
Abstract
Sequential interactions between several adhesion molecules and their ligands regulate lymphocyte circulation and leukocyte recruitment to inflammatory foci. Adhesion molecules are, therefore, central and critical components of the immune and inflammatory system. We review the evidence that tobacco smoking dysregulates specific components of the adhesion cascade, which may be a common factor in several smoking-induced diseases. Smoking causes inappropriate leukocyte activation, leukocyte-endothelial adhesion, and neutrophil entrapment in the microvasculature, which may help initiate local tissue destruction. Appropriate inflammatory reactions may thus be compromised. In addition to smoke-induced alterations to membrane bound endothelial and leukocyte adhesion molecule expression, which may help explain the above phenomena, smoking has a profound influence on circulating adhesion molecule profiles, most notably sICAM-1 and specific sCD44 variants. Elevated concentrations of soluble adhesion molecules may simply reflect ongoing inflammatory processes. However, increasing evidence suggests that specific soluble adhesion molecules are immunomodulatory, and that alterations to soluble adhesion molecule profiles may represent a significant risk factor for several diverse diseases. This evidence is discussed herein.
Similar content being viewed by others


Introduction
Smoking leads to a generalised leukocytosis [1–7]; influences the production of most immunoglobulin classes and sub-classes [8–11]; can induce T cell anergy (failure to respond to antigen) [12–14]; effect vascular dynamics [15–19]; cause inappropriate priming and activation of monocytes and neutrophils [4, 20, 21]; abnormal platelet aggregation [22, 23]; and a generalized increase in local and systemic inflammatory markers [21, 24, 25]. Therefore, there is considerable evidence that smoking exerts profound influences on multiple components of the immune and inflammatory system in humans, various aspects of which have been ably reviewed elsewhere [4, 14, 16, 26, 27]. This paper will focus on the influence of tobacco smoking on a group of molecules that function at the heart of the immune and inflammatory response – the cellular adhesion molecules.
Adhesion Molecule Overview
Adhesion molecules share the common physiological role of promoting cell-cell or cell-extracellular matrix adhesion, and many are multifunctional intracellular signalling molecules. The ability of lymphocytes (B cells and T cells) to circulate between the blood and secondary lymphoid tissue, and the movement of leukocytes (lymphocytes and other white blood cells, such as neutrophils and monocytes) from the systemic circulation to local sites of inflammation, are major and critical components of the immune response. Leukocytes must, therefore, interact with and cross endothelial barriers – either the high endothelial venules of the lymphatic system or the endothelial cells forming the microvasculature. The sequential processes of capture, rolling and adhesion of leukocytes to endothelial cells, and subsequent leukocyte extravasation and migration to specific sites, are mediated and controlled by a complex and overlapping series of interactions between adhesion molecules and their specific ligands – an adhesion cascade – as represented in Figure 1[28–31].

Essentially, leukocyte capture is mediated by P-selectin (CD62P) and L-selectin (CD62L). Rolling is mediated by P-selectin, L-selectin, and a third member of the selectin family of adhesion molecules, E-selectin (CD62E). Firm adhesion is mediated by interactions between intercellular adhesion molecule-1 (ICAM-1; CD54) and vascular cell adhesion molecule-1 (VCAM-1; CD106) and their ligands – the β2 integrins (particulary LFA-1 [αLβ2; CD11a/CD18] and Mac-1 [αMβ2; CD11b/CD18]) and very late-antigen-4 (VLA-4; CD49d), respectively – following further activation of the adhesion cascade by locally produced chemokines, and/or other stimuli, such as RANTES, fractaline, and stromal cell-derived factor-1α (SDF-1α) and SDF-1β, as reviewed in [31]. Extravasation through endothelial-endothelial cell junctions is less well understood. However, homophilic interactions of specific adhesion molecules [junctional adhesion molecules (JAM), platelet-endothelial cell adhesion molecule-1 (PECAM-1; CD31), and vascular endothelial cadherin (VE-cadherin; CDH5)] may play important roles in maintaining endothelial-endothelial cell adhesion, and these adhesion molecule interactions are probably disrupted in order to permit leukocyte transmigration [31–34]. Although the importance of another adhesion molecule, CD44, in lymphocyte homing has been known for some time, there is increasing evidence to suggest that CD44 is also involved in leukocyte adhesion and transmigration processes during inflammation [30, 35]. Glycosylation, and other post-translational modifications; expression densities of adhesion molecules, and their ligands, on leukocytes and endothelial cells; activation-induced alterations in the affinity of adhesion molecules for specific ligands; intracellular signal transduction pathways initiated on rolling; and co-operation between various adhesion molecules, all influence leukocyte/endothelial interactions, as reviewed elsewhere [28, 31, 36–42]. Significant interference with leukocyte transmigration due to environmental, acquired, or genetic factor(s) may be expected to have profound consequences. A classic example is leukocyte adhesion deficiency type I, a rare genetic disorder defined by insufficient or non-functional CD18, the common chain of the β2-integrins, resulting in compromised leukocyte trafficking, reflected in severe and recurrent bacterial infections, and delayed wound healing. This paper will focus on the evidence that tobacco smoking also influences the expression and release of specific adhesion molecules.
ICAM-1 and CD44
Of the major adhesion molecules, the evidence presented below will necessitate a particular focus on two – ICAM-1 and CD44. ICAM-1 is an integral membrane glycoprotein with five extracellular immunoglobulin-like domains and a short cytoplasmic tail, encoded by a 3.3 kb mRNA transcribed from a single gene with no alternate exons [43, 44]. ICAM-1 is expressed constitutively by a wide variety of cell types, including endothelial cells. Basal expression of ICAM-1 on endothelial cells, and some leukocytes, can be augmented following induction by TNF-α, IL-1β, and IFN-γ, and other inflammatory mediators [38, 45–49]. Recognised ligands of ICAM-1 include the β2-integrins of leukocytes, hyaluronate, sialophorin (CD43) and fibrinogen [50, 51], reflecting the major role played by ICAM-1 immune and inflammatory regulation.
By way of introduction to a complex adhesion molecule family, human CD44 is a widely expressed family of glycoproteins, encoded by a single gene containing 20 exons. Numerous CD44 isoforms are generated through alternate splicing of pre-mRNA. However, all CD44 isoforms share the same N- and C-terminal sequences. The haematopoetic, or standard form, of CD44 (CD44H; CD44s) is a 248 amino acid protein that contains no variant exon-encoded peptide sequences. Thus CD44H is the smallest CD44 isoform, with a molecular mass of 80–95 kDa, more than half of which is due to post-translational glycosylation events. Splice variation and significant post-translational modifications result in a multifunctional group of CD44 adhesion molecules, with important functions in embryogenesis, lymphocyte activation and homing, angiogenesis, leukocyte extravasation, anti-apoptosis signalling, presentation of growth factors and proteases, and cell migration and proliferation, including during tumor metastasis [30, 52–60]. Many of these activities appear to be mediated by interaction between the extracellular matrix component hyalouronate and CD44. However, several other CD44 ligands are recognised, including laminin, collagen, fibronectin, serglycin, osteopontin and aggrecan [57, 61, 62]. Many recent reviews address the structure, regulation, function, clinical significance, and therapeutic targeting of ICAM-1 [38, 50, 51, 63–67] and CD44 [30, 61–63, 68–74].
The Influence of Tobacco Smoking on Cell-Bound Adhesion Molecules
It has been known for some time that cigarette smoking can induce leukocyte-endothelial adhesion, microvascular and macrovascular entrapment of leukocytes, and leukocyte aggregation in humans and animal models [75–89]. Leukocyte-endothelial binding, and subsequent leukocyte-mediated tissue damage, is a central component of various smoking-associated inflammatory diseases, leading several groups to investigate the influence of tobacco use on adhesion molecule networks. This review will pay particular attention to chronic obstructive pulmonary disease (COPD), direct cigarette smoke exposure being most intense in the lungs; vascular diseases, vascular endothelial cells and circulating leukocytes being chronically exposed to systemically distributed components and metabolites of smoke; and chronic inflammatory periodontal disease (CIPD), with the periodontal tissues being both chronically exposed to systemic smoke components, and transiently exposed topically.
Chronic Obstructive Pulmonary Disease
COPD is characterised by chronic coughing, obstruction of the peripheral airways, and destruction of lung surfaces (emphysema). Although not all smokers develop COPD, 90% of those who do develop COPD are smokers. As much of the pulmonary tissue destruction in COPD is thought to be due to the recruitment and activation of inflammatory cells, several researchers have examined the influence of smoking on adhesion molecule expression and release by the endothelial cells comprising the pulmonary vasculature, and recruited alveolar inflammatory cells. Tobacco smoking results in a significantly increased number of immature neutrophils in the systemic circulation, characteristic of chronic stimulation of bone marrow [7]. Accordingly, there is a small, but significant, increase in circulating neutrophil L-selectin expression in smokers [7]. Immature neutrophils are preferentially sequestered in the lung microvasculature [90], and pulmonary neutrophil entrapment is a recognised phenomenon in smokers [85]. Additionally, absolute neutrophil and macrophage numbers are increased in the alveolar space of smokers [24, 88, 91–93]. These observations point to a smoking-induced dysregulation of adhesion molecule networks in the pulmonary environment.
In support of this, Schaberg et al. [88] noted that a significantly higher proportion of alveolar macrophages from smokers expressed β2-integrin subunits than alveolar macrophages obtained by pulmonary lavage from non-smokers. The increase in β2-integrin expression correlated with increased numbers of macrophages in the pulmonary environment. Increased binding of alveolar macrophages from smokers to TNF-α stimulated human umbilical vein endothelial cells (HUVECs) was mitigated by either pre-treatment of the endothelial cells with antibodies to ICAM-1, or pre-treating alveolar macrophages with antibodies to CD18. Therefore, smoking may contribute to increased ICAM-1/β2-integrin-dependent recruitment of inflammatory cells to the lungs. Lensmar et al. [94] reported that while the sputum of smokers (n = 9) contained more macrophages than the sputum of non-smokers (n = 7), the percentage of macrophages expressing ICAM-1 was significantly lower in smokers. However, upregulation of endothelial ICAM-1 expression is likely to be a more important limiting factor in terms of leukocyte recruitment.
