
Abstract
Background
Various typing methods have been developed for Neisseria gonorrhoeae, but none provide the combination of discrimination, reproducibility, portability, and genetic inference that allows the analysis of all aspects of the epidemiology of this pathogen from a single data set. Multilocus sequence typing (MLST) has been used successfully to characterize the related organisms Neisseria meningitidis and Neisseria lactamica. Here, the same seven locus Neisseria scheme was used to characterize a diverse collection of N. gonorrhoeae isolates to investigate whether this method would allow differentiation among isolates, and to distinguish these three species.
Results
A total of 149 gonococcal isolates were typed and submitted to the Neisseria MLST database. Although relatively few (27) polymorphisms were detected among the seven MLST loci, a total of 66 unique allele combinations (sequence types, STs), were observed, a number comparable to that seen among isolate collections of the more diverse meningococcus. Patterns of genetic variation were consistent with high levels of recombination generating this diversity. There was no evidence for geographical structuring among the isolates examined, with isolates collected in Liverpool, UK, showing levels of diversity similar to a global collection of isolates. There was, however, evidence that populations of N. meningitidis, N. gonorrhoeae and N. lactamica were distinct, with little support for frequent genetic recombination among these species, with the sequences from the gdh locus alone grouping the species into distinct clusters.
Conclusion
The seven loci Neisseria MLST scheme was readily adapted to N. gonorrhoeae isolates, providing a highly discriminatory typing method. In addition, these data permitted phylogenetic and population genetic inferences to be made, including direct comparisons with N. meningitidis and N. lactamica. Examination of these data demonstrated that alleles were rarely shared among the three species. Analysis of variation at a single locus, gdh, provided a rapid means of identifying misclassified isolates and determining whether mixed cultures were present.
Similar content being viewed by others

Background
Gonorrhoea, caused by the bacterium Neisseria gonorrhoeae, remains one of the most common sexually transmitted diseases contributing a substantial burden of morbidity, mortality and infertility worldwide. The disease is treatable and curable, but no vaccine is available. Consequently the control of this important disease depends on the identification and treatment of infected individuals and their contacts in transmission networks. High-resolution and reproducible typing methods for clinical isolates of the gonococcus are therefore central to the control of gonococcal infection. Knowledge of the gonococcal strains circulating both locally and globally, and of temporal changes in the prevalence of these strains, would identify transmission patterns and may assist in prevention and control of this disease.
Many typing schemes have been developed for N. gonorrhoeae but no single typing scheme has been generally adopted, and the lack of a single, generally accepted typing method has impeded the sharing of epidemiological data between laboratories. Auxotyping and serotyping are often applied to gonococci and these techniques are frequently combined, but they do not always provide sufficient resolution to distinguish between epidemiologically related and unrelated isolates [1].
Molecular based typing schemes [2–6] provide better discrimination among isolates. One method, multilocus enzyme electrophoresis (MLEE), which indexes variation in housekeeping genes, has been utilized to characterize gonococci, and has shown that strains with an AHU- (arginine, hypoxanthine and uracil requiring) auxotype are uniform, despite frequent recombination among gonococci [7]. AHU- isolates have been linked to disseminated gonococcal infection (DGI) [8], which is related to the penicillin sensitive phenotype usually found among these isolates [9].
Methods that use nucleotide sequencing, however, [10–13], are more portable, have greater definition, and make data storage in globally accessible databases via the internet easier. One method, based on the nucleotide sequence fragments from two gonococcal antigen genes under diversifying immune selection: por and tbpB (N. gonorrhoeae multi antigen sequence typing, NG-MAST) [14, 15], provides a high level of discrimination. However the NG-MAST database only includes genotypes, consisting of two number allelic profiles and the nucleotide sequences, with no isolate data available.
One established method for the characterization of bacteria is multilocus sequence typing (MLST), a development of MLEE, and a highly discriminatory system for indexing the relatedness among isolates based on genetic variation present in genes under stabilising selection for conservation of metabolic function [16]. It is employed for the characterisation of many bacterial species, including the closely related pathogen Neisseria meningitidis and the commensal Neisseria lactamica [16–21].
An intriguing feature of gonococcal biology is the very close relationship of this bacterium to N. meningitidis and N. lactamica, which also have an obligate association with humans, but inhabit the mucosal surface of the nasopharynx rather than the urogenital tract. Application of the same MLST scheme to N. gonorrhoeae, is therefore advantageous as it can be used to analyse genetic relationships among gonococcal isolates, as well as among the neisseriae [22]. Another advantage of MLST is its ability to discriminate among species, facilitating species identification and the detection of mixed bacterial cultures. This paper describes a N. gonorrhoeae typing scheme that exploits the existing globally accessible Neisseria MLST database [23, 24], which provides publicly available isolate information as well as nucleotide sequence data.
