD pathology and/or carriers of the APOE ϵ4 allele. Indeed, results from a masked long-term follow-up of the ADAPT cohort seemed to confirm this idea. The difficulty with early initiation of treatment, however, lies in recognizing those who have already experienced pre-symptomatic disease onset. In this respect, studies in asymptomatic carriers of mutations deterministic for early-onset familial AD may prove helpful, given the recent suggestion of the order in which biomarkers reach abnormal levels in this population [163], and that the clinical evolution of early-onset familial AD is highly predictable within a kindred, especially for age at onset [164]. Imaging of neuroinflammation in this population using PET, alongside CSF and plasma markers, may allow for a more integrative AD biomarker model [2, 165], particularly with respect to the interplay between glial activation, seeding of Aβ and hyperphosphorylated tau species, and cognitive decline. Moreover, TSPO ligands may prove sensitive to early AD pathophysiological changes in the form of toxic Aβ oligomers [166], changes that lie below the detection threshold of current in vivo AD biomarkers [2]. In the context of clinical trials using NSAIDs (compounds that retain therapeutic potential despite the general failure of trials conducted to date, particularly since they are relatively safe and highly available), PET could serve to demonstrate target engagement in addition to proving topographical information, critical information lacking from fluid- and plasma-based biomarkers. The reproducibility of TSPO binding, however, remains under debate [167, 168]. In this respect, clinical trials addressing this issue are currently underway, and will hopefully provide results clarifying the test-retest reliability of TSPO ligands [169, 170].
Concluding remarks and future directions
In general, the continued study of inflammatory mechanisms, including in particular the use of PET imaging for tracking neuroinflammatory changes, seems to have a promising role in AD. To date, radioisotopic probes targeting neuroinflammation have demonstrated encouraging results in preclinical and clinical studies, with these radiopharmaceuticals holding promise for inclusion as surrogate markers of disease progression in the next generation of clinical trials using anti-inflammatory therapies. Importantly, novel approaches aiming to augment the sensitivity of these PET imaging agents may be required, with inclusion of vascular [171] and genetic [172, 173] covariates likely to strengthen the value of PET outcomes.
Abbreviations
- [11C]-DED:
-
11C-deuterium-L-deprenyl
- [11C]PIB:
-
Pittsburgh compound B
- Aβ:
-
amyloid-beta
- AA:
-
arachidonic acid
- AD:
-
Alzheimer’s disease
- ADAPT:
-
Alzheimer's Disease Anti-inflammatory Prevention Trial
- APOE:
-
apolipoprotein E
- APP:
-
amyloid precursor protein
- CB2:
-
cannabinoid receptor type 2
- COX:
-
cyclooxygenase
- cPLA2:
-
cytosolic phospholipase A2
- CSF:
-
cerebrospinal fluid
- GFAP:
-
glial fibrillary acidic protein
- HAb:
-
high-affinity binders
- IL:
-
interleukin
- LAb:
-
low-affinity binders
- MAb:
-
mixed-affinity binders
- MAO-B:
-
monoamine oxidase B
- MCI:
-
mild cognitive impairment
- NSAID:
-
non-steroidal anti-inflammatory drug
- PBR:
-
peripheral benzodiazepine receptor
- PET:
-
positron emission tomography
- sPLA2:
-
secretory phospholipase A2
- Tg:
-
transgenic
- TNF:
-
tumor necrosis factor
- TSPO:
-
18 kDa translocator protein.
References
Hardy J, Selkoe DJ: The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002, 297: 353-356.
Jack CR, Knopman DS, Jagust WJ, Petersen RC, Weiner MW, Aisen PS, Shaw LM, Vemuri P, Wiste HJ, Weigand SD, Lesnick TG, Pankratz VS, Donohue MC, Trojanowski JQ: Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013, 12: 207-216.
Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, Iwatsubo T, Jack CR, Kaye J, Montine TJ, Park DC, Reiman EM, Rowe CC, Siemers E, Stern Y, Yaffe K, Carrillo MC, Thies B, Morrison-Bogorad M, Wagster MV, Phelps CH: Toward defining the preclinical stages of Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011, 7: 280-292.
Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, Gamst A, Holtzman DM, Jagust WJ, Petersen RC, Snyder PJ, Carrillo MC, Thies B, Phelps CH: The diagnosis of mild cognitive impairment due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011, 7: 270-279.
McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH: The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging-Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimers Dement. 2011, 7: 263-269.
Eikelenboom P, Stam FC: Immunoglobulins and complement factors in senile plaques. An immunoperoxidase study. Acta Neuropathol. 1982, 57: 239-242.
Eikelenboom P, Hack CE, Rozemuller JM, Stam FC: Complement activation in amyloid plaques in Alzheimer's dementia. Virchows Arch B Cell Pathol Incl Mol Pathol. 1989, 56: 259-262.
Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL, Araoz C: Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc Natl Acad Sci U S A. 1989, 86: 7611-7615.
Eikelenboom P, van Gool WA: Neuroinflammatory perspectives on the two faces of Alzheimer's disease. J Neural Transm. 2004, 111: 281-294.
Giulian D, Lachman LB: Interleukin-1 stimulation of astroglial proliferation after brain injury. Science. 1985, 228: 497-499.
Giulian D, Woodward J, Young DG, Krebs JF, Lachman LB: Interleukin-1 injected into mammalian brain stimulates astrogliosis and neovascularization. J Neurosci. 1988, 8: 2485-2490.
Goldgaber D, Harris HW, Hla T, Maciag T, Donnelly RJ, Jacobsen JS, Vitek MP, Gajdusek DC: Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc Natl Acad Sci U S A. 1989, 86: 7606-7610.
Perlmutter DH, Dinarello CA, Punsal PI, Colten HR: Cachectin/tumor necrosis factor regulates hepatic acute-phase gene expression. J Clin Invest. 1986, 78: 1349-1354.
Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K: Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci U S A. 1985, 82: 4245-4249.
Abraham CR, Selkoe DJ, Potter H: Immunochemical identification of the serine protease inhibitor alpha 1-antichymotrypsin in the brain amyloid deposits of Alzheimer's disease. Cell. 1988, 52: 487-501.
