
Abstract
Background
Snakebites are considered a neglected tropical disease that affects thousands of people worldwide. Although antivenom is the only treatment available, it is associated with several side effects. As an alternative, plants have been extensively studied in order to obtain an alternative treatment. In folk medicine, Azima tetracantha Lam. is usually used to treat snakebites. The present study aims to provide a scientific explanation for the use of this plant against snakebite. The extracts of shade dried leaves of A. tetracantha were tested for in vitro inhibitory activity on toxic venom enzymes like phosphomonoesterase, phosphodiesterase, acetylcholinesterase, hyaluronidase etc. from Bungarus caeruleus and Vipera russelli venoms.
Results
The ethylacetate extract rendered a significant inhibitory effect on the phosphomonoesterase, phosphodiesterase, phospholipase A2 and acetylcholinesterase enzymes.
Conclusions
The present study suggests that ethylacetate extract of A. tetracantha leaves possesses compounds that inhibit the activity of toxic enzymes from Bungarus caeruleus and Vipera russelli venom. Further pharmacological and in vivo studies would provide evidence that this substance may lead to a potential treatment against these venoms.
Similar content being viewed by others


Background
Snakebite remains a major health concern throughout the world especially in India, due to the high mortality rate in the country. Worldwide, snake envenomation incidence exceeds 5 million affected people per year [1]. In Asia, an estimated four million snakebites occur every year, of which approximately 50% of the victims are envenomed, resulting in 100,000 annual deaths [2]. In India alone about 35,000 to 50,000 deaths occur annually. The major families of snakes in India are Elapidae, Viperidae and Hydrophidae. The most common poisonous snakes in the country are cobra (Naja naja), krait (Bungarus caeruleus), Russell’s viper (Daboia russelli) and saw-scaled viper (Echis carinatus). The former two belong to Elapidae and the latter two belong to Viperidae [3]. The venoms of cobra and krait are neurotoxic, that is, they affect the victim’s central nervous system and cause heart failure. Their venom possesses several proteins, including cardiotoxins, neurotoxins and phospholipase A2, that are responsible for their toxicity. The venoms of Russell’s viper and saw-scaled viper are histotoxic and hemorrhagic, therefore they provoke hemorrhagic manifestations that include epistaxis and cardiac manifestations such as myocarditis and cardiac failure [4].
Presently, antivenom immunotherapy is the only treatment available against snake envenomation. The side effects of antivenom include anaphylactic shock, pyrogen reaction and serum sickness. These symptoms are possibly outcomes of the action of non-immunoglobulin proteins present in higher concentrations in antivenom [5]. In this perspective, several attempts have been made to develop snake venom antagonists from plants. In folk medicine, plant drug recipes are passed on to generations by oral tradition and are used as antidotes [6]. Considering the limitations of antivenom, investigations on plants are based on the fact that some of their extracts are rich in bioactive compounds with promising antivenom effects [7, 8].
Hemidesmus indicus root extract and methanolic leaf extract of Azadirachta indica have been proved to neutralize phospholipase A2 activity induced by Russell’s viper venom [9, 10]. Mimosa pudica has shown antihyaluronidase activity against Naja naja, Vipera russelli and Echis carinatus venoms [11]. Echis carinatus enzymatic effects have been inhibited by the extracts of Andrographis paniculata and Aristolochia indica. Enzymatic and pharmacological activities of phospholipase A2 induced by Vipera russelli venom have been inhibited by aristolochic acid from Aristolochia radix[12, 13].
In vitro tests with polyphenols from Areca catechu L. and Quercus infectoria Oliv. showed inhibition of phospholipase A2, proteases, hyaluronidase and L-amino acid oxidase of Naja naja kaouthia and Calloselasma rhodostoma venoms [14]. In addition, Tamarindus indica has shown potent venom neutralizing properties [15].
Azima tetracantha Lam. belongs to the Salvodoraceae family, it is known as Kundali in Ayurvedic medicine and also called uppimullu in kannada [16, 17]. Antidiarrheal activity has been reported in this plant and antimicrobial activity in its fruits [18, 19]. Moreover, there are reports that the juice of its leaves is efficient against toothache and earache [20]. In Indian tribes, leaf paste of A. tetracantha is used to treat snakebites [17]. The presence of dimeric piperdine alkaloids azimine, azacarpaine, carpaine, triterpenoids, isorhamnetin 3-rutinoside, neoascorbinogen and glucosinolates and novel fatty acids have been reported as some of the phytochemicals present in the plant [21–25]. The present study investigates the neutralizing activity of A. tetracantha plant extracts on krait and Russell’s viper venom toxic enzymes by in vitro methods.