Results
Diversity among alleles and sequence types
A total of 66 sequence types (STs) were identified among the 149 gonococcal isolates analysed. The number of alleles at each locus ranged from two at aroE to 10 at gdh (Table 1). Of the 66 unique STs, 35 STs were represented by single isolates, 29 STs were represented by two to six isolates, ST-1579 was represented by 10 isolates and ST-1595 was represented by 12 AHU- isolates. Another AHU- isolate was identified as ST-5688, which differed from the ST-1595 AHU- isolates by a single synonymous polymorphism in the gdh allele. There were eight STs among isolates collected from 10 cases of DGI. A larger study would be necessary to investigate any relationships between invasive isolates and ST.
The allele sequences for each ST, concatenated in frame, were used to indicate the polymorphic sites within each ST, demonstrating the diversity present (Figure 1). Data for these isolates were submitted to the Neisseria MLST database [23, 24] and were given ST designations and allele numbers in order of discovery, so that the first gonococcal ST identified in this study was designated ST-1579.

Polymorphic sites in concatenated gonococcal housekeeping gene sequences. The polymorphic sites are shown for each concatenated sequence of seven housekeeping gene fragments. The positions of the polymorphic sites, and the genes in which they occur are indicated along the top.
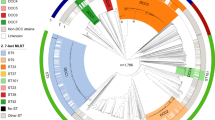
A neighbour-joining tree constructed from the concatenated allele sequences demonstrated the diversity of these isolates (Figure 2). Bacteria with the same STs were isolated in more than one location and some from more than one continent, while others demonstrated temporal persistence (Figure 2, Additional file 1).

The temporal and geographic distribution of 66 STs described for N. gonorrhoeae. A neighbour-joining tree was constructed from the concatenated MLST sequences from 66 STs obtained from 149 gonococcal isolates. Each circle denotes a particular ST and the countries and regions of isolation, if known are shown. Green circles indicate STs from isolates collected in Liverpool, UK between 1981 and 1991, pink circles indicate STs from isolates collected in Liverpool, UK between 2000 and 2001 and blue circles indicate STs from isolates collected elsewhere, including geographically and epidemiologically unlinked isolates. The two closely related STs obtained from the AHU- isolates are shown.
Comparisons of N. gonorrhoeae with N. meningitidis and N. lactamica
The allelic diversity within the 149 gonococcal isolates was compared with the diversity within 217 carried meningococci collected in the Czech Republic during 1993 [25] and a subset of 103 N. lactamica isolates collected as part of a longitudinal study of N. lactamica carriage in infants [20] (Table 1). The number of alleles and the percentage of polymorphic sites per allele were much greater for N. meningitidis and N. lactamica than for N. gonorrhoeae. The ratio of nonsynonymous to synonymous nucleotide substitutions (d N /d S ), calculated as an average over the entire MLST fragment for each locus was < 1 for each species, evidence that the loci used in the Neisseria MLST scheme were not subject to diversifying selection.
The number of gonococcal STs was compared to the number of STs among the Czech meningococcal carriage collection, the N. lactamica collection, and the collection of 107 meningococcal isolates used to develop the first MLST scheme and chosen to represent the diversity of the meningococcal population worldwide [16] (Table 2). The number of STs per 100 isolates among the gonococci (44) was comparable to the numbers of STs per 100 isolates among the carried meningococci (41) and the collection of 107 meningococci (47). The collection of 103 N. lactamica isolates had the highest number of STs per 100 isolates (67). When the collection of gonococcal isolates was divided into individual datasets, the dataset of 53 gonococcal isolates collected worldwide revealed 57 STs per 100 isolates, and the 38 gonococcal isolates collected in Liverpool between 2000 and 2001 comprised 55 STs per 100 isolates, demonstrating a greater number of STs per 100 isolates than in either meningococcal isolate collection.
In each of the collections analysed, many of the isolates had unique STs, with the percentage of unique STs among 53 gonococcal isolates collected worldwide (38%), comparable to that found among the collection of 107 meningococcal isolates (37%). The same percentage of unique alleles was found among 38 gonococcal isolates collected in Liverpool between 2000 and 2001, and the 217 Czech carried meningococci (29%).