McGeer PL, Akiyama H, Itagaki S, McGeer EG: Immune system response in Alzheimer's disease. Can J Neurol Sci. 1989, 16: 516-527.
McGeer PL, Akiyama H, Itagaki S, McGeer EG: Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci Lett. 1989, 107: 341-346.
Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, Civin WH, Brachova L, Bradt B, Ward P: Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci U S A. 1992, 89: 10016-10020.
Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, O'Banion MK, Pachter J, Pasinetti G, Plata-Salaman C, Rogers J, Rydel R, Shen Y, Streit W, Strohmeyer R, Tooyoma I, Van Muiswinkel FL, Veerhuis R, Walker D, Webster S, Wegrzyniak B, Wenk G, Wyss-Coray T: Inflammation and Alzheimer's disease. Neurobiol Aging. 2000, 21: 383-421.
Wyss-Coray T, Mucke L: Inflammation in neurodegenerative disease - a double-edged sword. Neuron. 2002, 35: 419-432.
Mrak RE, Griffin WS: Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging. 2005, 26: 349-354.
Hoozemans JJ, Veerhuis R, Rozemuller JM, Eikelenboom P: Soothing the inflamed brain: effect of non-steroidal anti-inflammatory drugs on Alzheimer's disease pathology. CNS Neurol Disord Drug Targets. 2011, 10: 57-67.
McGeer PL, Schulzer M, McGeer EG: Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer's disease: a review of 17 epidemiologic studies. Neurology. 1996, 47: 425-432.
in t’ Veld BA, Ruitenberg A, Hofman A, Launer LJ, van Duijn CM, Stijnen T, Breteler MM, Stricker BH: Nonsteroidal antiinflammatory drugs and the risk of Alzheimer's disease. N Engl J Med. 2001, 345: 1515-1521.
Hoozemans JJ, Veerhuis R, Rozemuller AJ, Eikelenboom P: Non-steroidal anti-inflammatory drugs and cyclooxygenase in Alzheimer's disease. Curr Drug Targets. 2003, 4: 461-468.
Arends YM, Duyckaerts C, Rozemuller JM, Eikelenboom P, Hauw JJ: Microglia, amyloid and dementia in alzheimer disease. A correlative study. Neurobiol Aging. 2000, 21: 39-47.
Vehmas AK, Kawas CH, Stewart WF, Troncoso JC: Immune reactive cells in senile plaques and cognitive decline in Alzheimer's disease. Neurobiol Aging. 2003, 24: 321-331.
Hoozemans JJ, van Haastert ES, Veerhuis R, Arendt T, Scheper W, Eikelenboom P, Rozemuller AJ: Maximal COX-2 and ppRb expression in neurons occurs during early Braak stages prior to the maximal activation of astrocytes and microglia in Alzheimer's disease. J Neuroinflammation. 2005, 2: 27.
Edison P, Archer HA, Gerhard A, Hinz R, Pavese N, Turkheimer FE, Hammers A, Tai YF, Fox N, Kennedy A, Rossor M, Brooks DJ: Microglia, amyloid, and cognition in Alzheimer's disease: An [11C](R)PK11195-PET and [11C]PIB-PET study. Neurobiol Dis. 2008, 32: 412-419.
Carter SF, Scholl M, Almkvist O, Wall A, Engler H, Langstrom B, Nordberg A: Evidence for astrocytosis in prodromal Alzheimer disease provided by 11C-deuterium-L-deprenyl: a multitracer PET paradigm combining 11C-Pittsburgh compound B and 18F-FDG. J Nucl Med. 2012, 53: 37-46.
Esposito G, Giovacchini G, Liow JS, Bhattacharjee AK, Greenstein D, Schapiro M, Hallett M, Herscovitch P, Eckelman WC, Carson RE, Rapoport SI: Imaging neuroinflammation in Alzheimer's disease with radiolabeled arachidonic acid and PET. J Nucl Med. 2008, 49: 1414-1421.
Hashimoto K, Inoue O, Suzuki K, Yamasaki T, Kojima M: Synthesis and evaluation of 11C-PK 11195 for in vivo study of peripheral-type benzodiazepine receptors using positron emission tomography. Ann Nucl Med. 1989, 3: 63-71.
Zhang MR, Kumata K, Maeda J, Yanamoto K, Hatori A, Okada M, Higuchi M, Obayashi S, Suhara T, Suzuki K: 11C-AC-5216: a novel PET ligand for peripheral benzodiazepine receptors in the primate brain. J Nucl Med. 2007, 48: 1853-1861.
Brown AK, Fujita M, Fujimura Y, Liow JS, Stabin M, Ryu YH, Imaizumi M, Hong J, Pike VW, Innis RB: Radiation dosimetry and biodistribution in monkey and man of 11C-PBR28: a PET radioligand to image inflammation. J Nucl Med. 2007, 48: 2072-2079.
Chauveau F, Van Camp N, Dolle F, Kuhnast B, Hinnen F, Damont A, Boutin H, James M, Kassiou M, Tavitian B: Comparative evaluation of the translocator protein radioligands 11C-DPA-713, 18F-DPA-714, and 11C-PK11195 in a rat model of acute neuroinflammation. J Nucl Med. 2009, 50: 468-476.
Doorduin J, Klein HC, Dierckx RA, James M, Kassiou M, de Vries EF: [11C]-DPA-713 and [18F]-DPA-714 as new PET tracers for TSPO: a comparison with [11C]-(R)-PK11195 in a rat model of herpes encephalitis. Mol Imaging Biol. 2009, 11: 386-398.
Tiwari AK, Yui J, Fujinaga M, Kumata K, Shimoda Y, Yamasaki T, Xie L, Hatori A, Maeda J, Nengaki N, Zhang MR: Characterization of a novel acetamidobenzoxazolone-based PET ligand for translocator protein (18 kDa) imaging of neuroinflammation in the brain. J Neurochem. 2014, 129: 712-720.
Yanamoto K, Yamasaki T, Kumata K, Yui J, Odawara C, Kawamura K, Hatori A, Inoue O, Yamaguchi M, Suzuki K, Zhang MR: Evaluation of N-benzyl-N-[11C]methyl-2-(7-methyl-8-oxo-2-phenyl-7,8-dihydro-9H-purin-9-yl)acetamide ([11C]DAC) as a novel translocator protein (18 kDa) radioligand in kainic acid-lesioned rat. Synapse. 2009, 63: 961-971.