In studies of the pulmonary endothelium, there is some evidence that ICAM-1 expression by bronchial vessels may not be affected by smoking [95]. However, Schaberg et al. [93] went on to observe a large increase in ICAM-1, but not P-selectin, E-selectin, or VCAM-1, expression by the endothelium of peripheral pulmonary vessels in smokers, compared to non-smokers. Schaberg et al. [93] also noted a strong correlation between cumulative tobacco smoke exposure (pack-years) and the percentage of ICAM-1-positive pulmonary vessels, consistent with a role for smoking in increased pulmonary recruitment of leukocytes through the dysregulation of adhesion molecule networks.
As only some smokers develop COPD, there is an obvious interest in defining the characteristics of smokers at enhanced risk of developing the disease [96]. To this end, Maestrelli et al. [97] reported that the numbers of neutrophils expressing CD11b and CD18, but not CD11a or CD11c, were increased in smoking subjects (n = 33) with airway obstruction, compared to smokers without airway obstruction, and hypothesised that CD11b/CD18 expression by sputum neutrophils may represent a marker for the development of chronic airway obstruction among smokers. Noguera et al. [98] investigated the characteristics of circulating neutrophils in subjects with COPD and in smokers and non-smokers without COPD. None of the COPD subjects were current smokers, although they had a total cumulative tobacco exposure similar to the smoking group (50 pack-years). There were no significant differences in Mac-1, LFA-1, or L-selectin expression by TNF-α stimulated or unstimulated neutrophils isolated from smokers or non-smokers with normal forced-expiratory volume. However, expression of Mac-1 on neutrophils was increased in those with COPD, compared to either healthy group (smokers or non-smokers), augmented by an increased respiratory burst, leading the authors to suggest that neutrophil dysfunction in COPD subjects may not be directly caused by smoking, but, rather, may represent a characteristic of COPD. It is also possible that smoking may not affect all smokers equally, and that a differential effect on CD18/CD11b expression could discriminate smokers at increased risk of developing COPD. However, a recent comparison of adhesion molecule expression profiles on leukocyte (L-selectin, VLA-4, and the three β2-integrin heterodimers), endothelial and epithelial (E-selectin, P-selectin, VCAM-1, ICAM-1, and ICAM-2) cell surfaces in freshly resected lungs from smokers with airways obstruction (n = 10) and smokers with normal lung function (n = 10) revealed no significant differences in adhesion molecule profiles between diseased and healthy groups [99]. The authors, therefore, concluded that development of airways obstruction in smokers could not be explained by differences in the expression of adhesion molecules known to be involved in the control of cell traffic in the lung.
Other investigations have monitored the effect of smoking on bronchial epithelial cells. Small-airway epithelial cells harvested from smokers exhibited significantly elevated ICAM-1 mRNA levels, compared to non-smokers [100]. Correspondingly, a dramatic increase in ICAM-1 release from the surface of cultured small-airway epithelial cells of smokers compared to cells from non-smokers (356 pg 106 cells-1 vs 113 pg 106cells-1) was noted. No such differences were noted in epithelial cells isolated from the main bronchi of smokers and non-smokers. Similarly, di Stefano et al. [95] found no difference in ICAM-1 expression on the bronchial epithelium of smokers and non-smokers. Cigarette smoke exposure resulted in a significant increase (up to 175%) in sICAM-1 release from primary explant cultures of human bronchial epithelial cells obtained from never-smokers, or smokers with COPD, but not in cultured bronchial epithelial cells from smokers with normal pulmonary function [101]. As similar results were noted for concentrations of the proinflammatory cytokine IL-1β, this suggests that not all smokers are equally susceptible to cigarette-smoke induced alterations to the pulmonary inflammatory response.
Vascular diseases
The pathogenesis of atherosclerosis, the precursor to most acute coronary syndromes and strokes, includes the activation of vascular endothelial cells, and the recruitment of inflammatory cells, predominantly macrophages, to the vessel wall [35, 102, 103]. Leukocyte-endothelial recruitment is adhesion molecule-dependent, and smoking is a major risk factor for vascular diseases. Therefore, attention has inevitably turned to examinations of the influence of tobacco smoking on adhesion molecule expression by vascular endothelial cells. Indeed, tobacco smoking has long been known to induce leukocyte-endothelial adhesion and leukocyte entrapment, as noted earlier.
Monocyte-endothelial interaction is clearly multifactorial. However, endothelial ICAM-1 expression is likely to exert a strong influence on such cell-cell adhesions. Adams et al. [102] were able to show upregulation of ICAM-1 (but not VCAM-1 or E-selectin) expression on HUVECs exposed to smokers' serum, and to confirm increased monocyte-endothelium adhesion in smokers selected to exhibit no other major risk factor for vascular disease. Nicotine has been shown to induce an increase in VCAM-1 mRNA synthesis, but not E-selectin, in primary human coronary artery endothelial cells, but a 1.6 fold elevation required 24 hr exposure to a concentration of nicotine as high as 10-5M (or 1.6 μg ml-1), which is higher than normal physiological exposure levels [17, 104, 105]. Weber et al. [89] determined that increased monocyte-endothelial adhesion in smokers is CD11b-dependent, through the use of a blocking anti-CD11b antibody. However, CD11b was not increased on the surface of freshly isolated monocytes. This indicates that smoking may lead to increased monocyte-HUVEC adhesion by influencing an endothelial Mac-1 ligand, with ICAM-1 the prime candidate, or that smoking may induce conformational, or other, alterations to Mac-1, leading to increased affinity for ICAM-1. Indeed, increased expression of CD11b may not be required or sufficient for increased leukocyte-endothelial adhesion [89, 106, 107].
Neutrophils contain large granular stores of CD11b that can be rapidly translocated to the surface on activation [108, 109]. Therefore, tobacco-induced translocation of CD11b could partly explain enhanced neutrophil adhesion in smokers. Recently, Koether et al. [20] exposed whole blood or neutrophils isolated from non-smokers to cigarette smoke condensate (CSC), and observed a rapid (minutes) 2.5 to 3 fold increase in CD18/CD11b expression on the neutrophil surface. Other studies have also shown an increased tobacco-induced expression of cell surface β2-integrins by leukocytes in in vitro studies [33, 80, 110]. Thus, up-regulation of CD11b/CD18 in primed neutrophils, coupled with endothelial ICAM-1 expression, would be expected to contribute to inappropriate neutrophil adhesion in smokers in cardiovascular, pulmonary, or other environments, with potential ramifications to local tissue integrity. However, this specific issue is not clearcut. Most in vivo, and some in vitro, studies in humans have not shown a difference between expression levels of β2-integrins on neutrophils and monocytes and of smokers and non-smokers [4, 7, 105, 111], which hints at a need for consideration of cell conditioning to tobacco smoke, and/or physiologically relevant dosing in in vitro models. Typical serum nicotine levels in smokers are in the region of 30 ng ml-1[17, 104, 105].
Shen et al. started the important task of dissecting the signalling pathways that underlie tobacco-induced monocyte-endothelial adhesion [33]. Cigarette smoke condensate (CSC) was shown to induce surface expression of ICAM-1, VCAM-1, and E-selectin on HUVECs and bovine aorta endothelial cells. Nicotine alone, even at 25 mg ml-1, did not influence endothelial adhesion molecule profiles. Inhibitors of protein synthesis (cycloheximide), transcription (actinomycin D) and protein kinase C (GF 109203X and chelerythrine), established that increased endothelial adhesion molecule expression required de novo adhesion molecule production, via a PKC-dependent pathway. NF-κB, a transcription factor, binds to a consensus site in specific adhesion molecule genes. Shen et al. [33] established the importance of CSC-induced NF-κB activation in the upregulation of endothelial adhesion molecule expression. Furthermore, CSC increased the migration of monocytic cells across HUVECs and bovine aorta endothelial cells, concomitant with an up to 10-fold increase in PECAM-1 phosphorylation, which was inhibited by PKC inhibitors. Unfortunately, such studies on the molecular mechanisms by which smoking may dysregulate the expression and release of specific adhesion molecules are rare, and there is an obvious need for a concerted research effort in this area.
Several groups have attempted to reduce smoking-induced leukocyte-endothelial adhesion in smokers by pharmacological intervention with anti-inflammatory agents. Smoking-induced monocyte-endothelial adhesion in humans was acutely abrogated to a significant degree by a single 7 g oral administration of L-arginine, but not vitamin C, suggesting a role for nitric oxide [102]. Weber et al. [89] showed that ten days of Vitamin C supplementation was successful in reducing monocyte-endothelial adhesion in smokers. Zapolska-Downar et al. [21] noted that oral administration of the non-steroidal anti-inflammatory drug ibuprofen reduced monocyte-HUVEC adhesion in smokers.
Adhesion molecule-mediated platelet aggregation represents a mechanism contributing to acute coronary events in smokers [22]. Accordingly, it has been shown that P-selectin expression is generally increased on the surface of platelets in smokers, a process that requires translocation of P-selectin from the intracellular α-granules [22, 112]. In vivo platelet activation, and the associated alterations in cell surface presentation of P-selectin molecules, can occur immediately after smoking a cigarette [22]. The increased surface of P-selectin on platelets is not affected by aspirin administration (2 weeks at 100 mg/day) [112].
Chronic inflammatory periodontal disease
Chronic inflammatory periodontal disease (CIPD) is a common disease of the supporting tissues of the teeth, resulting from a complex interaction between plaque bacteria and the host response. Like vascular disease and COPD, tobacco smoking is also a major risk factor for the development, progression, and exacerbation of periodontitis [113–116].