Genetic divergence and gene flow (F ST ) were calculated between 58 gonococcal isolates collected in Liverpool (1981–1989), 38 gonococcal isolates collected in Liverpool between 2000 and 2001, 53 gonococcal isolates collected worldwide, 217 Czech carried meningococci and 103 N. lactamica isolates (Table 3). Fixed differences were present between species but none among the three groups of gonococci. More polymorphisms were shared among the gonococcal groups than among the species, and the percentage nucleotide sequence divergence was greatest between species. The N. lactamica nucleotide sequences were the least similar to the gonococcal nucleotide sequences (9.46% divergence). The F ST value between the two gonococcal groups was close to zero (0.01, 0.02), whereas between gonococci and N. lactamica it was 0.79, and between gonococci and N. meningitidis it was 0.61. The three gonococcal isolate collections were not significantly different (p > 0.05), with no geographic or temporal structuring evident.
To determine whether a clustering algorithm would delineate the three species, a neighbour-joining tree was constructed with the concatenated nucleotide sequences from the 149 N. gonorrhoeae, the 103 N. lactamica, and the 217 carried meningococci (Figure 3a). This showed three distinct clusters, corresponding to each of the three species, supported by bootstrap values of 100%. Bootstrap values within the clusters were very variable (not shown), suggesting relationships were not well resolved, a finding consistent with high levels of within species recombination. Similar clustering was shown using split decomposition analysis (Figure 3b) [26], although with this method, N. gonorrhoeae appeared to form a distinct cluster within the diversity of the meningococcus. No alleles were common to more than one species when 149 gonococci, 324 meningococci, and 103 N. lactamica isolates were analysed.

MLST data resolves N. gonorrhoeae , N. meningitidis and N. lactamica into three clusters. (a) A neighbour-joining tree was constructed from the concatenated MLST sequences for each species. (b) A splits graph was constructed from the concatenated MLST sequences for each species. Bootstrap values are indicated for the main branches (2000 replications).
The alleles that make up the allelic profile of each ST were examined individually using neighbour-joining trees (data only shown for gdh). The tree for the alleles at the gdh locus resolved the species into three well-supported groups, producing a tree congruent with that obtained from the concatenated nucleotide sequences (Figure 4). The trees drawn from alleles at the other six loci did not resolve the three species into groups that were congruent with the concatenated nucleotide sequences, although the majority of alleles from the same species formed clusters, with the gonococcal alleles forming single tight groups for all seven loci.

Alleles from a single locus ( gdh ) resolved species specific clusters. A neighbour-joining tree was constructed from the gdh allele sequences for each species. Bootstrap values are indicated for the main branches (2000 replications).
Discussion
It has been suggested that MLST of the pathogen N. gonorrhoeae would not provide sufficient discrimination between strains [27], due to the uniformity of its housekeeping genes [28]. The present study has shown that N. gonorrhoeae can be typed effectively using the same MLST scheme employed to characterize N. meningitidis [16] and N. lactamica [20], with a genotypic diversity comparable to that found among meningococcal isolates [25]. Despite high levels of horizontal genetic exchange among gonococci [2], MLST is robust because it is based on data from seven genetic loci distributed around the chromosome and indexes variation that is subject to stabilising selection. It appears to provide a level of discrimination comparable to the NG-MAST typing scheme [15], although this has not been formally validated as different datasets have been used. MLST, however, has the advantage that isolate information is available alongside genotypic data in an established, publicly accessible database [23, 24]. Unlike schemes that rely on antigen gene variation [5, 15, 29], which is subject to diversifying immune selection, MLST data can also be used to examine the evolutionary relationships among strains.
A total of 149 gonococcal isolates were typed by MLST in the present study. While only 27 polymorphisms were detected among the seven loci, a total of 66 unique allele combinations, or STs, were recorded. The low level of nucleotide diversity among gonococci inevitably results in a tighter clustering of these isolates in phylogenetic trees compared to meningococcal and N. lactamica isolates when concatenated sequences are analysed. However, the use of allelic profiles demonstrates a comparable level of discriminatory power to MLST of N. meningitidis and N. lactamica. The gonococcal STs were well differentiated with some showing temporal and geographic persistence. For instance, isolates with ST-1596 and ST-1583 were first isolated in Liverpool during the 1980s and have since been isolated in 2000, suggesting they may have a fitness advantage that has enabled them to persist in the population for over a decade. A total of 31 STs were represented by more than one isolate, with one group, ST-1579 represented by 10 isolates from three different countries, suggesting that isolates were distributed widely and not structured geographically. No temporal structuring was evident either, as the isolates collected in Liverpool between 1981–1989 were not significantly different from those collected in the same location between 2000 and 2001 (p > 0.05). While there was no evidence of geographical or temporal structuring in the gonococcal populations, UK isolates predominated in this study and some were from undefined locations, which may have influenced the outcome of the analysis. Structuring may be evident if more geographically and temporally diverse isolate collections were examined by MLST.