Yasuno F, Kosaka J, Ota M, Higuchi M, Ito H, Fujimura Y, Nozaki S, Takahashi S, Mizukami K, Asada T, Suhara T: Increased binding of peripheral benzodiazepine receptor in mild cognitive impairment-dementia converters measured by positron emission tomography with [11C]DAA1106. Psychiatry Res. 2012, 203: 67-74.
Gulyas B, Halldin C, Vas A, Banati RB, Shchukin E, Finnema S, Tarkainen J, Tihanyi K, Szilagyi G, Farde L: [11C]vinpocetine: a prospective peripheral benzodiazepine receptor ligand for primate PET studies. J Neurol Sci. 2005, 229–230: 219-223.
Fujimura Y, Zoghbi SS, Simeon FG, Taku A, Pike VW, Innis RB, Fujita M: Quantification of translocator protein (18 kDa) in the human brain with PET and a novel radioligand, (18)F-PBR06. J Nucl Med. 2009, 50: 1047-1053.
Yui J, Maeda J, Kumata K, Kawamura K, Yanamoto K, Hatori A, Yamasaki T, Nengaki N, Higuchi M, Zhang MR: 18F-FEAC and 18F-FEDAC: PET of the monkey brain and imaging of translocator protein (18 kDa) in the infarcted rat brain. J Nucl Med. 2010, 51: 1301-1309.
Varrone A, Mattsson P, Forsberg A, Takano A, Nag S, Gulyas B, Borg J, Boellaard R, Al-Tawil N, Eriksdotter M, Zimmermann T, Schultze-Mosgau M, Thiele A, Hoffmann A, Lammertsma AA, Halldin C: In vivo imaging of the 18-kDa translocator protein (TSPO) with [18F]FEDAA1106 and PET does not show increased binding in Alzheimer's disease patients. Eur J Nucl Med Mol Imaging. 2013, 40: 921-931.
Evens N, Bosier B, Lavey BJ, Kozlowski JA, Vermaelen P, Baudemprez L, Busson R, Lambert DM, Van Laere K, Verbruggen AM, Bormans GM: Labelling and biological evaluation of [(11)C]methoxy-Sch225336: a radioligand for the cannabinoid-type 2 receptor. Nucl Med Biol. 2008, 35: 793-800.
Nagatsugi F, Hokazono J, Sasaki S, Maeda M: 20-[18F]fluoroarachidonic acid: tissue biodistribution and incorporation into phospholipids. Biol Pharm Bull. 1996, 19: 1316-1321.
Solito E, Sastre M: Microglia function in Alzheimer's disease. Front Pharmacol. 2012, 3: 14.
Chan WY, Kohsaka S, Rezaie P: The origin and cell lineage of microglia: new concepts. Brain Res Rev. 2007, 53: 344-354.
Nimmerjahn A, Kirchhoff F, Helmchen F: Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 2005, 308: 1314-1318.
Ransohoff RM, Perry VH: Microglial physiology: unique stimuli, specialized responses. Annu Rev Immunol. 2009, 27: 119-145.
Fetler L, Amigorena S: Neuroscience: Brain under surveillance: the microglia patrol. Science. 2005, 309: 392-393.
Rogers J, Luber-Narod J, Styren SD, Civin WH: Expression of immune system-associated antigens by cells of the human central nervous system: relationship to the pathology of Alzheimer's disease. Neurobiol Aging. 1988, 9: 339-349.
Styren SD, Civin WH, Rogers J: Molecular, cellular, and pathologic characterization of HLA-DR immunoreactivity in normal elderly and Alzheimer's disease brain. Exp Neurol. 1990, 110: 93-104.
Frautschy SA, Yang F, Irrizarry M, Hyman B, Saido TC, Hsiao K, Cole GM: Microglial response to amyloid plaques in APPsw transgenic mice. Am J Pathol. 1998, 152: 307-317.
Stalder M, Phinney A, Probst A, Sommer B, Staufenbiel M, Jucker M: Association of microglia with amyloid plaques in brains of APP23 transgenic mice. Am J Pathol. 1999, 154: 1673-1684.
Combs CK, Johnson DE, Cannady SB, Lehman TM, Landreth GE: Identification of microglial signal transduction pathways mediating a neurotoxic response to amyloidogenic fragments of beta-amyloid and prion proteins. J Neurosci. 1999, 19: 928-939.
Combs CK, Johnson DE, Karlo JC, Cannady SB, Landreth GE: Inflammatory mechanisms in Alzheimer's disease: inhibition of beta-amyloid-stimulated proinflammatory responses and neurotoxicity by PPARgamma agonists. J Neurosci. 2000, 20: 558-567.
Klegeris A, McGeer PL: Beta-amyloid protein enhances macrophage production of oxygen free radicals and glutamate. J Neurosci Res. 1997, 49: 229-235.
McDonald DR, Brunden KR, Landreth GE: Amyloid fibrils activate tyrosine kinase-dependent signaling and superoxide production in microglia. J Neurosci. 1997, 17: 2284-2294.
Prokop S, Miller KR, Heppner FL: Microglia actions in Alzheimer's disease. Acta Neuropathol. 2013, 126: 461-477.
Papadopoulos V, Baraldi M, Guilarte TR, Knudsen TB, Lacapere JJ, Lindemann P, Norenberg MD, Nutt D, Weizman A, Zhang MR, Gavish M: Translocator protein (18 kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci. 2006, 27: 402-409.
Venneti S, Lopresti BJ, Wiley CA: The peripheral benzodiazepine receptor (Translocator protein 18 kDa) in microglia: from pathology to imaging. Prog Neurobiol. 2006, 80: 308-322.
Chen MK, Guilarte TR: Translocator protein 18 kDa (TSPO): molecular sensor of brain injury and repair. Pharmacol Ther. 2008, 118: 1-17.
Anholt RR, Pedersen PL, De Souza EB, Snyder SH: The peripheral-type benzodiazepine receptor: localization to the mitochondrial outer membrane. J Biol Chem. 1986, 261: 576-583.