ICAM-1 expression is increased in the gingival vasculature during episodes of experimental gingivitis, -induced on cessation of normal oral hygiene procedures-coincident with peak measurements of IL-1 in the gingival crevicular fluid (GCF; a serum-derived fluid that exudes from the gingival sulcus) [117]. Rezavandi et al. [18] examined adhesion molecule profiles in periodontal tissues taken from smokers and non-smokers undergoing surgical treatment for CIPD. Compared to non-inflamed areas, the proportion of the microvasculature expressing ICAM-1 and E-selectin was significantly increased in inflamed areas of tissues, irrespective of smoking status. However, although smokers had thus retained the ability to upregulate adhesion molecule expression, an integral part of the inflammatory response, other signs of smoking-induced immune dysregulation in the gingival microvasculature were apparent. There was no statistically significant difference in the number of small blood vessels, detected by immunostaining of von Willebrand factor, in inflamed and non-inflamed areas of the gingival tissues of smokers. In other words, the normal angiogenic response that helps to define inflammation was compromised.
Rezavandi et al. [18] also noted that in histologically normal, non-inflamed periodontal tissues, the percentage of vessels expressing ICAM-1 was reduced in smokers. Thus, basal endothelial ICAM-1 expression levels may be compromised. Typical expression profiles of ICAM-1 in the gingival tissues of a smoker and non-smoker with CIPD are presented in Figure 2. As mentioned earlier, constituents of tobacco smoke can induce ICAM-1 expression on endothelial cells [33, 80, 93, 102]. One possible outcome of long-term tobacco smoke exposure is tolerance, with respect to ICAM-1 expression. It is also a possibility that the reduced level of ICAM-1 expression in normal gingival tissues could reflect ongoing shedding of membrane-bound ICAM-1. Smokers are, indeed, known to carry a high load of a soluble form(s) of ICAM-1 in the systemic circulation [105, 118], a topic we shall re-address shortly.

Expression of ICAM-1 in the gingival tissues of a smoker and non-smoker with chronic inflammatory periodontal disease. Typical histological sections of gingival tissue from a smoker (A) and a non-smoker (B) with CIPD. Tissue sections were labelled with monoclonal antibodies to ICAM-1, visualised with streptavidin-biotin peroxidase complex developed with diaminobenzidine/hydrogen peroxide, and counter-stained with haematoxylin. ICAM-1 positive cells (dark staining) are predominantly endothelial. The percentage of ICAM-1-positive vessels of the gingival microvasculature was determined following immunostaining of von Willebrand factor, an endothelial marker, in adjacent sections.
In contrast to the pulmonary alveolar space, the absolute number of neutrophils in the GCF or oral cavity fluids of smokers is comparable or lower in smokers, compared to non-smokers [119, 120]. This is despite the increased number of neutrophils in the systemic circulation of smokers [5, 7]. The reduced angiogenic response and compromised endothelial ICAM-1 expression profiles in the gingival tissues of smokers may help explain this initially surprising observation.
In summary, there is growing evidence that smoking influences tissue and cellular adhesion molecule expression profiles in several smoking-induced diseases: COPD; vascular diseases; and CIPD. However, such studies are at a preliminary stage, all tissues do not appear to respond equally, and the in vivo evidence for smoking-induced cell-associated adhesion molecule dysregulation has been obtained primarily with small numbers of patients. Further research into the influence of tobacco on adhesion molecules in smoking-induced inflammatory disease is warranted and necessary.
Soluble Adhesion Molecules
Soluble forms of many adhesion molecules are recognised (see Figure 3), but the relevance of these circulating forms of adhesion molecules is not clearly understood. Specific circulating adhesion molecules are proteolytically released from the cell surface of activated immune cells [45, 48, 121], therefore, these circulating adhesion molecules may simply reflect general immune function. However, there is convincing evidence that certain soluble adhesion molecules remain bioactive and have the potential to interfere with a variety of immunologic/inflammatory processes [29, 122]. As noted early on by Gearing and Newman [29], there are two obvious means by which soluble forms of adhesion molecules may have significant physiological influence – (i) by competitive inhibition of cell-cell interactions and, (ii) by binding to the appropriate ligand on the surface of a cell, thereby eliciting a response.

Release of soluble adhesion molecules.
sICAM-1
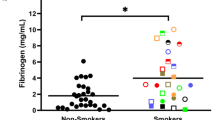
The evidence that tobacco smoking leads to a significant increase in circulating levels of sICAM-1 is overwhelming. In an investigation into the relationship between local (periodontal) and systemic inflammation, a significant elevation of sICAM-1 in smoking subjects, independent of periodontal status, was noted [118]. This observation has since been confirmed by many authors [105, 123–135]. In the pulmonary environment, involuntary cigarette smoke exposure ("environmental" or "passive" smoking) has been associated with significantly increased ICAM-1 concentrations in the bronchoalveolar lavage fluid of children [136]. Rumalla et al. [137] noticed a 10 fold elevation in sICAM-1 levels in the pulmonary lavage fluids of some individual smokers.
Cigarette smoking does not seem to lead to an acute increase in circulating sICAM-1 levels [105, 125]. Thus, raised circulating sICAM-1 levels in smokers could reflect tobacco-induced vascular damage, or other underlying pathologies. However, further studies have since established that the influence of smoking on sICAM-1 levels is dose-dependent and reversible. There is a strong correlation between sICAM-1 levels and several quantitative indices of recent tobacco smoke intake – expired-air CO levels [138]; plasma and serum cotinine concentrations [105, 138]; a composite index of tobacco intake [138]; and the systemic nicotine intake from a single cigarette [105]. Systemic sICAM-1 levels decline rapidly on smoking cessation to levels approaching that of non-smokers [125, 138]. Much of this recovery occurs in the first four weeks following biochemically-validated cessation (unpublished data). Taken as a whole these data provide strong evidence that tobacco smoking is a direct causative agent of a systemically increased sICAM-1 burden. The decline in sICAM-1 levels noted on cessation is not compromised by high-dosage nicotine replacement therapy, in the form of transdermal nicotine patches (unpublished data). Therefore, it is a component (or metabolite) of tobacco smoke other than nicotine (or cotinine) that is responsible for elevated sICAM-1 concentrations in smokers. Typical concentrations of circulating adhesion molecules are presented in Table 1.
sICAM-1 and disease
A growing number of studies report a significant association between elevated sICAM-1 levels, and the presence of most vascular diseases; the prediction of future adverse events; and poor prognosis [128, 131, 134, 142–149]. Thus, elevated sICAM-1 levels may have serious consequences. Serum sICAM-1 concentrations are also reported to be significantly elevated in subjects with other diverse disease entities, including non-small-cell lung cancer and other malignancies [127, 150], diabetes [29, 151], cystic fibrosis [152], inflammatory bowel diseases [153], bronchial asthma [154], allergic alveolitis [155, 156], and several other inflammatory diseases. However, a majority of such studies have not considered smoking habits. Therefore some degree of re-evaluation of sICAM-1 as a disease marker may be required in light of the profound, and reversible, influence of tobacco smoke on sICAM-1.
Blann et al. [157] reported that increased sICAM-1 concentration was only a weak predictor of disease progression in peripheral atherosclerosis, when the study population was balanced for age, gender, and smoking status. Wallen et al. [134] observed that while elevated sICAM-1 levels in those with angina pectoris was associated with cardiovascular death, or non-fatal myocardial infarction (354 ng ml-1, n = 7) compared to those who remained event free (282 ng ml-1, n = 86), clinical risk factors, including smoking (57% vs 21%), were more prevalent in those with a poor outcome. Additionally, mean sICAM-1 was significantly raised in smokers, compared to never-smokers, to a comparable extent to that noted between coronary event and event free groups. Similarly, O'Malley et al. [130] observed a significant increase in sICAM-1 levels in a group of individuals with ischaemic heart disease, both at time of presentation with chest pain, and three months later, compared to healthy controls. Therefore, sICAM-1 levels where not influenced by the acute event. However, for the purposes of this review it is most interesting that sICAM-1 elevation was confounded by cigarette smoking. Fassbender et al. [126] monitored soluble adhesion molecule profiles in 173 subjects with cerebrovascular disaeases, and 67 controls. Although, there was an increase in sICAM-1 levels in the total population with cerebrovascular disease compared to controls (275 ng ml-1 vs 260 ng ml-1, respectively), the diseased population contained twice the number of smokers, and the authors note that sICAM-1 levels were significantly increased in smoking subjects. Again, Rifai et al. [158] examined sICAM-1 levels in the plasma of 100 men with angiographically documented coronary heart disease, and 100 healthy controls matched for age and smoking status. Under this experimental strategy, there was no significant difference in median sICAM-1 concentrations between the diseased (335 ng ml-1) and healthy groups (339 ng ml-1).
Some studies certainly suggest that elevated sICAM-1 is a suitable marker of vascular disease status and a risk factor for future acute vascular events [131, 146, 149], and that these significant associations are not confounded by smoking habits. Nevertheless, it has been clearly established that a dramatic, dose-dependent increase in circulating sICAM-1 levels is one consequence of tobacco smoking. Therefore smokers will be more likely to be in the upper quartile of sICAM-1 levels, where increased risk of vascular disease is most evident [146]. Indeed, the influence of smoking on sICAM-1 concentration is commonly reported to be as great, or greater [118, 126, 158] as that ascribed to disease [126, 130, 158].