MLST provides a useful tool to study both the local and global distribution of isolates such as those with the AHU- phenotype, making it possible to track particular variants and examine transmission patterns. Of the 13 AHU- gonococci in the present study, 12 had identical genotypes with one differing by a single synonymous mutation at one locus. This illustrates the close relationship of this group and the ability of MLST to differentiate isolates with this auxotype. Further validation of the method would be required before MLST was used to resolve questions related to an outbreak situation and it may be necessary to complement it with antigen gene sequencing, as used in meningococcal epidemiology [30].
As the gonococcal MLST scheme uses nucleotide sequence data from exactly the same gene fragments as the meningococcal scheme, it can be used to compare MLST data from different Neisseria species, allowing phylogenetic and population genetic inferences to be made. The gonococcal MLST data were compared to data from studies of N. meningitidis and N. lactamica isolate collections previously published by the authors [16, 20, 25]. The use of these data, as opposed to the entire Neisseria MLST database was preferred as they had been extensively characterized and their provenance could be confirmed.
Like the N. lactamica and N. meningitidis alleles, the d N /d S ratios of the gonococcal alleles suggest that these loci evolve slowly and are not affected by diversifying selection, making them suitable for analysing evolutionary relationships among these species. However, this ratio could be affected by the small number of polymorphisms present within the collection. The MLST data for the three individual species were examined using: (1) the allelic profiles, (2) the individual alleles at each locus, and (3) the concatenated sequences for each allelic profile. When the MLST profiles were compared, the STs were unique to each species. The allele sequences were also species specific and no alleles were common among the neisseriae when 149 gonococcal, 324 meningococcal, and 103 N. lactamica isolates were examined. When additional N. lactamica isolates from the carriage study [20] and from German and Czech collections (unpublished data, not shown) were included in the analysis, only two alleles were common to more than one species. These alleles, at the pgm locus, were present among both N. gonorrhoeae and N. lactamica isolates. Alleles at this locus are among the least variable in gonococci. Thus, it seems more probable that these common alleles are a result of shared ancestry rather than interspecies recombination.
One of the advantages of a common MLST scheme for the neisseriae is that it can be used to distinguish between the Neisseria species and to identify unknown or misclassified isolates. Both neighbour-joining and split decomposition methods, using the concatenated MLST data, clustered the isolates into three distinct groups. The clustering of the STs into groups suggests that minimal recombination occurs among the housekeeping genes of these three Neisseria. This is confirmed by the F ST analysis, which suggests low levels of recombination among the species, the high number of fixed differences, the low number of shared polymorphisms, and the lack of alleles shared among species. Interestingly, the split decomposition analysis clustered the gonococcal sequences within the diversity of the meningococcus, reflecting the close ancestral relationship between these bacteria [31].
Although genetic recombination has been reported among N. gonorrhoeae, N. lactamica and N. meningitidis [32, 33], the physical and temporal separation of these species within the human host is likely to contribute to a low frequency of interspecies recombination. N. gonorrhoeae, which colonises the urogenital tract, is rarely found in children as it is sexually transmitted and is only occasionally found in adult throats; N. meningitidis is carried in the throats of approximately 10% of the adult population [34] but is rarely carried by young children and found infrequently in the urogenital tract; N. lactamica is carried by only about 2% of adults [35] but is highly prevalent in young children with carriage rates of around 40 % [36, 37]. While this limits opportunity for interspecies recombination, it does not affect intraspecies recombination, which may occur frequently creating an increasing number of STs from the available pool of alleles for each species, as has been observed in meningococci [25].
The results of the present study are inconsistent with a previous report that the Neisseria housekeeping alleles used in MLST were widely distributed among the neisseriae due to frequent interspecies recombination [38]. This was an in silico study that compared sets of 500 meningococcal STs downloaded from the Neisseria MLST database with all STs assigned to other named Neisseria species in the same database. The data that were analysed were not verified experimentally and access to the original samples was not requested. The present study did not include any of the apparently hybrid STs as they did not form part of the coherent populations analysed. We investigated these apparent hybrid STs present in the database for which samples were available. In all cases these were STs generated from historical freeze-dried cultures from which it was impossible to grow live organisms. Further analysis of the DNA samples suggested these were from mixed cultures and that the hybrid STs were a consequence of differential amplification of some loci. In conclusion, the present study finds little experimental support for extensive interspecies recombination among housekeeping genes in the Neisseria.