Cymerman U, Pazos A, Palacios JM: Evidence for species differences in ‘peripheral’ benzodiazepine receptors: an autoradiographic study. Neurosci Lett. 1986, 66: 153-158.
Ching AS, Kuhnast B, Damont A, Roeda D, Tavitian B, Dolle F: Current paradigm of the 18-kDa translocator protein (TSPO) as a molecular target for PET imaging in neuroinflammation and neurodegenerative diseases. Insights Imaging. 2012, 3: 111-119.
Jacobs AH, Tavitian B, INMiND consortium: Noninvasive molecular imaging of neuroinflammation. J Cereb Blood Flow Metab. 2012, 32: 1393-1415.
Venneti S, Lopresti BJ, Wang G, Hamilton RL, Mathis CA, Klunk WE, Apte UM, Wiley CA: PK11195 labels activated microglia in Alzheimer's disease and in vivo in a mouse model using PET. Neurobiol Aging. 2009, 30: 1217-1226.
Zhang MR, Maeda J, Ogawa M, Noguchi J, Ito T, Yoshida Y, Okauchi T, Obayashi S, Suhara T, Suzuki K: Development of a new radioligand, N-(5-fluoro-2-phenoxyphenyl)-N-(2-[18F]fluoroethyl-5-methoxybenzyl)acetamide, for pet imaging of peripheral benzodiazepine receptor in primate brain. J Med Chem. 2004, 47: 2228-2235.
Maeda J, Zhang MR, Okauchi T, Ji B, Ono M, Hattori S, Kumata K, Iwata N, Saido TC, Trojanowski JQ, Lee VM, Staufenbiel M, Tomiyama T, Mori H, Fukumura T, Suhara T, Higuchi M: In vivo positron emission tomographic imaging of glial responses to amyloid-beta and tau pathologies in mouse models of Alzheimer's disease and related disorders. J Neurosci. 2011, 31: 4720-4730.
Sturchler-Pierrat C, Abramowski D, Duke M, Wiederhold KH, Mistl C, Rothacher S, Ledermann B, Burki K, Frey P, Paganetti PA, Waridel C, Calhoun ME, Jucker M, Probst A, Staufenbiel M, Sommer B: Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci U S A. 1997, 94: 13287-13292.
Cagnin A, Brooks DJ, Kennedy AM, Gunn RN, Myers R, Turkheimer FE, Jones T, Banati RB: In-vivo measurement of activated microglia in dementia. Lancet. 2001, 358: 461-467.
Schuitemaker A, Kropholler MA, Boellaard R, van der Flier WM, Kloet RW, van der Doef TF, Knol DL, Windhorst AD, Luurtsema G, Barkhof F, Jonker C, Lammertsma AA, Scheltens P, van Berckel BN: Microglial activation in Alzheimer's disease: an (R)-[11C]PK11195 positron emission tomography study. Neurobiol Aging. 2013, 34: 128-136.
Wiley CA, Lopresti BJ, Venneti S, Price J, Klunk WE, DeKosky ST, Mathis CA: Carbon 11-labeled Pittsburgh Compound B and carbon 11-labeled (R)-PK11195 positron emission tomographic imaging in Alzheimer disease. Arch Neurol. 2009, 66: 60-67.
Okello A, Edison P, Archer HA, Turkheimer FE, Kennedy J, Bullock R, Walker Z, Kennedy A, Fox N, Rossor M, Brooks DJ: Microglial activation and amyloid deposition in mild cognitive impairment: a PET study. Neurology. 2009, 72: 56-62.
Yasuno F, Ota M, Kosaka J, Ito H, Higuchi M, Doronbekov TK, Nozaki S, Fujimura Y, Koeda M, Asada T, Suhara T: Increased binding of peripheral benzodiazepine receptor in Alzheimer's disease measured by positron emission tomography with [11C]DAA1106. Biol Psychiatry. 2008, 64: 835-841.
Kreisl WC, Jenko KJ, Hines CS, Lyoo CH, Corona W, Morse CL, Zoghbi SS, Hyde T, Kleinman JE, Pike VW, et al: A genetic polymorphism for translocator protein 18 kDa affects both in vitro and in vivo radioligand binding in human brain to this putative biomarker of neuroinflammation. J Cereb Blood Flow Metab. 2013, 33: 53-58.
Suribhatla S, Baillon S, Dennis M, Marudkar M, Muhammad S, Munro D, Spreadbury C, Lindesay J: Neuropsychological performance in early and late onset Alzheimer's disease: comparisons in a memory clinic population. Int J Geriatr Psychiatry. 2004, 19: 1140-1147.
Shiino A, Watanabe T, Maeda K, Kotani E, Akiguchi I, Matsuda M: Four subgroups of Alzheimer's disease based on patterns of atrophy using VBM and a unique pattern for early onset disease. Neuroimage. 2006, 33: 17-26.
Karas G, Scheltens P, Rombouts S, van Schijndel R, Klein M, Jones B, van der Flier W, Vrenken H, Barkhof F: Precuneus atrophy in early-onset Alzheimer's disease: a morphometric structural MRI study. Neuroradiology. 2007, 49: 967-976.
Licht EA, McMurtray AM, Saul RE, Mendez MF: Cognitive differences between early- and late-onset Alzheimer's disease. Am J Alzheimers Dis Other Demen. 2007, 22: 218-222.
Owen DR, Howell OW, Tang SP, Wells LA, Bennacef I, Bergstrom M, Gunn RN, Rabiner EA, Wilkins MR, Reynolds R, Matthews PM, Parker CA: Two binding sites for [3H]PBR28 in human brain: implications for TSPO PET imaging of neuroinflammation. J Cereb Blood Flow Metab. 2010, 30: 1608-1618.
Owen DR, Gunn RN, Rabiner EA, Bennacef I, Fujita M, Kreisl WC, Innis RB, Pike VW, Reynolds R, Matthews PM, Parker CA: Mixed-affinity binding in humans with 18-kDa translocator protein ligands. J Nucl Med. 2011, 52: 24-32.