Recently, Becker et al. [149] have suggested that the significant relationship between elevated systemic sICAM-1 and vascular disease is independent of renal function and major risk factors such as clinical or sub-clinical atherosclerosis (prior vascular disease or ankle-brachial pressure index), endothelial activation (von Willebrand factor), inflammation (C-reactive protein), or elevated sVCAM-1. Thus a causative link between sICAM-1 and vascular disease has yet to be firmly established. While it is likely that underlying inflammatory disease(s) will contribute to some extent to the systemic sICAM-1 load, the possibility that the tobacco-induced sICAM-1 burden may directly influence the immune response, and thus represent a common factor in tobacco-induced diseases, is deserving of attention. To this end, the immunomodulatory effects attributed to sICAM-1 that may be relevant to various disease processes are summarised in Figure 4. Briefly, stimulation of proteloytic enzyme release from granulocytes, which has been proposed as a mediator of multiple organ failure [159], could make a major contribution to tissue breakdown; stimulation of inflammatory mediator release may be expected to potentiate the inflammatory response; competitive inhibition of leukocyte-endothelial interactions may be expected to compromise migration of activated leukocytes; inhibition of immune surveillance could abet tumor cells in evading detection; and promotion of angiogenesis may aid tumor growth, and have obvious relevance to other angiogenesis-dependent diseases. Detailed explanations of these proposed bioactive sICAM-1 properties can be obtained from the references provided in the figure legend [47, 159–177].

The antioxidant capacity of smokers is compromised [178]. The influence of antioxidant therapy on smoking-induced endothelial-leukocyte interactions was discussed briefly earlier. We are currently in the final stages of a study examining the short-term influence of high-dose vitamin C on circulating ICAM-1 levels. However, since embarking on this study an independent research group [133] has reported that the elevation of serum sICAM-1 in smokers is not abrogated by long-term treatment with a different class of antioxidant molecule-α-tocopherol (2 years at 400 IU dL/day). sICAM-1 levels were reduced in healthy men by glucocorticoid (dexamethasone) treatment [179].
Molecular characteristics of sICAM-1
sICAM-1 exists as a 80–105 kDa monomeric protein [50, 51, 180, 181], and as an unconfirmed immunoreactively distinct, and essentially uncharacterised, 130 kDa protein [182]. Evidence for alternate splicing resulting in two mRNA species directly encoding either sICAM-1 or membrane-bound ICAM-1 in humans has been presented [183]. However, the majority of studies have found no evidence for alternate mRNA species in humans, and the predominant theory is that sICAM-1 is released from the cell surface following proteolysis of the cell-bound form, at a defined site [45, 48, 49, 184]. Dimeric ICAM-1 exhibits significantly higher affinity for its major ligand (LFA-1) than monomeric ICAM-1 [185, 186]. Young et al. [187] purified sICAM-1 from normal human serum and the urine of patients with bladder cancer by immunoaffinity chromatography and detected only monomeric sICAM-1, which could not block LFA-1/ICAM-1-dependent cell-mediated cytolosis of bladder tumour target cells. However, others have described sICAM-1 dimers and multimers in vivo [180, 181]. Transcriptional regulation of ICAM-1 is particularly complex, with unusual post-transcriptional control mechanisms described [50, 51, 66]. Cell-specific regulatory mechanisms and cell-specific post-translational modifications of ICAM-1 proteins may affect ICAM-1 function [184, 188, 189]. However, to the best of our knowledge, the character, and source, of ICAM-1 molecules elevated in smokers has not yet been investigated. Clearly, clarification of the specific molecular characteristics of sICAM-1 in smokers would represent a huge step towards an understanding the potential role of sICAM-1 in smoking-induced diseases. Equally, there is a pressing need to understand the mechanisms by which components, or metabolites, of tobacco smoke induce the upregulation and shedding of sICAM-1 molecules from the endothelium, and other cellular sources. Interestingly, despite elevated systemic sICAM-1 concentration noted in smokers, there is a consistent decrease, approaching four fold, in GCF ICAM-1 levels in smokers with CIPD [190]. No difference between serum and GCF sICAM-1 concentration is apparent in non-smokers. Thus, ICAM-1 molecules are inhibited in their passage from the periodontal microvasculature or through the periodontal tissues. This indirectly implies the sICAM-1 molecules in smokers are active, and may be interacting with to be determined ligands. However, a reduced angiogenic response in smokers is another possible explanation for these observations.
sCD44
In an investigation designed to examine the potential of specific reputed tumour-associated circulating sCD44 isoforms to act as biomarkers in certain malignancies, Kittl et al. [140] observed a significant elevation in the mean concentration of sCD44 containing the product of exon 5 (sCD44v5) and sCD44 containing the product of exon 6 (sCD44v6), in self-reported smokers, compared to self-reported non-smokers. Total sCD44 has since been shown to be elevated in the blood of smokers, compared to non-smokers [191], to a small, but significant, extent. Like sICAM-1, the elevations in CD44v5 and v6, but not total CD44, are dose-dependent and reversible [141]. Smoking cessation led to a reduction in concentrations of both sCD44v5 and sCD44v6 (-13 ng ml-1 and -62 ng ml-1, repectively) to levels approaching those reported in non-smokers [140, 141]. Additionally, sCD44 recovery was not influenced by transdermal nicotine replacement therapy (unpublished results).
Alterations to circulating sCD44 profiles patterns have been extensively attributed diagnostic and prognostic potential in several malignancies, including, but not limited to, non-Hodgkin's lymphoma, breast, gastric, and colon cancer [61, 72, 192–199]. Soluble CD44 is also known to be raised in subjects with particular inflammatory conditions [200–202]. However, the majority of studies that have examined the potential utility of sCD44 isoforms as disease biomarkers have not considered smoking status. In light of the recent evidence that smoking exerts a profound dose-dependent and reversible influence on systemic concentrations of specific sCD44 variant molecules, then it may be necessary to re-evaluate ascribed diagnostic and prognostic specificities in certain inflammatory and malignant diseases. Cellular and tissue CD44 isoform expression patterns have also been widely proposed as markers of tumor growth, metastatic potential, and poor prognosis in several malignancies, including lung cancer [53, 60, 203–210]. Alternations to CD44 profiles have also been noted in non-cancerous, but pre-neoplastic, lung tissues [210]. There is, however, evidence that tobacco can induce alterations to cell-bound CD44 profiles in normal cells. Nicotine and its primary metabolite – cotinine – have both been shown to alter CD44 expression profiles on lung microvascular endothelial cells (LEISVO), and bone marrow-derived (STR-12) endothelial cells [211]. Therefore, it may be useful to address the influence of tobacco on the expression of membrane-bound CD44 isoforms in situ in a definitive manner in future studies.
Soluble CD44 was first reported in serum over twenty years ago [212]. Despite this, the physiological relevance of sCD44 molecules is unclear. However, by analogy with sICAM-1, it is entirely possible that circulating CD44 molecules could be immunomodulatory. There is some evidence to support this theory. In an in vitro system, sCD44 (in liposomes) was able to partially suppress T cell activation [213]. Recently, a novel sCD44 splice variant (CD44RC) was cloned that dramatically enhanced the hyaluronan binding activity of cell surface CD44 [214]. This unusual and unexpected observation has been suggested to result from CD44RC binding to chondroitin sulfate side-chains attached to cell surface CD44. Therefore, a multivalent complex with increased avidity for hyaluronan is generated. In addition to this functional originality, CD44RC is produced in a, so far, unique manner – splicing of the 3' end of CD44 exon 2 into an internal site within exon 18, resulting in an altered reading frame and, thus, a novel CD44 isoform. Other studies have suggested that sCD44 variants can bind hyaluronate, and that the affinity of binding is determined by specific glycosylations [196, 215]. Specific sCD44 molecules can bind fibronectin [196]. sCD44, in serum, has also been shown to inhibit human peripheral blood lymphocyte binding to endothelial cells in frozen tonsil sections [196]. Thus, specific sCD44 molecules are bioactive and, potentially, immunomodulatory.
Metastasis to the lung following intravenous delivery of TA3/St murine mammary carcinoma cells is prevented when the carcinoma cells are transfected with sCD44-encoding cDNA, where sCD44 probably acts as a competitive inhibitor of cell surface CD44-hyaluronate interactions [216]. Indeed, sCD44 transfection was subsequently shown to prevent hyaluronate-mediated clustering of CD44 on tumour cell surface [60]. Peterson et al. [217] inhibited murine mammary carcinoma cell growth through transfection with cDNA encoding hyaluronate-binding sCD44v8-v10 or sCD44v6-v10. Other studies have also indicated that sCD44 may interfere with the growth and metastasis of specific tumour cells in mice [217–220]. Suppression of tumour formation by a human sCD44 fusion protein has again been demonstrated lately [221], and a soluble CD44-immunoglobulin fusion protein was able to block the migration of a CD44H-transfected human melanoma cell line across a hyaluronate-coated surface [222]. Interestingly, CD44 may act as a tumor cell surface anchor for MMP-9, a matrix metalloproteinase, which may contribute to collagen degradation and contribute to tumor invasiveness [60]. sCD44 can disrupt CD44/MMP-9 clusters and inhibit tumor metastasis in vivo [60].
Alternate splicing of the CD44 pre-mRNA permits the subsequent translation of a plethora of potential CD44 variant proteins, whose function may be further modified by extensive and varied post-translational modifications. The molecular characteristics of smoking-influenced CD44 proteins have yet to be ascertained. CD44 has proposed roles in leukocyte-endothelial interactions, there is increasing evidence of several potential mechanisms by which CD44 may play an aetiological role in certain cancers and vascular diseases [35, 53, 55, 59, 60], there are reputed diagnostic associations between CD44 profiles and various diseases, and there are undoubted connections between tobacco use and specific cancers and certain life-threatening inflammatory processes. Consideration of the available data shows that there is a need to further develop our understanding of the influence of tobacco smoking on the CD44 gene, and encoded variant CD44 protein family, both as a means of properly attributing any diagnostic and prognostic significance to CD44 molecules, and perhaps, in order to unravel any mechanisms of tobacco-induced disease that may be, partially at least, CD44-mediated.