The lack of congruence among all of the phylogenetic trees may be a consequence of either shared ancestry or infrequent genetic exchange among the species. The relatively short lengths of the individual sequences used would also reduce any phylogenetic signal and therefore concatenated sequences were used to improve resolution. Only the tree for the alleles at the gdh locus produced a tree congruent with that obtained from the concatenated nucleotide sequences. The gdh locus may have evolved more rapidly than the other loci as these species diverged away from the ancestral population, creating gdh alleles that appear highly distinct for each species as shown in the gdh gene tree.
The species specificity of the gdh alleles and the congruence of the gdh gene tree with that produced from the concatenated sequences suggest that analysing sequences at this locus alone may be useful in differentiating among these three species and might help identify misclassified isolates. Occasionally Neisseria are misidentified [39, 40], therefore a typing tool that can be exploited to differentiate species using either MLST profiles, allele sequences at particular loci or the concatenated gene sequences, could prove extremely helpful alongside traditional microbiological methods. This is especially important if commensal species are misidentified as N. gonorrhoeae, which could lead to serious social, legal and medical consequences [41].
Although a number of other commensal neisseriae have been typed (unpublished results from the MLST database [23, 24]), these were not included in this study as too few isolates of these species have been typed for robust, meaningful analyses. MLST of representative collections of these other commensals, in particular Neisseria polysaccharea and Neisseria cinerea, which are closely related to the pathogenic Neisseria [42] would be advantageous, as knowledge of the genotypes of these species could be applied to species definitions and could facilitate identification of misclassified isolates.
Conclusion
This analysis has shown that MLST can be used effectively to characterise N. gonorrhoeae collections, obtained both locally and globally, and has demonstrated a level of discrimination that appears comparable to that determined for the meningococcus using MLST [16, 25] and the gonococcus using the NG-MAST scheme [15]. As an identical scheme has been used to characterize both N. meningitidis and N. lactamica, these data can be exploited to help define the three species, using either STs, individual alleles, in particular those at the gdh locus, or by concatenating the MLST data.
Methods
Bacterial isolates
A total of 149 gonococcal DNA samples were analysed, including 58 from Liverpool, collected between 1981 and 1991 and 38 from Liverpool collected between 2000 and 2001, one of which was known to be AHU-. A collection of 33 samples were obtained from isolates provided by the Genitourinary Infections Reference Laboratory, Gonococcus Reference Unit, Public Health Laboratory, Bristol, UK, and consisted of isolates that were geographically and epidemiologically unlinked. These included 12 isolates from cases of uncomplicated gonorrhoea from the UK (Colchester, London-Central Middlesex, South Wales, Epsom, Bristol, East London, Tyneside, Greenwich, Nottingham, Haverfordwest, and Birmingham), 11 isolates from cases of uncomplicated gonorrhoea from elsewhere in the world (Thailand, USA, South Africa, Pakistan, Uzbekistan, Hong Kong, Ireland, Bangladesh, Russia, Taiwan, and Spain), and 10 isolates from different and unrelated cases of DGI (five blood cultures and five joint fluid isolates). The remaining DNA samples were obtained from five isolates collected in Africa (two from Malawi and three from Nigeria), a collection of 12 AHU- isolates, and three reference isolates (FA19, FA1090, F62). Information regarding these isolates is available from the Neisseria MLST database [23, 24].
DNA preparation
DNA was extracted from 100 μl of boiled cell suspensions obtained from gonococci collected in Liverpool with the Isoquick nucleic acid extraction kit (ISC Bioexpress, Kaysville, UT, USA), used in accordance with the manufacturer's instructions. Samples collected elsewhere were provided as pure chromosomal DNA.
MLST
PCR amplifications and sequencing of the seven Neisseria MLST housekeeping gene fragments: abcZ, adk, aroE, fumC, gdh, pdhC, and pgm were undertaken with the oligonucleotide primers detailed in Table 4 using the protocol described previously [43]. All nucleotide sequences were determined directly from the PCR products. Briefly, sequence templates were generated using the PCR, and purified by precipitation with polyethylene glycol and sodium chloride [44]. The termination products were generated by cycle sequencing with the appropriate primers and BigDye terminators (Applied Biosystems). The products were then separated with an ABI prism 3700 automated DNA analyser. The sequence of each strand was determined at least once, and the resultant DNA sequences were assembled using the STADEN suite of computer programs [45]. Allele numbers and sequence types (STs) were assigned by querying the Neisseria MLST database [23, 24].