Owen DR, Yeo AJ, Gunn RN, Song K, Wadsworth G, Lewis A, Rhodes C, Pulford DJ, Bennacef I, Parker CA, StJean PL, Cardon LR, Mooser VE, Matthews PM, Rabiner EA, Rubio JP: An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab. 2012, 32: 1-5.
Mizrahi R, Rusjan PM, Kennedy J, Pollock B, Mulsant B, Suridjan I, De Luca V, Wilson AA, Houle S: Translocator protein (18 kDa) polymorphism (rs6971) explains in-vivo brain binding affinity of the PET radioligand [18F]-FEPPA. J Cereb Blood Flow Metab. 2012, 32: 968-972.
Yoder KK, Nho K, Risacher SL, Kim S, Shen L, Saykin AJ: Influence of TSPO genotype on 11C-PBR28 standardized uptake values. J Nucl Med. 2013, 54: 1320-1322.
Gong JP, Onaivi ES, Ishiguro H, Liu QR, Tagliaferro PA, Brusco A, Uhl GR: Cannabinoid CB2 receptors: immunohistochemical localization in rat brain. Brain Res. 2006, 1071: 10-23.
Van Sickle MD, Duncan M, Kingsley PJ, Mouihate A, Urbani P, Mackie K, Stella N, Makriyannis A, Piomelli D, Davison JS, Marnett LJ, Di Marzo V, Pittman QJ, Patel KD, Sharkey KA: Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science. 2005, 310: 329-332.
Esposito G, Iuvone T, Savani C, Scuderi C, De Filippis D, Papa M, Di Marzo V, Steardo L: Opposing control of cannabinoid receptor stimulation on amyloid-beta-induced reactive gliosis: in vitro and in vivo evidence. J Pharmacol Exp Ther. 2007, 322: 1144-1152.
Benito C, Tolon RM, Pazos MR, Nunez E, Castillo AI, Romero J: Cannabinoid CB2 receptors in human brain inflammation. Br J Pharmacol. 2008, 153: 277-285.
Horti AG, Gao Y, Ravert HT, Finley P, Valentine H, Wong DF, Endres CJ, Savonenko AV, Dannals RF: Synthesis and biodistribution of [11C]A-836339, a new potential radioligand for PET imaging of cannabinoid type 2 receptors (CB2). Bioorg Med Chem. 2010, 18: 5202-5207.
Martin-Moreno AM, Brera B, Spuch C, Carro E, Garcia-Garcia L, Delgado M, Pozo MA, Innamorato NG, Cuadrado A, de Ceballos ML: Prolonged oral cannabinoid administration prevents neuroinflammation, lowers beta-amyloid levels and improves cognitive performance in Tg APP 2576 mice. J Neuroinflammation. 2012, 9: 8.
Wu J, Bie B, Yang H, Xu JJ, Brown DL, Naguib M: Activation of the CB2 receptor system reverses amyloid-induced memory deficiency. Neurobiol Aging. 2013, 34: 791-804.
Koppel J, Vingtdeux V, Marambaud P, d’Abramo C, Jimenez H, Stauber M, Friedman R, Davies P: CB2 receptor deficiency increases amyloid pathology and alters tau processing in a transgenic mouse model of Alzheimer's disease. Mol Med. 2014, 20: 29-36.
Sofroniew MV, Vinters HV: Astrocytes: biology and pathology. Acta Neuropathol. 2010, 119: 7-35.
Allen NJ, Bennett ML, Foo LC, Wang GX, Chakraborty C, Smith SJ, Barres BA: Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature. 2012, 486: 410-414.
Hughes EG, Elmariah SB, Balice-Gordon RJ: Astrocyte secreted proteins selectively increase hippocampal GABAergic axon length, branching, and synaptogenesis. Mol Cell Neurosci. 2010, 43: 136-145.
Ullian EM, Sapperstein SK, Christopherson KS, Barres BA: Control of synapse number by glia. Science. 2001, 291: 657-661.
Sofroniew MV: Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32: 638-647.
Verkhratsky A, Olabarria M, Noristani HN, Yeh CY, Rodriguez JJ: Astrocytes in Alzheimer's disease. Neurotherapeutics. 2010, 7: 399-412.
Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D: Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J Neuroimmunol. 1989, 24: 173-182.
Hoozemans JJ, Rozemuller JM, van Haastert ES, Veerhuis R, Eikelenboom P: Cyclooxygenase-1 and −2 in the different stages of Alzheimer's disease pathology. Curr Pharm Des. 2008, 14: 1419-1427.
Jantzen PT, Connor KE, DiCarlo G, Wenk GL, Wallace JL, Rojiani AM, Coppola D, Morgan D, Gordon MN: Microglial activation and beta-amyloid deposit reduction caused by a nitric oxide-releasing nonsteroidal anti-inflammatory drug in amyloid precursor protein plus presenilin-1 transgenic mice. J Nucl Med. 2002, 22: 2246-2254.
Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM: Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Nucl Med. 2000, 20: 5709-5714.
Lim GP, Yang F, Chu T, Gahtan E, Ubeda O, Beech W, Overmier JB, Hsiao-Ashec K, Frautschy SA, Cole GM: Ibuprofen effects on Alzheimer pathology and open field activity in APPsw transgenic mice. Neurobiol Aging. 2001, 22: 983-991.
Yan Q, Zhang J, Liu H, Babu-Khan S, Vassar R, Biere AL, Citron M, Landreth G: Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer's disease. J Nucl Med. 2003, 23: 7504-7509.
Kuchibhotla KV, Lattarulo CR, Hyman BT, Bacskai BJ: Synchronous hyperactivity and intercellular calcium waves in astrocytes in Alzheimer mice. Science. 2009, 323: 1211-1215.
Santillo AF, Gambini JP, Lannfelt L, Langstrom B, Ulla-Marja L, Kilander L, Engler H: In vivo imaging of astrocytosis in Alzheimer's disease: an 11C-L-deuteriodeprenyl and PIB PET study. Eur J Nucl Med Mol Imaging. 2011, 38: 2202-2208.
Fowler JS, Logan J, Volkow ND, Wang GJ: Translational neuroimaging: positron emission tomography studies of monoamine oxidase. Mol Imaging Biol. 2005, 7: 377-387.