Other soluble adhesion molecules
Blann et al. [157] reported that, compared to appropriate control groups, sVCAM-1 is significantly elevated in the serum of smokers with peripheral artery disease, but not healthy smokers, implying an indirect relationship. Elsewhere, much of the available evidence suggests that tobacco smoking does not influence sVCAM-1 concentrations [134, 142, 223]. Osterud et al. [224] found that sVCAM-1 levels were actually lower in male smokers, compared to non-smokers, but this was not true for females, or combined genders. Equally, and on balance, smoking does not appear to influence systemic concentrations of the selectins [105, 112, 118, 151, 225–227], or PECAM-1 [105, 224, 228]. However, there is some evidence to suggest that smoking could influence sP-selectin levels. Blann et al. [229] observed a significant rise sP-selectin levels in smokers. However, the smokers and non-smoking subjects were not matched for age or disease status (deep venous thrombosis), and the difference was not impressive. This is broadly in agreement with previous reports by the same authors, who have acknowledged that any potential relationship between smoking and sP-selectin is weak [124, 230–232]. Osterud et al. [224] found a small, but significant, rise in sP-selectin in the serum of women smokers, compared to non-smokers, that was not apparent in the male or in the total study population. Limited evidence suggests that smoking could influence circulating sE-selectin concentrations. Kitamura et al. [233] observed a significant increase in serum sE-selectin levels in heavy smokers (20–40 cigarettes day-1; 92 ng ml-1), compared to non-smokers (67 ng ml-1). However, this observation was made in subjects with pustulosis palmaris et plantaris, a dermatological disease that seems to exert a strong influence on sE-selectin in serum, and it is not clear how the incidence of this confounding condition was distributed between the smoking and non-smoking groups. Thus, the balance of evidence suggests that if circulating profiles of other major adhesion molecules are indeed influenced by tobacco smoking, then this influence is certainly not as profound as the dramatic effects seen in sICAM-1 and some sCD44 variants.
Concluding Remarks
Research into the relationship between tobacco use and adhesion molecule networks is at an early stage. Smoking directly causes an increase in soluble ICAM-1 and specific CD44 variant concentrations in the systemic circulation, diluting several previous diagnostic and prognostic attributions. Both sICAM-1 and CD44 are considered to possess several immunomodulatory properties. Clarification of the specific sources and molecular characteristics of smoking-influenced sICAM-1 and sCD44 protein should provide insight into potential functions of soluble adhesion molecules in tobacco-induced diseases. The influence of tobacco on the cellular and tissue distribution of cell-bound adhesion molecules is less clear. Smoking status should certainly be considered in studies that examine potential prognostic and diagnostic assignations of significance to circulating adhesion molecule profiles and membrane-bound expression in cells and tissues. This does not occur in the majority of studies, even in diseases were the aetiological significance of smoking is unequivocal, such as neoplastic and malignant lung tissues and COPD. Although there is extensive, and growing, knowledge of the molecular mechanisms that regulate transcription, expression, and shedding of ICAM-1, CD44, and other adhesion molecules, studies that address how tobacco smoke may influence these control networks are minimal. Adhesion molecules function at the heart of the immune and inflammatory response. Dysregulation of these critical molecules may represent a common mechanism(s) underlying susceptibility to a variety smoking-induced diseases.
Abbreviations
- CIPD:
-
chronic inflammatory periodontal disease
- COPD:
-
chronic obstructive pulmonary disease
- GCF:
-
gingival crevicular fluid
- HUVEC:
-
human umbilical vein endothelial cells
- ICAM-1:
-
intercellular adhesion molecule-1 (CD54)
- LFA-1:
-
lymphocyte functional antigen-1 (CD11a/CD18; (αLβ2)
- Mac-1:
-
CD11b/CD18 (αMβ2)
- PECAM-1:
-
platelet-endothelial cell adhesion molecule-1
- VCAM-1:
-
vascular cell adhesion molecule-1 (CD106).
References
Corre F, Lellouch J, Schwartz D: Smoking and leucocytecounts. Results of an epidemiological survey. Lancet. 1971, 2 (7725): 632-634.
Finkelstein EI, Nardini M, Vliet van der A: Inhibition of neutrophil apoptosis by acrolein: a mechanism of tobacco-related lung disease?. American Journal of Physiology. 2001, 281: L732-739.
Hughes DA, Haslam PL, Townsend PJ, Turner-Warwick M: Numerical and functional alteration in circulatory lymphocytes in cigarette smokers. Clinical and Experimental Immunology. 1985, 61: 459-466.
Pitzer JE, Del Zoppo GJ, Schmid-Schonbein GW: Neutrophil activation in smokers. Biorheology. 1996, 33: 45-58.
Schwartz J, Weiss ST: Cigarette smoking and peripheral blood leukocyte differentials. Annals of Epidemiology. 1994, 4: 236-242.
Tanigawa T, Araki S, Nakata A, Sakurai S: Increase in the helper inducer (CD4+CD29+) T lymphocytes in smokers. Industrial Health. 1998, 36: 78-81.
van Eeden SF, Hogg JC: The response of human bone marrow to chronic cigarette smoking. European Respiratory Journal. 2000, 15: 915-921.
Bahna SL, Heiner DC, Myhre BA: Changes in serum IgD in cigarette smokers. Clinical and Experimental Immunology. 1983, 51: 624-630.
Bahna SL, Heiner DC, Myrhe BA: Immunoglobulin E pattern in cigarette smokers. Journal of Allergy and Clinical Immunology. 1983, 38: 57-64.
Burrows B, Halonen M, Barbee RA, Lebowitx MD: The relationship of serum IgE to cigarette smoking. American Reviews in Respiratory Disease. 1981, 124: 523-525.
Gunsolley JC, Pandey JP, Quinn SM, Tew J, Schenkein HA: The effect of race, smoking and immunoglobulin allotypes on IgG subclass concentrations. Journal of Periodontal Research. 1997, 32: 381-387.
Geng Y, Savage SM, Johnson LJ, Seagrave J, Sopori ML: Effects of nicotine on the immune response. I. Chronic exposure to nicotine impairs antigen receptor-mediated signal transduction in lymphocytes. Toxicology and Applied Pharmacology. 1995, 135: 268-278.
Geng Y, Savage SM, Razani-Boroujerdi S, Sopori ML: Effects of nicotine on the immune response. II. Chronic nicotine treatment induces T cell anergy. Journal of Immunology. 1996, 156: 2384-2390.
Sopori ML, Kozak W: Immunomodulatory effects of cigarette smoke. Journal of Neuroimmunology. 1998, 83: 148-156.
Celermajer DS, Adams MR, Clarkson P, Robinson J, McCredie R, Donald A, Deanfield JE: Passive smoking and impaired endothelium-dependent arterial dilatation in healthy young adults. New England Journal of Medicine. 1996, 334: 150-154.
Ernst E: Haemorheological consequences of chronic cigarette smoking. Journal of Cardiovascular Risk. 1995, 2: 435-439.
Meekin TN, Wilson RF, Scott DA, Ide M, Palmer RM: Laser Doppler flowmeter measurement of relative gingival and forehead skin blood flow in light and heavy smokers during and after smoking. Journal of Clinical Periodontology. 2000, 27: 236-242.
Rezavandi K, Palmer RM, Odell EW, Scott DA, Wilson RF: Expression of E-Selectin and ICAM-1 in gingival tissues of smokers and non-smokers with periodontitis. Journal of Oral Pathology and Medicine. 2002, 31: 59-64.
Smith CJ, Fischer TH: Particulate and vapor phase constituents of cigarette mainstream smoke and risk of myocardial infarction. Atherosclerosis. 2001, 158: 257-267.
Koethe SM, Kuhnmuench JR, Becker CG: Neutrophil priming by cigarette smoke condensate and a tobacco anti-idiotypic antibody. American Journal of Pathology. 2000, 157: 1735-1743.
Zapolska-Downar D, Naruszewicz M, Zapolski-Downar A, Markiewski M, Bukowska H, Millo B: Ibuprofen inhibits adhesiveness of monocytes to endothelium and reduces cellular oxidative stress in smokers and non-smokers. European Journal of Clinical Investigation. 2000, 30: 1002-1010.
Nair S, Kulkarni S, Camoens HM, Ghosh K, Mohanty D: Changes in platelet glycoprotein receptors after smoking – a flow cytometric study. Platelets. 2001, 12: 20-26.
Smith CJ, Fischer TH: Particulate and vapor phase constituents of cigarette mainstream smoke and risk of myocardial infarction. Atherosclerosis. 2001, 158: 257-267.
Kuschner WG, D'Alessandro A, Wong H, Blanc PD: Dose-dependent cigarette smoking-related inflammatory responses in healthy adults. European Respiratory Journal. 1996, 9: 1989-1994.
Tappia PS, Troughton KL, Langley-Evans SC, Grimble RF: Cigarette smoking influences cytokine production and antioxidant defense. Clinical Science. 1995, 88: 485-489.
Barbour SE, Nakashima K, Zhang JB, Tangada S, Hahn CL, Schenkein HA, Tew JG: Tobacco and smoking: environmental factors that modify the host response (immune system) and have an impact on periodontal health. Critical Reviews in Oral Biology and Medicine. 1997, 8: 437-460.
Lehr HA: Microcirculatory dysfunction induced by cigarette smoking. Microcirculation. 2000, 7: 367-384.
Díaz-González F, Sánchez-Madrid F: Inhibition of leukocyte adhesion: an alternative mechanism of action for anti-inflammatory drugs. Immunology Today. 1998, 19: 169-172.
Gearing AJH, Newman W: Circulating adhesion molecules in disease. Immunology Today. 1994, 14: 506-512.
Johnson P, Maiti A, Brown KL, Li R: A role for the cell adhesion molecule CD44 and sulfation in leukocyte-endothelial cell adhesion during an inflammatory response?. Biochemical Pharmacology. 2000, 59: 455-465.
Johnson-Léger C, Aurraund-Lions M, Imhof BA: The parting of the endothelium: miracle, or simply a junctional affair?. Journal of Cell Science. 2000, 113: 921-933.