Data analysis
The computer program START, version 1.05 [46] was utilised to examine the number of polymorphic sites and the ratios of nonsynonymous to synonymous nucleotide substitutions (d N /d S ) among the alleles. Nucleotide sequences from the seven loci were concatenated in-frame to produce single sequences of length 3282 bp for each ST, using the concatenation tool found at the PubMLST website [23, 24]. DnaSP, version 4 [47], was used to calculate shared polymorphisms and fixed differences [48] between the isolate collections, and F ST values [49] were calculated using Arlequin, version 2, [50]. The F ST statistic measures the extent of genetic differentiation and computes an average level of gene flow, so that an F ST value of zero would indicate free genetic recombination, whereas an F ST value of one would indicate that recombination is unlikely). Neighbour-joining trees were drawn from the concatenated MLST alleles and the individual allele sequences using MEGA, version 2.1 [51], which was also used to measure nucleotide sequence divergence. All three coding positions were examined and the Kimura 2-parameter distance correction [52] was applied. The concatenated sequence data were also visualised using split decomposition analysis, using hamming distances with SplitsTree, version 3.1 [53]. The reliability of the inferred phylogenies was evaluated using bootstrap tests (2000 replications).
References
Sarafian SK, Knapp JS: Molecular epidemiology of gonorrhea. Clin Microbiol Revs. 1989, 2 (Suppl): S49-55.
O'Rourke M, Stevens E: Genetic structure of Neisseria gonorrhoeae populations: a non-clonal pathogen. J Gen Microbiol. 1993, 139: 2603-2611.
Cooke SJ, de la Paz H, La Poh C, Ison CA, Heckels JE: Variation within serovars of Neisseria gonorrhoeae detected by structural analysis of outer-membrane protein PIB and by pulsed-field gel electrophoresis. Microbiology. 1997, 143: 1415-1422.
Ng LK, Dillon JR: Typing by serovar, antibiogram, plasmid content, riboprobing, and isoenzyme typing to determine whether Neisseria gonorrhoeae isolates requiring proline, citrulline, and uracil for growth are clonal. J Clin Microbiol. 1993, 31: 1555-1561.
O'Rourke M, Ison CA, Renton AM, Spratt BG: Opa-typing: a high resolution tool for studying the epidemiology of gonorrhoea. Mol Microbiol. 1995, 17: 865-875.
Thompson DK, Deal CD, Ison CA, Zenilman JM, Bash MC: A typing system for Neisseria gonorrhoeae based on biotinylated oligonucleotide probes to PIB gene variable regions. J Infect Dis. 2000, 181: 1652-1660.
Gutjahr TS, O'Rourke M, Ison CA, Spratt BG: Arginine-, hypoxanthine-, uracil-requiring isolates of Neisseria gonorrhoeae are a clonal lineage within a non-clonal population. Microbiology. 1997, 143: 633-640.
Knapp JS, Holmes KK: Disseminated gonococcal infections caused by Neisseria gonorrhoeae with unique nutritional requirements. J Infect Dis. 1975, 132: 204-208.
Eisenstein BI, Lee TJ, Sparling PF: Penicillin sensitivity and serum resistance are independent attributes of strains of Neisseria gonorrhoeae causing disseminated gonococcal infection. Infect Immun. 1977, 15: 834-841.
Hobbs MM, Alcorn TM, Davis RH, Fischer W, Thomas JC, Martin I, Ison C, Sparling PF, Cohen MS: Molecular typing of Neisseria gonorrhoeae causing repeated infections: evolution of porin during passage within a community. J Infect Dis. 1999, 179: 371-381.
Posada D, Crandall KA, Nguyen M, Demma JC, Viscidi RP: Population genetics of the porB gene of Neisseria gonorrhoeae : different dynamics in different homology groups. Mol Biol Evol. 2000, 17: 423-436.
Unemo M, Olcen P, Albert J, Fredlund H: Comparison of serologic and genetic porB-based typing of Neisseria gonorrhoeae : consequences for future characterization. J Clin Microbiol. 2003, 41: 4141-4147.
Viscidi RP, Demma JC: Genetic diversity of Neisseria gonorrhoeae housekeeping genes. J Clin Microbiol. 2003, 41: 197-204.
Neisseria gonorrhoeae Multi Antigen Sequence Typing Database. [http://www.ng-mast.net/]
Martin IM, Ison CA, Aanensen DM, Fenton KA, Spratt BG: Rapid sequence-based identification of gonococcal transmission clusters in a large metropolitan area. J Infect Dis. 2004, 189: 1497-1505.