Methods
Venom
The lyophilized venoms of Bungarus caeruleus and Vipera russelli were obtained from Irula Snake Catcher’s Cooperative Society, Kancheepuram, Chennai. Snake venom (5 mg/mL) was dissolved in physiological saline and centrifuged at 2000 g for ten minutes. The supernatant was used for further analysis and stored at 4°C. The protein concentration was estimated according to the method of Lowry et al. [26].
5′ Adenosine mono phosphate (5′ AMP), disodium-p-nitrophenol phosphate, L-leucine, diansidine hydrochloride, horseradish peroxidase, 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB), acetylthiocholine iodide, hyaluronic acid, cetyltrimethylammonium bromide, lecithin were purchased from Himedia Laboratories (India) and casein from Sigma Aldrich Laboratories (USA). All the other reagents were of analytical grade.
Plants and extraction
Fresh leaves of A. tetracantha were collected in September in Kurubarahatty village, Chitradurga district, Karnataka, India. The plant specimens were identified and authenticated by the National Ayurveda Dietetics Research Institute, Bangalore, Karnataka (Drug Authentication/SMPU/NADRI/BNG/2013-2014/765). The leaves were thoroughly washed in order to remove adhering dust, shade dried and ground into powder for further use.
Powdered material was prepared in soxhlet apparatus using petroleum ether (60-80°C), hexane, chloroform, ethyl acetate, methanol and water. The extraction procedure was carried out until the solvent becomes colorless in the soxhlet loop. The extracts were dried using rotary vaccum evaporator and the residue was expressed in terms of dry weight, which was used for further analysis.
About 50 g of leaf powder was soaked in 150 mL of ethanol overnight and filtered. The residue left after filtration was suspended in the same amount of ethanol and left for 48 hours. The two filtrates were mixed, dried using rotatory vacuum evaporator and the residue was stored for further use.
Qualitative analysis of extracts
Phytochemical analysis of the extracts was performed to detect the presence of constituents such as alkaloids, terpenoids, flavonoids, phenols, saponins, carbohydrates and other metabolites [27, 28].
Thin layer chromatography of extracts
The extracts were examined by thin layer chromatography (TLC) on analytical plates over silica gel (TLC grade, Merck, India). The different solvent systems used for separation were petroleum ether and ethylacetate, hexane and ethylacetate in three different ratios (9:1, 8:2, 7:3); methanol, water, formic acid in the ratio 18:9:1 and chloroform alone. In each case the spots were visualized under ultraviolet light (UV), exposed to iodine vapors and sprayed with vanillin sulfuric acid and ferric chloride solution.
Enzyme inhibition studies (In vitro enzyme inhibition assays)
Protease
Protease assay of crude venom was performed according to the method of Greenberg [29]. The reaction mixture composed of 0.5% casein, 1.0 mL of Tris–HCl buffer (pH 8.0), 0.5 mL of 0.25% crude venom and the reaction mixture incubated for four hours at 37°C. At the end of four hours, the reaction was stopped by adding trichloroacetic acid (TCA) and filtered. The filtrate (1.0 mL) was used for protein estimation by the method of Lowry et al.[26] using L-tyrosine as a standard. In the above investigation, one unit of enzyme activity was defined as the amount that yielded 0.02 μmole of tyrosine/hour under experimental conditions described. For the inhibition studies, venom was preincubated with the extracts for 30 minutes at 37°C.
5′ nucleotidase
5′ Nucleotidase was assayed by the method of Rowe et al.[30]. The substrate solution contained 1 mL of Tris–HCl buffer (pH 8.0), 0.1 mL of 0.1 M magnesium chloride and 0.8 mL of 0.15% 5′AMP followed by 0.25 mL of 0.1% crude venom and incubated at 37°C for 15 minutes. At the end of incubation time, the reaction was quenched by adding TCA and filtered. The filtrate was assayed for inorganic phosphate by the method of Fiske and Subbarow [31] at 625 nm using potassium dihydrogen phosphate as standard. In this analysis, one unit of enzyme activity was defined as the amount that yielded 0.01 μmole of inorganic phosphate/minute under the experimental conditions. For the inhibition studies, venom was preincubated with the extracts for 30 minutes at 37°C.