Muller WA, Weigl SA, Deng X, Phillips DM: PECAM-1 is required for transendothelial migration of leukocytes. Journal of Experimental Medicine. 1993, 178: 449-460.
Shen Y, Rattan V, Sultana C, Kalra VK: Cigarette smoke condensate-induced adhesion molecule expression and transendothelial migration of monocytes. American Journal of Physiology. 1996, 270: H1624-1633.
Vaporciyan AA, DeLisser HM, Yan HC, Mendiguren II, Thom SR, Jones ML, Ward PA, Albelda SM: Involvement of platelet-endothelial cell adhesion molecule-1 in neutrophil recruitment in vivo. Science. 1993, 262: 1580-1582.
Cuff CA, Kothapalli D, Azonobi I, Chun S, Zhang Y, Belkin R, Yeh C, Secreto A, Assoian RK, Rader DJ, Pure E: The adhesion receptor CD44 promotes atherosclerosis by mediating inflammatory cell recruitment and vascular cell activation. Journal of Clinical Investigation. 2001, 108: 1031-1040.
Etzioni A, Doerschuk CM, Harlan JM: Of mice and men: leukocyte and adhesion molecule deficiencies. Blood. 1999, 94: 3281-3288.
Hellewell PG: Adhesion molecule strategies. Pulmonary Pharmacology and Therapeutics. 1999, 12: 137-141.
Hubbard AK, Rothlein R: Intercellular adhesion molecule-1 (ICAM-1) expression and cell signaling cascades. Free Radical Biology and Medicine. 2000, 28: 1379-1386.
Prosper F, Verfaille CM: Regulation of hematopoiesis through adhesion receptors. Journal of Leukocyte Biology. 2001, 69: 307-316.
Salmi M, Jalkenen S: How do lymphocytes know where to go: current concepts and enigmas of lymphocyte homing. Advances in Immunology. 1997, 64: 139-218.
Shimizu Y, Rose DM, Ginsberg MH: Integrins in the immune system. American Journal of Physiology. 1999, 265: 325-380.
Steeber DA, Tedder TF: Adhesion molecule cascades direct lymphocyte recirculation and leukocyte migration during inflammation. Immunologic Research. 2001, 22: 299-317.
Simmons D, Makgoba MW, Seed B: ICAM, an adhesion ligand of LFA-1, is homologous to the neural cell adhesion molecule NCAM. Nature. 1988, 331: 624-627.
Staunton DE, Marlin SD, Stratowa C, Dustin ML, Springer TA: Primary structure of ICAM-1 demonstrates interaction between members of the immunoglobulin and integrin supergene families. Cell. 1988, 52: 925-933.
Budnik A, Grewe M, Gyufko K, Krutmann J: Analysis of the production of soluble ICAM-1 molecules by human cells. Experimental Hematology. 1996, 24: 352-359.
Cartwright JE, Whitley GS, Johnstone AP: The expression and release of adhesion molecules by human endothelial cell lines and their consequent binding of lymphocytes. Experimental Cell Research. 1995, 217: 329-335.
Hashimoto M, Shingu M, Ezaki I, Nobunaga M, Minamihara M, Kato K, Sumioki H: Production of soluble ICAM-1 from human endothelial cells induced by IL-1 beta and TNF-alpha. Inflammation. 1994, 18: 163-173.
Lyons PD, Benveniste EN: Cleavage of membrane-associated ICAM-1 from astrocytes: involvement of a metalloprotease. Glia. 1998, 22: 103-112.
Pigott R, Dillon LP, Hemingway IH, Gearing AJ: Soluble forms of E-selectin, ICAM-1 and VCAM-1 are present in the supernatants of cytokine activated cultured endothelial cells. Biochemical and Biophysical Research Communications. 1992, 187: 584-589.
Ohh M, Takei F: New insights into the regulation of ICAM-1 gene expression. Leukaemia and Lymphoma. 1996, 20: 223-228.
Stolpe Van de A, Saag Van der PT: Intercellular adhesion molecule-1. Journal of Molecular Medicine. 1996, 74: 13-33.
Bennett KL, Jackson DG, Simon JC, Tanczos E, Peach R, Modrell B, Stamenkovic I, Plowman G, Aruffo A: CD44 isoforms containing exon V3 are responsible for the presentation of heparin-binding growth factor. Journal of Cell Biology. 1995, 128: 687-698.
Bourguignon LY: CD44-mediated oncogenic signaling and cytoskeleton activation during mammary tumor progression. Journal of Mammary Gland Biology and Neoplasia. 2001, 6: 287-297.
Dohadwala M, Luo J, Zhu L, Lin Y, Dougherty GJ, Sharma S, Huang M, Pold M, Batra RK, Dubinett SM: Non-small cell lung cancer cyclooxygenase-2-dependent invasion is mediated by CD44. Journal of Biological Chemistry. 2001, 276: 20809-20812.
Lin YH, Yang-Yen HF: The osteopontin-Cd44 survival signal involves activation of the phosphatidylinositol-3-kinase/akt signaling pathway. Journal of Biological Chemistry. 2001, 276: 46024-46030.
Lisignoli G, Grassi F, Zini N, Toneguzzi S, Piacentini A, Guidolin D, Bevilacqua C, Facchini A: Anti-Fas-induced apoptosis in chondrocytes reduced by hyaluronan: evidence for CD44 and CD54 (intercellular adhesion molecule 1) involvement. Arthritis and Rheumatology. 2001, 44: 1800-1807.
Ponta H, Wainwright D, Herrlich P: The CD44 protein family. International Journal of Biochemistry and Cell Biology. 1998, 30: 299-305.
Screaton GR, Bell MV, Jackson DG, Cornelis FB, Gerth U, Bell JI: Genomic structure of DNA encoding the lymphocyte homing receptor CD44 reveals at least 12 alternatively spliced exons. Proceedings of the National Academy of Science USA. 1992, 89: 12160-12164.
Yasuda M, Tanaka Y, Fujii K, Yasumoto K: CD44 stimulation down-regulates Fas expression and Fas-mediated apoptosis of lung cancer cells. International Immunology. 2001, 13: 1309-1319.
Yu Q, Stamenkovic I: Localization of matrix metallproteinase 9 to the cell surface provides a mechanism for CD44-mediated tumor invasion. Genes and Development. 1999, 13: 35-48.
Lesley J, Hyman R, Kincade PW: CD44 and its interaction with extracellular matrix. Advances in Immunology. 1993, 54: 271-335.
Lesley J, Hyman R, English N, Catterall JB, Turner GA: CD44 in inflammation and metastasis. Glycoconjugate Journal. 1997, 14: 611-622.
Entwistle J, Hall CL, Turley EA: HA receptors: regulators of signalling to the cytoskeleton. Journal of Cellular Biochemistry. 1996, 61: 569-577.
Hayflick JS, Kilgannon P, Gallitin WM: The intercellular adhesion molecule (ICAM) family of proteins. New members and novel functions. Immunologic Research. 1998, 17: 313-327.
Henry SP, Templin MV, Gillett N, Rojko J, Levin AA: Correlation of toxicity and pharmacokinetic properties of a phosphorothioate oligonucleotide designed to inhibit ICAM-1. Toxicologic Pathology. 1999, 27: 95-100.
Roebuck KA, Finnegan A: Regulation of intercellular adhesion molecule-1 (CD54) gene expression. Journal of Leukocyte Biology. 1999, 66: 876-888.
Watanabe T, Fan J: Atherosclerosis and inflammation mononuclear cell recruitment and adhesion molecules with reference to the implication of ICAM-1/LFA-1 pathway in atherogenesis. International Journal of Cardiology. 1998, 66: S45-53.
Bajorath J: Molecular organization, structural features, and ligand binding characteristics of CD44, a highly variable cell surface glycoprotein with multiple functions. Proteins. 2000, 39: 103-111.
Goodison S, Urquidi V, Tarin D: CD44 cell adhesion molecules. Molecular Pathology. 1999, 52: 189-196.
Ilangumaran S, Borisch B, Hoessli DC: Signal transduction via CD44: role of plasma membrane microdomains. Leukaemia and Lymphoma. 1999, 35: 455-469.
Kincade PW, Zheng Z, Katoh S, Hanson L: The importance of the cellular environment to function of the CD44 matrix receptor. Current Opinion in Cell Biology. 1997, 635-642.
Naot D, Sionov RV, Ish-Shalom D: CD44: Structure, function and association with the malignant process. Advances in Cancer Research. 1997, 71: 241-319.
Pure E, Cuff CA: A crucial role for CD44 in inflammation. Trends in Molecular Medicine. 2001, 7: 213-221.
Wielenga VJM, Neut van der R, Offerhaus GJA, Pals ST: CD44 glycoproteins in colorectal cancer: expression, function, and prognostic value. Advances in Cancer Research. 2000, 77: 169-187.
Bosken CH, Doerschuk CM, English D, Hogg JC: Neutrophil kinetics during active cigarette smoking in rabbits. Journal of Applied Physiology. 1991, 71: 630-637.
Bosken CH, Hards J, Gatter K, Hogg JC: Characterization of the inflammatory reaction in the peripheral airways of cigarette smokers using immunocytochemistry. American Review of Respiratory Diseases. 1992, 145: 911-917.
Bridges AB, Hill A, Belch JJ: Cigarette smoking increases white blood cell aggregation in whole blood. Journal of the Royal Society of Medicine. 1993, 86: 139-140.
Dougan PS, Edwards JD, Zhan X, Wilde M, Agrawal DK: Cigarette smoking increases monocyte adherence to cultured endothelial cell monolayer. Biochemical and Biophysical Research Communications. 1994, 203: 929-934.
Hunninghake GW, Crystal RG: Cigarette smoking and lung destruction. Accumulation of neutrophils in the lungs of cigarette smokers. American Review of Respiratory Diseases. 1983, 128: 833-838.