Maiden MCJ, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, Zhang Q, Zhou J, Zurth K, Caugant DA, et al: Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci USA. 1998, 95: 3140-3145.
Dingle KE, Colles FM, Wareing DRA, Ure R, Fox AJ, Bolton FJ, Bootsma HJ, Willems RJL, Urwin R, Maiden MCJ: Multilocus sequence typing system for Campylobacter jejuni. J Clin Microbiol. 2001, 39: 14-23.
Jones N, Bohnsack JF, Takahashi S, Oliver KA, Chan MS, Kunst F, Glaser P, Rusniok C, Crook DW, Harding RM, et al: Multilocus sequence typing system for group B Streptococcus. J Clin Microbiol. 2003, 41: 2530-2536.
Priest FG, Barker M, Baillie LW, Holmes EC, Maiden MC: Population structure and evolution of the Bacillus cereus group. J Bacteriol. 2004, 186: 7959-7970.
Bennett JS, Griffiths DT, McCarthy ND, Sleeman KL, Jolley KA, Crook DW, Maiden MC: Genetic diversity and carriage dynamics of Neisseria lactamica in infants. Infect Immun. 2005, 73: 2424-2432.
Pullinger GD, Lopez-Benavides M, Coffey TJ, Williamson JH, Cursons RT, Summers E, Lacy-Hulbert J, Maiden MC, Leigh JA: Application of Streptococcus uberis multilocus sequence typing: analysis of the population structure detected among environmental and bovine isolates from New Zealand and the United Kingdom. Appl Environ Microbiol. 2006, 72: 1429-1436.
Bennett JS, Jolley KA, Maiden MCJ: Is the gonococcus a clonal complex of the meningococcus?. Proceedings of the 13th International Pathogenic Neisseria Conference. 2002, Oslo, Norway: 42. Nordberg Aksidenstrykkeri AS
Neisseria MLST website. [http://pubmlst.org/neisseria/]
Jolley KA, Chan MS, Maiden MC: mlstdbNet – distributed multi-locus sequence typing (MLST) databases. BMC Bioinformatics. 2004, 5: 86-
Jolley KA, Kalmusova J, Feil EJ, Gupta S, Musilek M, Kriz P, Maiden MC: Carried meningococci in the Czech Republic: a diverse recombining population. J Clin Microbiol. 2000, 38: 4492-4498.
Bandelt HJ, Dress AW: Split decomposition: a new and useful approach to phylogenetic analysis of distance data. Mol Phylogenet Evol. 1992, 1: 242-252.
Palmer HM, Arnold C: Genotyping Neisseria gonorrhoeae using fluorescent amplified fragment length polymorphism analysis. J Clin Microbiol. 2001, 39: 2325-9.
Spratt BG, Maiden MCJ: Bacterial population genetics, evolution and epidemiology. Proc R Soc Lond B Biol Sci. 1999, 354: 701-710.
Bash MC, Zhu P, Gulati S, McKnew D, Rice PA, Lynn F: por Variable-region typing by DNA probe hybridization is broadly applicable to epidemiologic studies of Neisseria gonorrhoeae. J Clin Microbiol. 2005, 43: 1522-1530.
Feavers IM, Gray SJ, Urwin R, Russell JE, Bygraves JA, Kaczmarski EB, Maiden MCJ: Multilocus sequence typing and antigen gene sequencing in the investigation of a meningococcal disease outbreak. J Clin Microbiol. 1999, 37 (12): 3883-3887.
Feavers IM, Maiden MCJ: A gonococcal porA pseudogene: implications for understanding the evolution and pathogenicity of Neisseria gonorrhoeae. Mol Microbiol. 1998, 30: 647-656.
Saez-Nieto JA, Lujan R, Martinez-Suarez JV, Berron S, Vazquez JA, Vinas M, Campos J: Neisseria lactamica and Neisseria polysaccharea as possible sources of meningococcal beta-lactam resistance by genetic transformation. Antimicrob Agents Chemother. 1990, 34: 2269-2272.
Vázquez JA, Berron S, O'Rourke M, Carpenter G, Feil E, Smith NH, Spratt BG: Interspecies recombination in nature: a meningococcus that has acquired a gonococcal PIB porin. Mol Microbiol. 1995, 15: 1001-1007.
Caugant DA, Høiby EA, Magnus P, Scheel O, Hoel T, Bjune G, Wedege E, Eng J, Frøholm LO: Asymptomatic carriage of Neisseria meningitidis in a randomly sampled population. J Clin Microbiol. 1994, 32: 323-330.