Phosphomonoesterase
The phosphomonoesterase activity was determined by the method of Bessey et al.[32] with slight modifications. The reaction mixture included 1.0 mL of Tris–HCl buffer (pH 8.0), 1.0 mL of disodium-p- nitrophenol phosphate, 0.5 mL of 0.25% crude venom and was incubated at 37°C for three hours. The absorbance was measured at 425 nm. p-Nitrophenol was used as the standard. One unit of enzyme activity was defined as the amount that yielded 0.1 μmole of p-nitrophenol/hour under the experimental conditions. For inhibition studies, venom was preincubated with the extracts for 30 minutes at 37°C.
Phosphodiesterase
Phosphodiesterase activity was determined by a method modified from Lo et al. [33]. The assay mixture contained 0.1 mL of venom solution, 0.5 mL of 0.0025 M Na-p-nitrophenyl phosphate, 0.3 mL of 0.01 M MgSO4 and 0.5 mL of 0.17 M Tris–HCl (pH 8.0). The absorbance was measured at 400 nm. Phosphodiesterase activity was expressed in nanomoles of product released/minute. Molar extinction coefficient at 400 nm was 8100 Cm−1 M−1[34]. For the inhibition studies, the venom was preincubated with the extracts for 30 minutes at 37°C.
L-amino acid oxidase
The L-amino acid oxidase activity was carried out according to Li et al. [35]. Reaction mixture consisted of 1.0 mL of L-leucine, 2.0 mL of Tris–HCl buffer (pH 8.0), 0.25 mL of 0.1% dianisidine hydrochloride, 0.15 mL of 0.1% horseradish peroxidase and 0.04 mL of 0.5% crude venom solution. It was allowed to stand for ten minutes at room temperature and then the absorbance was measured at 415 nm. One unit (U) was defined as the amount of enzyme that catalyzed the formation of 1 μmol H2O2 per minute. For the inhibition studies, venom was preincubated with extracts for 30 minutes at 37°C.
Acetylcholinesterase
Acetylcholine esterase activity was assayed following Ellman et al. method [36]. The reaction mixture comprised 3.0 mL of the phosphate buffer (pH 8.0), 10 μL of DTNB (10 mmole/L) and 20 μL of acetylethiocholine iodide (158.5 mmol/L). A total of 50 μL of 0.1% crude venom and 3 mL of buffer solution was incubated at room temperature for five minutes. Then, 10 μL of DTNB (a strong oxidizing agent) and 20 μL of substrate acetylethiocholine iodide were added in order to reach a final concentration of 1 mmole/L. The increase in absorbance at 412 nm was measured on a double beam spectrophotometer against control mixture prepared at the same time. However, in the latter case, 50 μL of enzyme was replaced with 50 μL of buffer solution. For the inhibition studies, venom was preincubated with extracts for 30 minutes at 37°C.
Hyaluronidase
Hyaluronidase assay of crude venom was determined turbidometrically by the method of Pukrittayakamee et al. [37]. The assay mixture contained buffer Tris–HCl (pH 8.0), 50 μg of hyaluronic acid (0.5 mg/mL in buffer) and enzymes in a final volume of 1.0 mL. The mixture was incubated for 15 minutes at 37°C and the reaction was quenched by the addition of 2 mL of 2.5% (w/v) cetyltrimethylammonium bromide in 2% NaOH (w/v). The absorbance was read at 400 nm (within ten minutes) against a control solution containing 1 mL of the same buffer and 2 mL of 2.5% (w/v) cetyltrimethylammonium bromide in 2% NaOH (w/v). Turbidity reducing activity was expressed as a percentage of the remaining hyaluronic acid, taking the absorbance of a tube in which no enzyme was added as 100%. One unit was defined as the amount of enzyme that provoked 50% turbidity reduction. Specific activity was defined as turbidity reducing units per milligram of enzyme. For the inhibition studies, venom was preincubated with extracts for 30 minutes at 37°C.