Kalra VK, Ying Y, Deemer K, Natarajan R, Nadler JL, Coates TD: Mechanism of cigarette smoke condensate induced adhesion of human monocytes to cultured endothelial cells. Journal of Cell Physiology. 1994, 160: 154-162.
Kilburn KH, McKenzie W: Leukocyte recruitment to airways by cigarette smoke and particle phase in contrast to cytotoxicity of vapor. Science. 1975, 189: 634-637.
Klut ME, Doerschuk CM, Van Eeden SF, Burns AR, Hogg JC: Activation of neutrophils within pulmonary microvessels of rabbits exposed to cigarette smoke. American Journal of Respiratory Cell and Molecular Biology. 1993, 9: 82-89.
Lehr HA, Kress E, Menger MD, Friedl HP, Hubner C, Arfors KE, Messmer K: Cigarette smoke elicits leukocyte adhesion to endothelium in hamsters: inhibition by CuZn-SOD. Free Radical Biology and Medicine. 1993, 14: 573-581.
Lehr HA, Kress E, Menger MD: Involvement of 5-lipoxygenase products in cigarette smoke-induced leukocyte/endothelium interaction in hamsters. International Journal of Microcirculation: Clinical and Experimental. 1993, 12: 61-73.
MacNee W, Wiggs B, Belzberg AS, Hogg JC: The effect of cigarette smoking on neutrophil kinetics in human lungs. New England Journal of Medicine. 1989, 321: 924-928.
Ricevuti G, Mazzone A, Mazzucchelli I, Fossati G, Pasotti D, Cavigliano P, Rolandi L, Viarengo G, Rossi M, Notario A: Phagocyte activation in coronary artery disease. FEMS Microbiology and Immunology. 1992, 5: 271-278.
Rylander R: Pulmonary cell responses to inhaled cigarette smoke. Archives of Environmental Health. 1974, 29: 329-333.
Schaberg T, Lauer C, Lode H, Fischer J, Haller H: Increased number of alveolar macrophages expressing adhesion molecules of the leukocyte adhesion molecule family in smoking subjects. Association with cell-binding ability and superoxide anion production. American Review of Respiratory Diseases. 1992, 146: 1287-1293.
Weber C, Erl W, Weber K, Weber PC: Increased adhesiveness of isolated monocytes to endothelium is prevented by vitamin C intake in smokers. Circulation. 1996, 93: 1488-1492.
Terashima T, Klut ME, English D, Hards J, Hogg JC, van Eeden SF: Cigarette smoking causes sequestration of polymorphonuclear leukocytes released from the bone marrow in lung microvessels. American Journal of Respiratory Cell and Molecular Biology. 1999, 20: 171-177.
Costabel U, Guzman J: Effect of smoking on bronchoalveolar lavage constituents. European Respiratory Journal. 1992, 5: 776-779.
Hoogsteden HC, van Hal PT, Wijkhuijs JM, Hop W, Verkaik AP, Hilvering C: Expression of the CD11/CD18 cell surface adhesion glycoprotein family on alveolar macrophages in smokers and nonsmokers. Chest. 1991, 100: 1567-1571.
Schaberg T, Rau M, Oerter R, Liebers U, Rahn W, Kaiser D, Witt C, Lode H: Expression of adhesion molecules in peripheral pulmonary vessels from smokers and nonsmokers. Lung. 1996, 174: 71-81.
Lensmar C, Elmberger G, Skold M, Eklund A: Smoking alters the phenotype of macrophages in induced sputum. Respiratory Medicine. 1998, 92: 415-420.
Di Stefano A, Maestrelli P, Roggeri A, Turato G, Calabro S, Potena A, Mapp CE, Ciaccia A, Covacev L, Fabbri LM: Upregulation of adhesion molecules in the bronchial mucosa of subjects with chronic obstructive bronchitis. American Journal of Respiratory and Critical Care Medicine. 1994, 149: 803-810.
Hogg JC: Identifying smokers at risk for developing airway obstruction. Chest. 1998, 114: 355.
Maestrelli P, Calcagni PG, Saetta M, Bertin T, Mapp CE, Sanna A, Veriter C, Fabbri LM, Stanescu D: Integrin upregulation on sputum neutrophils in smokers with chronic airway obstruction. American Journal of Respiratory and Critical Care Medicine. 1996, 154: 1296-1300.
Noguera A, Batle S, Miralles C, Iglesias J, Busquets X, MacNee W, Agusti AGN: Enhanced neutrophil response in chronic obstructive pulmonary disease. Thorax. 2001, 56: 432-437.
Gonzalez S, Hards J, van Eeden S, Hogg JC: The expression of adhesion molecules in cigarette smoke-induced airways obstruction. European Respiratory Journal. 1996, 9: 1995-2001.
Takizawa H, Tanaka M, Takami K, Ohtoshi T, Ito K, Satoh M, Okada Y, Yamasawa F, Umeda A: Increased expression of inflammatory mediators in small-airway epithelium from tobacco smokers. American Journal of Physiology: Lung Cellular and Molecular Physiology. 2000, 278: L906-L913.
Rusznak C, Mills PR, Devalia JL, Sapsford RJ, Davies RJ, Lozewicz S: Effect of cigarette smoke on the permeability and IL-1beta and sICAM-1 release from cultured human bronchial epithelial cells of never-smokers, smokers, and patients with chronic obstructive pulmonary disease. American Journal of Respiratory Cell and Molecular Biology. 2000, 23: 530-536.
Adams MR, Jessup W, Celermajer DS: Cigarette smoking is associated with increased human monocyte adhesion to endothelial cells: reversibility with oral L-arginine but not vitamin C. Journal of the American College of Cardiology. 1997, 29: 491-497.
Kevil CG, Patel RP, Bullard DC: Essential role of ICAM-1 in mediating monocyte adhesion to aortic endothelial cells. American Journal of Physiology. 2001, 281: C1442-1447.
Russell MA, Jarvis M, Iyer R, Feyerabend C: Relation of nicotine yield of cigarettes to blood nicotine concentrations in smokers. British Medical Journal. 1980, 280: 972-976.
Scott DA, Todd DH, Wilson RF, Coward PY, Odell EW, Poston RN, Matthews JP, Palmer RM: The acute influence of tobacco smoking on adhesion molecule expression on monocytes and neutrophils and on circulating adhesion molecule levels in vivo. Addiction Biology. 2000, 5: 195-205.
Lopez AF, Williamson DJ, Gamble JR, Begley CG, Harlan JM, Klebanoff SJ, Waltersdorph A, Wong G, Clark SC, Vadas MA: Recombinant human granulocyte-macrophage colony-stimulating factor stimulates in vitro mature human neutrophil and eosinophil function, surface receptor expression, and survival. Journal of Clinical Investigation. 1986, 78: 1220-1228.
Spark JI, Scott DJA, Chetter IC, Guillou PJ, Kester RC: Does soluble intercellular adhesion molecule-1 (ICAM-1) affect neutrophil activation and adhesion following ischaemia-reperfusion?. European Journal of Vascular and Endovascular Surgery. 1999, 17: 115-120.
Bainton DF, Miller LJ, Kishimoto TK, Springer TA: Leukocyte adhesion receptors are stored in peroxidase-negative granules of human neutrophils. Journal of Experimental Medicine. 1987, 166: 1641-1653.
Miller LJ, Bainton DF, Borregaard N, Springer TA: Stimulated mobilization of monocyte Mac-1 and p150,95 adhesion proteins from an intracellular vesicular compartment to the cell surface. Journal of Clinical Investigation. 1987, 80: 535-544.
Ryder MI, Fujitaki R, Lebus S, Mahboub M, Faia B, Muhaimin D, Hamada M, Hyun W: Alterations of neutrophil L-selection and CD18 expression by tobacco smoke: implications for periodontal diseases. Journal of Periodontal Research. 1998, 33: 359-368.
Selby C, Drost E, Brown D, Howie S, MacNee W: Inhibition of neutrophil adherence and movement by acute cigarette smoke exposure. Experimental Lung Research. 1992, 18: 813-827.
Pernerstorfer T, Stohlawetz P, Stummvoll G, Kapiotis S, Szekeres T, Eichler HG, Jilma B: Low-dose aspirin does not lower in vivo platelet activation in healthy smokers. British Journal of Haematology. 1998, 102: 1229-1231.
Ismail AI, Bert BA, Ekland SA: Epidemiologic patterns of smoking and periodontal disease in the United States. Journal of the American Dental Association. 1983, 106: 617-621.
Machtei EE, Dunford R, Hausmann E, Grossi SG, Powell J, Cummins D, Zambon JJ, Genco RJ: Longitudinal study of prognostic factors in established periodontitis patients. Journal of Clinical Periodontology. 1997, 24: 102-109.
Scott DA, Palmer RM, Stapleton JA: Validation of smoking status in clinical research into inflammatory periodontal disease. Journal of Clinical Periodontology. 2001, 28: 715-22.
Tomar SL, Asma S: Smoking-attributable periodontitis in the United States: findings from NHANES III. National Health and Nutrition Examination Survey. Journal of Periodontology. 2000, 71: 743-751.
Kinane DF, Adonogianaki E, Moughal N, Winstanley FP, Mooney J, Thornhill M: Immunocytochemical characterization of cellular infiltrate, related endothelial changes and determination of GCF acute-phase proteins during human experimental gingivitis. Journal of Periodontal Research. 1991, 26: 286-288.
Koundouros E, Odell E, Coward PY, Wilson RF, Palmer RM: Soluble adhesion molecules in serum of smokers and non-smokers, with and without periodontitis. Journal of Periodontal Research. 1996, 31: 596-599.
Alavi AL, Palmer RM, Odell EW, Coward PY, Wilson RF: Elastase in gingival crevicular fluid from smokers and non-smokers with chronic inflammatory periodontal disease. Oral Diseases. 1995, 1: 103-105.