Cartwright KAV, Stuart JM, Jones DM, Noah ND: The Stonehouse survey: nasopharyngeal carriage of meningococci and Neisseria lactamica. Epidemiol Infect. 1987, 99: 591-601.
Olsen SF, Djurhuus B, Rasmussen K, Joensen HD, Larsen SO, Zoffman H, Lind I: Pharyngeal carriage of Neisseria meningitidis and Neisseria lactamica in households with infants within areas with high and low incidences of meningococcal disease. Epidemiol Infect. 1991, 106: 445-457.
Blakebrough IS, Greenwood BM, Whittle HC, Bradley AK, Gilles HM: The epidemiology of infections due to Neisseria meningitidis and Neisseria lactamica in a northern Nigerian community. J Infect Dis. 1982, 146: 626-637.
Hanage WP, Fraser C, Spratt BG: Fuzzy species among recombinogenic bacteria. BMC Biology. 2005, 3: 6-
Cann KJ, Rogers TR: The phenotypic relationship of Neisseria polysaccharea to commensal and pathogenic Neisseria spp. J Med Microbiol. 1989, 29: 251-254.
Knapp JS, Hook EW: Prevalence and persistence of Neisseria cinerea and other Neisseria spp. in adults. J Clin Microbiol. 1988, 26: 896-900.
Dossett JH, Appelbaum PC, Knapp JS, Totten PA: Proctitis associated with Neisseria cinerea misidentified as Neisseria gonorrhoeae in a child. J Clin Microbiol. 1985, 21: 575-577.
Guibourdenche M, Popoff MY, Riou JY: Deoxyribonucleic acid relatedness among Neisseria gonorrhoeae, N. meningitidis,N. lactamica, N. cinerea and "Neisseria polysaccharea". Ann Inst Pasteur Microbiol. 1986, 137B: 177-185.
Jolley KA: Multi-locus sequence typing. Meningococcal Disease: Methods and Protocols. Edited by: Pollard AJ, Maiden MC. 2001, Totowa, New Jersey: Humana Press, 173-186.
Embley TM: The linear PCR reaction: a simple and robust method for sequencing amplified rRNA genes. Lett Appl Microbiol. 1991, 13: 171-174.
Staden R: The Staden sequence analysis package. Mol Biotechnol. 1996, 5: 233-241.
Jolley KA, Feil EJ, Chan MS, Maiden MC: Sequence type analysis and recombinational tests (START). Bioinformatics. 2001, 17: 1230-1231.
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R: DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003, 19: 2496-2497.
Hey J: The structure of genealogies and the distribution of fixed differences between DNA sequence samples from natural populations. Genetics. 1991, 128: 831-840.
Hudson RR, Slatkin M, Maddison WP: Estimation of levels of gene flow from DNA sequence data. Genetics. 1992, 132: 583-589.
Schneider S, Roessli D, Excoffier L: Arlequin Version 2.000: A Software for Population Genetic Data Analysis. 2000, Geneva: University of Geneva
Kumar S, Tamura K, Jakobsen IB, Nei M: MEGA2: molecular evolutionary genetics analysis software. Bioinformatics. 2001, 17: 1244-1245.
Kimura M: A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980, 16: 111-120.
Huson DH: SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics. 1998, 14: 68-73.
Acknowledgements
We thank Mary O'Leary (University of Pittsburgh) for assistance with data analysis. This publication made use of the Neisseria Multilocus Sequence Typing website developed by Keith Jolley and Man-Suen Chan and sited at the University of Oxford [24]. The development of the site has been funded by the Wellcome Trust and European Union. JSB, KAJ and MCJM were funded by the Wellcome Trust.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
JSB designed the study, undertook MLST of the N. gonorrhoeae isolates, analysed and interpreted the results, and drafted the manuscript. KAJ reviewed the manuscript and maintains the Neisseria MLST website and database. MCJM conceived of the study and contributed to the drafting of the manuscript. IMF contributed to the drafting of the manuscript. CAH, NJS and PFS provided DNA samples from gonococcal isolate collections and provided information relating to these isolates, where available.
Electronic supplementary material
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Bennett, J.S., Jolley, K.A., Sparling, P.F. et al. Species status of Neisseria gonorrhoeae: evolutionary and epidemiological inferences from multilocus sequence typing . BMC Biol 5, 35 (2007). https://doi.org/10.1186/1741-7007-5-35
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1741-7007-5-35