Phospholipase A2
Phospholipase A2 assay was determined according to the acidimetric method of Tan and Tan [38] with slight modification. Briefly, a lecithin suspension was prepared with 1% lecithin, 18 mM calcium chloride, and 8.1 mM sodium deoxycholate in equal proportions. The pH of the suspension was adjusted to 8.0 with 1 M sodium hydroxide, and stirred for ten minutes to ensure homogenous mixing. An amount of 0.1 mL of venom solution/fraction was added to 15 mL of egg yolk suspension to initiate the hydrolysis. The initial decrease in pH was measured. A decrease of 1 pH unit corresponded to 133 μmoles of fatty acid release. Enzyme activity was expressed as μmoles of fatty acid released/minute [34]. For the inhibition studies, venom was preincubated with extracts for 30 minutes at 37°C.
Results and discussion
The extracts were dried using a vacuum evaporator and weight was expressed in terms of dry weight and tabulated in Table 1. The qualitative phytochemical analysis of plant extracts showed significant presence of the metabolites (Table 2). The analysis showed that alkaloids were present only in methanolic extracts which were devoid of carbohydrates, glycosides and resins. The study by Maruthi et al. [39] showed similar results except for the presence of carbohydrates. Another study by Muthuswamy et al.[40] reported the presence of alkaloids in ethylacetate extract. Changes in the presence of metabolites in various extracts could be due to the time and season in which leaves were collected.
The thin layer chromatography profiling of all seven extracts provides an insight of the number of phytochemicals present in the extracts. Each extract was separated in a different solvent system according to their polarity and in different ratios of solvent systems. The components in petroleum ether, chloroform and ethylacetate extracts were well separated in petroleum ether and ethylacetate solvent system in the ratio 9:1 and 8:2 respectively. The spots in methanolic extract were well separated in chloroform alone and aqueous extract in methanol, water and formic acid in the ratio 18:9:1. Spots were well defined in ethanolic and hexane extracts with the hexane and ethylacetate solvent system in the ratio 9:1. The visualization of spots was better under UV compared to rest of the agents used. The Figure 1 shows the results under UV light.

Thin layer chromatography of the extracts visualized under ultraviolet light. Lane 1: petroleum ether extract, lane 2: hexane extract, lane 3: chloroform extract, lane 4: ethyl acetate extract, lane 5: methanol extract, lane 6: water extract, and lane 7: ethanol extract.
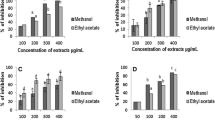
Enzymatic and inhibition studies revealed that the ethylacetate extract of the plant was able to inhibit phosphodiesterase (Figure 2), phosphomonoesterase (Figure 3), acetylcholinesterase (Figure 4), 5′ nucleotidase (Figure 5), phospholipase A2 (Figure 6) and hyaluronidase (Figure 7) enzymes present in the venom which renders it as an active extract. The extract is able to inhibit these toxic enzymes in both venoms at 100 μg/mL concentration. The protease and L-amino acid oxidase enzymes were not inhibited by any of the extracts in both venoms. Phospholipase A2 and hyaluronidase of Vipera russelli and Bungarus caeruleus was not inhibited by any of the extracts respectively. The chloroform extract also shows promising results but the acetylcholinesterase was well inhibited in ethyl acetate extract, as it is one of the most toxic enzymes present in venoms of these snakes.

In vitro inhibition studies of Azima tetracantha plant extracts against the phosphodiesterase enzyme of (A) Vipera russelli and (B) Bungarus caeruleus venom expressed as percent inhibition.

In vitro inhibition studies of Azima tetracantha plant extracts against the phosphomonoesterase enzyme of (A) Vipera russelli and (B) Bungarus caeruleus venom expressed as percent inhibition.

In vitro inhibition studies of Azima tetracantha plant extracts against the acetylcholinesterase enzyme of (A) Vipera russelli and (B) Bungarus caeruleus venom expressed as percent inhibition.

In vitro inhibition studies of Azima tetracantha plant extracts against the 5′ nucleotidase enzyme of (A) Vipera russelli and (B) Bungarus caeruleus venom expressed as percent inhibition.

In vitro inhibition studies of Azima tetracantha plant extracts against phospholipase A 2 enzyme of Bungarus caeruleus venom expressed as percent inhibition.