Saura J, Bleuel Z, Ulrich J, Mendelowitsch A, Chen K, Shih JC, Malherbe P, Da Prada M, Richards JG: Molecular neuroanatomy of human monoamine oxidases A and B revealed by quantitative enzyme radioautography and in situ hybridization histochemistry. Neuroscience. 1996, 70: 755-774.
Fowler JS, MacGregor RR, Wolf AP, Arnett CD, Dewey SL, Schlyer D, Christman D, Logan J, Smith M, Sachs H, Aquilonius SM, Bjurling P, Halldin C, Hartvig P, Leenders KL, Lundqvist H, Oreland L, Stalnacke CG, Langstrom B: Mapping human brain monoamine oxidase A and B with 11C-labeled suicide inactivators and PET. Science. 1987, 235: 481-485.
Fowler JS, Volkow ND, Wang GJ, Logan J, Pappas N, Shea C, MacGregor R: Age-related increases in brain monoamine oxidase B in living healthy human subjects. Neurobiol Aging. 1997, 18: 431-435.
Saura J, Luque JM, Cesura AM, Da Prada M, Chan-Palay V, Huber G, Loffler J, Richards JG: Increased monoamine oxidase B activity in plaque-associated astrocytes of Alzheimer brains revealed by quantitative enzyme radioautography. Neuroscience. 1994, 62: 15-30.
Smith MA, Rottkamp CA, Nunomura A, Raina AK, Perry G: Oxidative stress in Alzheimer's disease. Biochim Biophys Acta. 2000, 1502: 139-144.
Lichtenstein MP, Carriba P, Masgrau R, Pujol A, Galea E: Staging anti-inflammatory therapy in Alzheimer's disease. Front Aging Neurosci. 2010, 2: 142.
Gulyas B, Pavlova E, Kasa P, Gulya K, Bakota L, Varszegi S, Keller E, Horvath MC, Nag S, Hermecz I, Magyar K, Halldin C: Activated MAO-B in the brain of Alzheimer patients, demonstrated by [11C]-L-deprenyl using whole hemisphere autoradiography. Neurochem Int. 2011, 58: 60-68.
Engler H, Lundberg PO, Ekbom K, Nennesmo I, Nilsson A, Bergstrom M, Tsukada H, Hartvig P, Langstrom B: Multitracer study with positron emission tomography in Creutzfeldt-Jakob disease. Eur J Nucl Med Mol Imaging. 2003, 30: 85-95.
Fowler JS, Volkow ND, Logan J, Schlyer DJ, MacGregor RR, Wang GJ, Wolf AP, Pappas N, Alexoff D, Shea C, Gatley SJ, Dorflinger E, Yoo K, Morawsky L, Fazzini E: Monoamine oxidase B (MAO B) inhibitor therapy in Parkinson's disease: the degree and reversibility of human brain MAO B inhibition by Ro 19 6327. Neurology. 1993, 43: 1984-1992.
Johnson KA, Mueller ST, Walshe TM, English RJ, Holman BL: Cerebral perfusion imaging in Alzheimer's disease. Use of single photon emission computed tomography and iofetamine hydrochloride I 123. Arch Neurol. 1987, 44: 165-168.
Powers WJ, Perlmutter JS, Videen TO, Herscovitch P, Griffeth LK, Royal HD, Siegel BA, Morris JC, Berg L: Blinded clinical evaluation of positron emission tomography for diagnosis of probable Alzheimer's disease. Neurology. 1992, 42: 765-770.
Yoshiura T, Hiwatashi A, Noguchi T, Yamashita K, Ohyagi Y, Monji A, Nagao E, Kamano H, Togao O, Honda H: Arterial spin labelling at 3-T MR imaging for detection of individuals with Alzheimer's disease. Eur Radiol. 2009, 19: 2819-2825.
Bartenstein P, Minoshima S, Hirsch C, Buch K, Willoch F, Mosch D, Schad D, Schwaiger M, Kurz A: Quantitative assessment of cerebral blood flow in patients with Alzheimer's disease by SPECT. J Nucl Med. 1997, 38: 1095-1101.
Sun GY, Horrocks LA, Farooqui AA: The roles of NADPH oxidase and phospholipases A2 in oxidative and inflammatory responses in neurodegenerative diseases. J Neurochem. 2007, 103: 1-16.
Shimizu T, Wolfe LS: Arachidonic acid cascade and signal transduction. J Neurochem. 1990, 55: 1-15.
Balsinde J, Winstead MV, Dennis EA: Phospholipase A(2) regulation of arachidonic acid mobilization. FEBS Lett. 2002, 531: 2-6.
Ricciotti E, FitzGerald GA: Prostaglandins and inflammation. Arterioscler Thromb Vasc Biol. 2011, 31: 986-1000.
Park JY, Pillinger MH, Abramson SB: Prostaglandin E2 synthesis and secretion: the role of PGE2 synthases. Clin Immunol. 2006, 119: 229-240.
Dennis EA: Diversity of group types, regulation, and function of phospholipase A2. J Biol Chem. 1994, 269: 13057-13060.
Weichel O, Hilgert M, Chatterjee SS, Lehr M, Klein J: Bilobalide, a constituent of Ginkgo biloba, inhibits NMDA-induced phospholipase A2 activation and phospholipid breakdown in rat hippocampus. Naunyn Schmiedebergs Arch Pharmacol. 1999, 360: 609-615.
Mattson MP, Chan SL: Neuronal and glial calcium signaling in Alzheimer's disease. Cell Calcium. 2003, 34: 385-397.
Lehtonen JY, Holopainen JM, Kinnunen PK: Activation of phospholipase A2 by amyloid beta-peptides in vitro. Biochemistry. 1996, 35: 9407-9414.
Greenamyre JT, Maragos WF, Albin RL, Penney JB, Young AB: Glutamate transmission and toxicity in Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 1988, 12: 421-430.
Montine KS, Quinn JF, Zhang J, Fessel JP, Roberts LJ, Morrow JD, Montine TJ: Isoprostanes and related products of lipid peroxidation in neurodegenerative diseases. Chem Phys Lipids. 2004, 128: 117-124.
McGeer PL, McGeer E, Rogers J, Sibley J: Anti-inflammatory drugs and Alzheimer disease. Lancet. 1990, 335: 1037.
Imbimbo BP, Solfrizzi V, Panza F: Are NSAIDs useful to treat Alzheimer's disease or mild cognitive impairment?. Front Aging Neurosci. 2010, 2: doi:10.3389/fnagi.2010.00019
Szekely CA, Thorne JE, Zandi PP, Ek M, Messias E, Breitner JC, Goodman SN: Nonsteroidal anti-inflammatory drugs for the prevention of Alzheimer's disease: a systematic review. Neuroepidemiology. 2004, 23: 159-169.
Zandi PP, Anthony JC, Hayden KM, Mehta K, Mayer L, Breitner JC, Cache County Study I: Reduced incidence of AD with NSAID but not H2 receptor antagonists: the Cache County Study. Neurology. 2002, 59: 880-886.
Szekely CA, Breitner JC, Fitzpatrick AL, Rea TD, Psaty BM, Kuller LH, Zandi PP: NSAID use and dementia risk in the Cardiovascular Health Study: role of APOE and NSAID type. Neurology. 2008, 70: 17-24.
Breitner JC, Haneuse SJ, Walker R, Dublin S, Crane PK, Gray SL, Larson EB: Risk of dementia and AD with prior exposure to NSAIDs in an elderly community-based cohort. Neurology. 2009, 72: 1899-1905.
Hayden KM, Zandi PP, Khachaturian AS, Szekely CA, Fotuhi M, Norton MC, Tschanz JT, Pieper CF, Corcoran C, Lyketsos CG, Breitner JC, Welsh-Bohmer KA: Does NSAID use modify cognitive trajectories in the elderly? The Cache County study. Neurology. 2007, 69: 275-282.
Cornelius C, Fastbom J, Winblad B, Viitanen M: Aspirin, NSAIDs, risk of dementia, and influence of the apolipoprotein E epsilon 4 allele in an elderly population. Neuroepidemiology. 2004, 23: 135-143.
Stewart WF, Kawas C, Corrada M, Metter EJ: Risk of Alzheimer's disease and duration of NSAID use. Neurology. 1997, 48: 626-632.
Van Dam D, Coen K, De Deyn PP: Ibuprofen modifies cognitive disease progression in an Alzheimer's mouse model. J Psychopharmacol. 2010, 24: 383-388.
McKee AC, Carreras I, Hossain L, Ryu H, Klein WL, Oddo S, LaFerla FM, Jenkins BG, Kowall NW, Dedeoglu A: Ibuprofen reduces Abeta, hyperphosphorylated tau and memory deficits in Alzheimer mice. Brain Res. 2008, 1207: 225-236.
Kotilinek LA, Westerman MA, Wang Q, Panizzon K, Lim GP, Simonyi A, Lesne S, Falinska A, Younkin LH, Younkin SG, Rowan M, Cleary J, Wallis RA, Sun GY, Cole G, Frautschy S, Anwyl R, Ashe KH: Cyclooxygenase-2 inhibition improves amyloid-beta-mediated suppression of memory and synaptic plasticity. Brain. 2008, 131: 651-664.
Weggen S, Eriksen JL, Das P, Sagi SA, Wang R, Pietrzik CU, Findlay KA, Smith TE, Murphy MP, Bulter T, Bulter T, Kang DE, Marquez-Sterling N, Golde TE, Koo EH: A subset of NSAIDs lower amyloidogenic Abeta42 independently of cyclooxygenase activity. Nature. 2001, 414: 212-216.
Avramovich Y, Amit T, Youdim MB: Non-steroidal anti-inflammatory drugs stimulate secretion of non-amyloidogenic precursor protein. J Biol Chem. 2002, 277: 31466-31473.
Sastre M, Dewachter I, Rossner S, Bogdanovic N, Rosen E, Borghgraef P, Evert BO, Dumitrescu-Ozimek L, Thal DR, Landreth G, Walter J, Klockgether T, van Leuven F, Heneka MT: Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPARgamma. Proc Natl Acad Sci U S A. 2006, 103: 443-448.
Morihara T, Teter B, Yang F, Lim GP, Boudinot S, Boudinot FD, Frautschy SA, Cole GM: Ibuprofen suppresses interleukin-1beta induction of pro-amyloidogenic alpha1-antichymotrypsin to ameliorate beta-amyloid (Abeta) pathology in Alzheimer's models. Neuropsychopharmacology. 2005, 30: 1111-1120.
Szekely CA, Green RC, Breitner JC, Ostbye T, Beiser AS, Corrada MM, Dodge HH, Ganguli M, Kawas CH, Kuller LH, Psaty BM, Resnick SM, Wolf PA, Zonderman AB, Welsh-Bohmer KA, Zandi PP: No advantage of A beta 42-lowering NSAIDs for prevention of Alzheimer dementia in six pooled cohort studies. Neurology. 2008, 70: 2291-2298.
Thomas T, Nadackal TG, Thomas K: Aspirin and non-steroidal anti-inflammatory drugs inhibit amyloid-beta aggregation. Neuroreport. 2001, 12: 3263-3267.
Kukar TL, Ladd TB, Bann MA, Fraering PC, Narlawar R, Maharvi GM, Healy B, Chapman R, Welzel AT, Price RW, Moore B, Rangachari V, Cusack B, Eriksen J, Jansen-West K, Verbeeck C, Yager D, Eckman C, Ye W, Sagi S, Cottrell BA, Torpey J, Rosenberry TL, Fauq A, Wolfe MS, Schmidt B, Walsh DM, Koo EH, Golde TE: Substrate-targeting gamma-secretase modulators. Nature. 2008, 453: 925-929.
Alafuzoff I, Overmyer M, Helisalmi S, Soininen H: Lower counts of astroglia and activated microglia in patients with Alzheimer's disease with regular use of non-steroidal anti-inflammatory drugs. J Alzheimers Dis. 2000, 2: 37-46.
Halliday GM, Shepherd CE, McCann H, Reid WG, Grayson DA, Broe GA, Kril JJ: Effect of anti-inflammatory medications on neuropathological findings in Alzheimer disease. Arch Neurol. 2000, 57: 831-836.