In vitro inhibition studies of Azima tetracantha plant extracts against the hyaluronidase enzyme of Vipera russelli venom expressed as percent inhibition.
A previous work by Dhananjaya et al.[41] has reported the inhibition of enzymes present in Russell’s viper venom by Mangifera indica stem bark extract at lower concentrations. The studies by Gopi et al.[42] have also reported the inhibition of toxic enzymes present in the Naja naja venom by Andrographis paniculata active methanolic extract. The present study was performed using a concentration of 100 μg/mL, but the enzyme inhibiting concentration would be as lower as 10 to 500 μL of the diluted extract. Therefore, the actual concentration required to inhibit the enzymes completely should be studied with different concentrations of the active extract.
Conclusion
In India, snakebite is a major health problem that leads to several deaths annually. Vipera russelli and Bungarus caeruleus are the most common snakes found throughout the country and a large number of deaths occur due to envenomation by these snakes. Snake antivenom remains the only specific treatment against envenomation by snakes. It is usually derived from horse sera, therefore it contains animal immunoglobulins (that frequently cause complement-mediated side effects), and other proteins (that may cause serum sickness and, occasionally, anaphylactic shock). Although the use of plants against snakebites has been long observed, more scientific attention has been given since to it in the recent 20 years [43]. Several Indian medicinal plants are employed by folk medicine for the treatment of snakebites [6]. In the current study, we examined the antivenom potential of A. tetracantha Lam. plant extracts. The inhibition potential of plant extracts in relation to snake toxic enzymes was assayed. The studies were carried out by incubating the venom with plant extracts prior to analysis. The results showed that plant extracts were capable of inhibiting such enzymes. The ethylacetate extract, particularly, showed promising inhibition activity against phosphomonoesterase, phosphodiesterase, acetylcholinesterase, phospholipase A2, 5′ nucleotidase and hyaluronidase enzymes, whereas the protease L-amino acid oxidase could not be inhibited. The inhibitory activities of plant extracts against snake venoms should be further confirmed by in vivo studies using animal models and by pharmacological analysis in vitro including neutralization of fibrinogenolytic activity, neutralization of procoagulant activity and neutralization of hemolytic activity. The authors believe that plant extracts could lead to the development of new drugs against the venom of Indian snakes.
Ethics committee approval
Venom used in the present study was purchased from a government licensed institute of Tamilnadu state, India, that follows proper procedures regarding animal handling. Doctoral committee of Center for Post Graduate studies and Research, Jain University, Banglore.
References
Chippaux JP: Snake-bites: appraisal of the global situation. Bull World Health Organ 1998, 76(5):515–524.
Sharma SK, Chappuis F, Jha N, Bovier PA, Loutan L, Koirala S: Impact of snake bites and determinants of fatal outcomes in southeastern Nepal. Am J Trop Med Hyg 2004, 71(2):234–238.
Bawaskar HS: Snake venoms and antivenoms: critical supply issues. J Assoc Physicians India 2001, 52: 11–13.
Meenatchisundaram S, Michael A: Snake bite and therapeutic measures: Indian scenario. Indian J Sci Technol 2009, 2(10):69–73.
Devi CM, Bai MV, Lal AV, Umashankar PR, Krishnan LK: An improved method for isolation of anti-viper venom antibodies from chicken egg yolk. J Biochem Biophys Methods 2002, 51(2):129–138. 10.1016/S0165-022X(02)00002-7
Alam MI, Gomes A: Snake venom neutralization by Indian medicinal plants ( Vitex negundo and Emblica officinalis ) root extracts. J Ethnopharmacol 2003, 86(1):75–80. 10.1016/S0378-8741(03)00049-7
Martz W: Plants with a reputation against snakebites. Toxicon 1992, 30(10):1131–1142. 10.1016/0041-0101(92)90429-9
Soares AM, Ticli TK, Marcussi S, Lourenço MV, Januário AH, Sampaio SV, Giglio JR, Lomonte B, Pereira PS: Medicinal plants with inhibitory properties against snake venoms. Curr Med Chem 2005, 12(22):2625–2641. 10.2174/092986705774370655
Chatterjee I, Chakravarty AK, Gomes A: Daboia russelli and Naja kaouthia venom neutralization by lupeol acetate isolated from the root extract of Indian sarsaparilla Hemidesmus indicus R.Br. J Ethnopharmacol 2006, 106(1):38–43. 10.1016/j.jep.2005.11.031
Mukherjee AK, Doley R, Saikia D: Isolation of a snake venom phospholipase A2 (PLA2) inhibitor (AIPLAI) from leaves of azadirachta indica (neem): mechanism of PLA2 inhibition by AIPLAI in vitro condition. Toxicon 2008, 51(8):1548–1553. 10.1016/j.toxicon.2008.03.021
Girish KS, Mohanakumari HP, Nagaraju S, Vishwanath BS, Kemparaju K: Hyaluronidase and protease activities from Indian snake venoms: neutralization by Mimosa pudica root extract. Fitoterapia 2004, 75(3–4):378–380.
Vishwanath BS, Kini RM, Gowda TV: Characterization of three edema-inducing phospholipase A2 enzymes from habu ( Trimeresurus flavoviridis ) venom and their interaction with the alkaloid aristolochic acid. Toxicon 1987, 25(5):501–515. 10.1016/0041-0101(87)90286-8
Vishwanath BS, Gowda TV: Interaction of aristolochic acid with Vipera russelli phospholipase A2: its effect on enzymatic and pathological activities. Toxicon 1987, 25(9):929–937. 10.1016/0041-0101(87)90155-3
Leanpolchareanchai J, Pithayanukul P, Bavovada R: Anti-necrosis potential of polyphenols against snake venoms. Immunopharmacol Immunotoxicol 2009, 31(4):556–562. 10.3109/08923970902821702
Ushanandini S, Nagaraju S, Harish Kumar K, Vedavathi M, Machiah DK, Kemparaju K, Vishwanath BS, Gowda TV, Girish KS: The anti-snake venom properties of Tamarindus indica (leguminosae) seed extract. Phytother Res 2006, 20(10):851–858. 10.1002/ptr.1951
Kirtikar KR, Basu BD: Indian Medicinal Plants. Volume 7. Deharadun: Oriental Enterprises; 2001:2130–2133.
Hiremath VT, Taranath TC: Traditional phytotherapy for snake bites by tribes of Chitradurga district, Karnataka, India. Ethnobot Leaflets 2010, 14: 120–125.
Al-Fatimi M, Wurster M, Schoder G, Lindequist U: Antioxidant, antimicrobial and cytotoxic activities of selected medicinal plants from Yemen. J Ethnopharmacol 2007, 111(3):657–666. 10.1016/j.jep.2007.01.018
Senthamarai R, Kavimani S, Jayakar B, Jayalakshmi A, Jayasakthi S, Sethuramani A: Diuretic activity of Azima tetracantha in rats. Indian Drugs 1996, 33(9):478–479.
Hema TA, Shiny M, Parvathy J: Antimicrobial activity of leaves of Azima tetracantha against clinical pathogens. Int J Pharm Pharm Sci 2012, 4(4):317–319.
Hebbar SS, Harsha VH, Shripathi V, Hegde GR: Ethnomedicine of Dharwad district in Karnataka, India-plants used in oral health care. J Ethnopharmacol 2004, 94(2–3):261–266.
Rall GJH, Smalberger TM, de Waal HL, Arndt RR: Dimeric piperidene alkaloids from Azima tetracantha Lam: Azimine, azcarpine and carpaine. Tetrahedron Lett 1967, 9(7):896.
Rao EV, Prasad Rao PRS: Occurrence of triterpenoids in Azima tetracantha . Curr Sci 1978, 47(22):857.
Williams UV, Nagarajan S: Isorhamnetin 3-O-rutinoside from leaves of Azima tetracantha Lam. Indian J Chem, B-Org Chem Incl Med Chem 1988, 27: 397.
Daulatabad CD, Desai VA, Hosamani KM, Jamkhandi AM: Novel fatty acids in Azima tetracantha seed oil. J Am Oil Chem Soc 1991, 68(12):978–979. 10.1007/BF02657547
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ: Protein measurement with the Folin phenol reagent. J Biol Chem 1951, 193(1):265–275.
Kemp W: Qualitative organic analysis: spectrochemical techniques. 2nd edition. New York: McGraw- Hill; 1986:197.
Sazada S, Verma A, Rather AA, Jabeen F, Meghvansi MK: Preliminary phytochemicals analysis of some important medicinal and aromatic plants. Adv Biol Res 2009, 3(5–6):188–195.