Aisen PS, Davis KL: Inflammatory mechanisms in Alzheimer's disease: implications for therapy. Am J Psychiatry. 1994, 151: 1105-1113.
Rogers J, Kirby LC, Hempelman SR, Berry DL, McGeer PL, Kaszniak AW, Zalinski J, Cofield M, Mansukhani L, Willson P, Kogan F: Clinical trial of indomethacin in Alzheimer's disease. Neurology. 1993, 43: 1609-1611.
Scharf S, Mander A, Ugoni A, Vajda F, Christophidis N: A double-blind, placebo-controlled trial of diclofenac/misoprostol in Alzheimer's disease. Neurology. 1999, 53: 197-201.
Aisen PS, Schmeidler J, Pasinetti GM: Randomized pilot study of nimesulide treatment in Alzheimer's disease. Neurology. 2002, 58: 1050-1054.
Soininen H, West C, Robbins J, Niculescu L: Long-term efficacy and safety of celecoxib in Alzheimer's disease. Dement Geriatr Cogn Disord. 2007, 23: 8-21.
Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, Farlow MR, Jin S, Thomas RG, Thal LJ, Alzheimer's Disease Cooperative Study: Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003, 289: 2819-2826.
Reines SA, Block GA, Morris JC, Liu G, Nessly ML, Lines CR, Norman BA, Baranak CC, Rofecoxib Protocol 091 Study Group: Rofecoxib: no effect on Alzheimer's disease in a 1-year, randomized, blinded, controlled study. Neurology. 2004, 62: 66-71.
Pasqualetti P, Bonomini C, Dal Forno G, Paulon L, Sinforiani E, Marra C, Rossini PM, Zanetti O: A randomized controlled study on effects of ibuprofen on cognitive progression of Alzheimer's disease. Aging Clin Exp Res. 2009, 21: 102-110.
ADAPT Research Group: Cardiovascular and cerebrovascular events in the randomized, controlled Alzheimer's Disease Anti-Inflammatory Prevention Trial (ADAPT). PLoS Clin Trials. 2006, 1: e33.
Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC, Dominantly Inherited Alzheimer Network: Clinical and biomarker changes in dominantly inherited Alzheimer's disease. N Engl J Med. 2012, 367: 795-804.
Wu L, Rosa-Neto P, Hsiung GY, Sadovnick AD, Masellis M, Black SE, Jia J, Gauthier S: Early-onset familial Alzheimer's disease (EOFAD). Can J Neurol Sci. 2012, 39: 436-445.
Jack CR, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ: Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol. 2010, 9: 119-128.
Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL: Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998, 95: 6448-6453.
Jucaite A, Cselenyi Z, Arvidsson A, Ahlberg G, Julin P, Varnas K, Stenkrona P, Andersson J, Halldin C, Farde L: Kinetic analysis and test-retest variability of the radioligand [11C](R)-PK11195 binding to TSPO in the human brain - a PET study in control subjects. EJNMMI Res. 2012, 2: 15.
Turkheimer FE, Edison P, Pavese N, Roncaroli F, Anderson AN, Hammers A, Gerhard A, Hinz R, Tai YF, Brooks DJ: Reference and target region modeling of [11C]-(R)-PK11195 brain studies. J Nucl Med. 2007, 48: 158-167.
Reproducibility of the 11C-PBR28 PET Signal. [http://www.clinicaltrials.gov/ct2/show/NCT02086240]
Study of [11C]PBR28 TSPO PET as a disease marker in MS patients. [http://public.ukcrn.org.uk/search/StudyDetail.aspx?StudyID=16651]
Tomasi G, Edison P, Bertoldo A, Roncaroli F, Singh P, Gerhard A, Cobelli C, Brooks DJ, Turkheimer FE: Novel reference region model reveals increased microglial and reduced vascular binding of 11C-(R)-PK11195 in patients with Alzheimer's disease. J Nucl Med. 2008, 49: 1249-1256.
Lambert JC, Heath S, Even G, Campion D, Sleegers K, Hiltunen M, Combarros O, Zelenika D, Bullido MJ, Tavernier B, Letenneur L, Bettens K, Berr C, Pasquier F, Fiévet N, Barberger-Gateau P, Engelborghs S, De Deyn P, Mateo I, Franck A, Helisalmi S, Porcellini E, Hanon O, de Pancorbo MM, Lendon C, Dufouil C, Jaillard C, Leveillard T, Alvarez V, Bosco P, European Alzheimer's Disease Initiative Investigators, et al: Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009, 41: 1094-1099.
Kamboh MI, Demirci FY, Wang X, Minster RL, Carrasquillo MM, Pankratz VS, Younkin SG, Saykin AJ, Jun G, Baldwin C, Logue MW, Buros J, Farrer L, Pericak-Vance MA, Haines JL, Sweet RA, Ganguli M, Feingold E, Dekosky ST, Lopez OL, Barmada MM, Alzheimer's Disease Neuroimaging Initiative: Genome-wide association study of Alzheimer's disease. Transl Psychiatry. 2012, 2: e117.
Acknowledgments
This work was supported by the Canadian Institutes of Health Research (CIHR; MOP-11-51-31), Alzheimer's Association (NIRG-08-92090), Fonds de Recherche du Québec - Santé (FRSQ; Chercheur Boursier), the Allan Tiffin Trust (Infrastructure), the Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq, Brazil), Fundação de Amparo à Pesquisa do Rio Grande do Sul (Fapergs, Brazil), and INCT for Excitotoxicity and Neuroprotection/CNPq. The authors wish to thank Dr Paul Edison and Drs William Kreisl and Robert Innis for providing Figures 2 and 3, respectively.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
ERZ and AL were responsible for the conception and design of the review, and for drafting and revising the manuscript. ALB, JB, SG and PRN were responsible for revising the manuscript. All authors read and approved the final manuscript.
Eduardo Rigon Zimmer, Antoine Leuzy contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Zimmer, E.R., Leuzy, A., Benedet, A.L. et al. Tracking neuroinflammation in Alzheimer’s disease: the role of positron emission tomography imaging. J Neuroinflammation 11, 120 (2014). https://doi.org/10.1186/1742-2094-11-120
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1742-2094-11-120