Greenberg DM: Plant proteolytic enzymes. In Methods in Enzymology. Edited by: Colowick SP, Kalpan NO. New York: Academic; 1955:54–64.
Rowe M, de Gast GC, Platts-Mills TA, Asherson GL, Webster AD, Johnson SM: Lymphocyte 5′-nucleotidase in primary hypogammaglobulinaemia and cord blood. Clin Exp Immunol 1980, 39(2):337–343.
Fiske CH, Subbarow Y: The colorimetric determination of phosphorus. J Biol Chem 1925, 66: 375–400.
Bessey OA, Lowry OH, Brock MJ: A method for the rapid determination of alkaline phosphates with five cubic millimeters of serum. J Biol Chem 1946, 164: 321–329.
Lo TB, Chen YH, Lee CY: Chemical studies of Formosan cobra ( Naja naja atra ) venom. Part 1. Chromatographic separation of crude venom on CM-Sepadex and preliminary characterization of its components. J Chin Chem Soc 1966, 13(1):165–177.
Yap MKK, Tan NH, Fung SY: Biochemical and toxinological characterization of Naja sumatrana (Equatorial spitting cobra) venom. J Venom Anim Toxins incl Trop Dis 2011, 17(4):451–459. Available at: http://www.scielo.br/pdf/jvatitd/v17n4/12.pdf
Li ZY, Yu TF, Lian EC: Purification and characterization of L-amino acid oxidase from king cobra ( Ophiophagus hannah ) venom and its effects on human platelet aggregation. Toxicon 1994, 32(11):1349–1358. 10.1016/0041-0101(94)90407-3
Ellman GL, Courtney KD, Andres V Jr, Feather-Stone RM: A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961, 7(1):88–95.
Pukrittayakamee S, Warrell DA, Desakorn V, McMichael AJ, White NJ, Bunnag D: The hyaluronidase activities of some Southeast Asian snake venoms. Toxicon 1988, 26(7):629–637. 10.1016/0041-0101(88)90245-0
Tan NH, Tan CS: Acidimetric assay for phospholipase A using egg yolk suspension as substrate. Anal Biochem 1988, 170(2):282–288. 10.1016/0003-2697(88)90632-X
Maruthi ET, Ramesh CK, Mahmood R: Evaluation of anthelmintic and antimicrobial activities of Azima tetracantha Lam. Int J Pharm Sci 2010, 2(1):375–381.
Muthuswamy P, Elakkiya S, Manjupriya K, Deepa K, Ramachandran S, Shanumgapandiyan P: Preliminary phytochemical and in vitro antioxidant perspectives of the leaf extracts of Azima Tetracantha lam (Family: Salvadoraceae). Int J Pharm Biol Sci 2012, 3(1):50.
Dhananjaya BL, Zameer F, Girish KS, D’Souza CJ: Anti-venom potential of aqueous extract of stem bark of Mangifera indica L. against Daboia russellii (Russell’s viper) venom. Indian J Biochem Biophys 2011, 48(3)):175–183.
Gopi K, Renu K, Raj M, Kumar D, Muthuvelan B: The neutralization effect of methanol extract of Andrographis paniculata on Indian cobra Naja naja snake venom. J Pharm Res 2011, 4(4):1010–1012.
Fattepur SR, Gawade SP: Preliminary screening of herbal plant extracts for anti-venom activity against common sea snake ( Enhydrina schistosa ). Pharmacogn Mag 2007, 3(9):56–60.
Acknowledgments
The authors would like to thank Jain University for the funding of this research.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that there are no competing interests.
Authors’ contributions
BJ and MSV worked on the extraction and characterization of phytochemicals in the laboratory. SSM is the corresponding author and designer of the research. KKM helped in the characterization of phytochemicals and enzyme assays by providing suitable suggestions and also helped in characterization of plant species. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Janardhan, B., Shrikanth, V.M., Mirajkar, K.K. et al. In vitro screening and evaluation of antivenom phytochemicals from Azima tetracantha Lam. leaves against Bungarus caeruleus and Vipera russelli. J Venom Anim Toxins Incl Trop Dis 20, 12 (2014). https://doi.org/10.1186/1678-9199-20-12
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1678-9199-20-12