Pauletto NC, Liede K, Nieminen A, Larjava H, Uitto V-J: Effect of cigarette smoking on oral elastase activity in adult periodontitis patients. Journal of Periodontology. 2000, 71: 58-62.
Fors BP, Goodarzi K, von Andrian UH: L-selectin shedding is independent of its subsurface structures and topographic distribution. Journal of Immunology. 2001, 167: 3642-3651.
Hafezi-Moghadam A, Thomas KL, Prorock AJ, Huo Y, Ley K: L-selectin shedding regulates leukocyte recruitment. Journal of Experimental Medicine. 2001, 193: 863-872.
Bergmann S, Siekmeier R, Mix C, Jaross W: Even moderate cigarette smoking influences the pattern of circulating monocytes and the concentration of sICAM-1. Respiration Physiology. 1998, 114: 269-275.
Blann AD, Steele C, McCollum CN: The influence of smoking on soluble adhesion molecules and endothelial cell markers. Thrombosis Research. 1997, 85: 433-438.
Blann AD, Kirkpatrick U, Devine C, Naser S, McCollum CN: The influence of acute smoking on leucocytes, platelets and the endothelium. Atherosclerosis. 1998, 141: 133-139.
Fassbender K, Bertsch T, Mielke O, Muhlhauser F, Hennerici M: Adhesion molecules in cerebrovascular diseases. Evidence for an inflammatory endothelial activation in cerebral large-and small-vessel disease. Stroke. 1999, 30: 1647-1650.
Grothey A, Heistermann P, Philippou S, Voigtmann R: Serum levels of soluble intercellular adhesion molecule-1 (ICAM-1, CD54) in patients with non-small-cell lung cancer: correlation with histological expression of ICAM-1 and tumour stage. British Journal of Cancer. 1998, 77: 801-807.
Hwang SJ, Ballantyne CM, Sharrett AR, Smith LC, Davis CE, Gotto AM, Boerwinkle E: Circulating adhesion molecules VCAM-1, ICAM-1, and E-selectin in carotid atherosclerosis and incident coronary heart disease cases: the Atherosclerosis Risk Communities (ARIC) study. Circulation. 1997, 96: 4219-4225.
Noguchi T, Tsujisaki M, Imai K, Dodo M, Tabuchi Y, Nakajima T, Kajita A, Hayashi I, Sugiura T, Kumahara Y: Relationship among risk factors of atherosclerosis, leukocyte count, and soluble intercellular adhesion molecule-1. Internal Medicine. 1998, 37: 123-126.
O'Malley T, Ludlam CA, Riemermsa RA, Fox KA: Early increase in levels of soluble inter-cellular adhesion molecule-1 (sICAM-1). Potential risk factor for the acute coronary syndromes. European Heart Journal. 2001, 22: 1226-1234.
Rohde LE, Lee RT, Rivero J, Jamacochian M, Arroyo LH, Briggs W, Rifai N, Libby P, Creager MA, Ridker PM: Circulating cell adhesion molecules are correlated with ultrasound-based assessment of carotid atherosclerosis. Arteriosclerosis, Thrombosis and Vascular Biology. 1998, 18: 1765-1770.
Rohde LE, Hennekens CH, Ridker PM: Cross-sectional study of soluble intercellular adhesion molecule-1 and cardiovascular risk factors in apparently healthy men. Arteriosclerosis, Thrombosis and Vascular Biology. 1999, 19: 1595-1599.
van Tits LJ, de Waart F, Hak-Lemmers HL, van Heijst P, de Graaf J, Demacker PN, Stalenhoef AF: Effects of alpha-tocopherol on superoxide production and plasma intercellular adhesion molecule-1 and antibodies to oxidized LDL in chronic smokers. Free Radicals in Biology and Medicine. 2001, 30: 1122-1129.
Wallen NH, Held C, Rehnqvist N, Hjemdahl P: Elevated serum intercellular adhesion molecule-1 and vascular adhesion molecule-1 among patients with stable angina pectoris who suffer cardiovascular death or non-fatal myocardial infarction. European Heart Journal. 1999, 20: 1039-1043.
Zoppini G, Targher G, Cacciatori V, Guerriero A, Muggeo M: Chronic cigarette smoking is associated with increased plasma circulating intercellular adhesion molecule 1 levels in young type 1 diabetic patients. Diabetes Care. 1999, 22: 1871-1874.
Grigg J, Riedler J, Robertson CF: Soluble intercellular adhesion molecule-1 in the bronchoalveolar lavage fluid of normal children exposed to parental cigarette smoke. European Respiratory Journal. 1999, 13: 810-813.
Rumalla A, Herndon B, Pyszczynski D, Suvarna P: Does pulmonary airway inflammation relate to intercellular adhesion molecule (ICAM-1) in bronchoalveolar lavage specimens? A pilot study. Molecular Medicine. 1997, 94: 186-189.
Scott DA, Stapleton JA, Coward PY, Wilson RF, Sutherland G, Palmer RM, Gustavsson G: Dramatic decline in circulating intercellular adhesion molecule-1 concentration on quitting tobacco smoking. Blood Cells Molecules and Diseases. 2000, 26: 255-258.
Tonnesen P, Paoletti P, Gustavsson G, Russell MA, Saracci R, Gulsvik A, Rijcken B, Sawe U: Higher dosage nicotine patches increase one-year smoking cessation rates: results from the European CEASE trial. European Respiratory Journal. 1999, 13: 238-246.
Kittl EM, Ruckser R, Rech-Weichselbraun I, Hinterberger W, Bauer K: Significant elevation of tumour-associated isoforms of soluble CD44 in serum of normal individuals caused by cigarette smoking. European Journal of Clinical Chemistry and Clinical Biochemistry. 1997, 35: 81-84.
Scott DA, Stapleton JA, Wilson RF, Sutherland G, Palmer RM, Coward PY, Gustavsson G, Odell EW, Poston RN: Plasma concentrations of reputed tumor-associated soluble CD44 isoforms (v5 and v6) in smokers are dose-related and decline on smoking cessation. Cancer Epidemiology, Biomarkers and Prevention. 2000, 9: 1211-1214.
Blann AD, McCollum CN: Circulating endothelial cell/leukocyte adhesion molecules in atherosclerosis. Thrombosis and Haemostasis. 1994, 72: 151-154.
Altered levels of soluble adhesion molecules in rheumatoid arthritis, vasculitis and systemic sclerosis. British Journal of Rheumatology. 1995, 34: 814-819.
Ghaisis NK, Chandreshwar NS, Foley B, Goggins M, Crean P, Kelly A, Kelleher D, Walsh M: Elevated levels of circulating soluble adhesion molecules in peripheral blood of patients with unstable angina. American Journal of Cardiology. 1997, 80: 617-619.
Morisaki N, Saito I, Tamura K, Tashiro J, Masuda M, Kanzaki T, Watanabe S, Masuda Y, Saito Y: New indices of ischemic heart disease and aging: studies on the serum levels of soluble intercellular adhesion molecule-1 (ICAM-1) and soluble vascular cell adhesion molecule-1 (VCAM-1) in patients with hypercholesterolemia and ischemic heart disease. Atherosclerosis. 1997, 131: 43-48.
Ridker PM, Hennekens CH, Roitman-Johnson B, Stampfer MJ, Allen J: Plasma concentration of soluble intercellular adhesion molecule-1 and risks of future myocardial infarction in apparently healthy men. Lancet. 1998, 351: 88-92.
Shyu KG, Chang H, Lin CC, Kuan P: Circulating intercellular adhesion molecule-1 and E-selectin in patients with acute coronary syndrome. Chest. 1996, 109: 1627-1630.
Squadrito F, Saitta A, Altavilla D, Ioculano M, Canale P, Campo GM, Squadrito G, Di Tano G, Mazzu A, Caputi AP: Thrombolytic therapy with urokinase reduces increased circulating endothelial adhesion molecules in acute myocardial infarction. Inflammation Research. 1996, 45: 14-19.
Becker A, van Hinsbergh VWM, Jager A, Kostense PJ, Dekker JM, Nijpels G, Heine RJ, Bouter LM, Stehouwer CDA: Why is soluble intercellular adhesion molecule-1 related to cardiovascular mortality?. European Journal of Clinical Investigation. 2002, 32: 1-8.
Banks RE, Gearing AJH, Hemingway IK, Norfolk DR, Perren TJ, Selby PJ: Circulating intercellular adhesion molecule-1 (ICAM-1), E-selectin and vascular cell adhesion molecule-1 (VCAM-1) in human malignancies. British Journal of Cancer. 1993, 68: 122-124.
Steiner M, Reinhardt KM, Krammer B, Ernst B, Blann AD: Increased levels of soluble adhesion molecules in type 2 (non-insulin dependent) diabetes mellitus are independent of glycaemic control. Thombosis and Haemostasis. 1994, 72: 979-984.
De Rose V, Oliva A, Messore B, Grosso B, Mollar C, Pozzi E: Circulating adhesion molecules in cystic fibrosis. American Journal of Respiratory and Critical Care Medicine. 1998, 157: 1234-1239.
Goke M, Hoffmann JC, Evers J, Kruger H, Manns MP: Elevated serum concentrations of soluble selectin and immunoglobulin type adhesion molecules in patients with inflammatory bowel disease. Journal of Gastroenterology. 1997, 32: 480-486.
Kobayashi T, Hashimoto S, Isma K, Amemiya E, Yamaguchi M, Horie T: Elevation of serum soluble intercellular adhesion molecule-1 (sICAM-1) and sE-selectin levels in bronchial asthma. Clinical and Experimental Immunology. 1994, 96: 110-115.
Pforte A, Schiessler A, Gais P, von Kress S, Beer B, Riethmuller G, Ziegler-Heitbrock HW: Expression of the adhesion molecule ICAM-1 on alveolar macrophages and in serum in extrinsic allergic alveolitis. Respiration. 1993, 60: 221-226.
Shijubo N, Imai K, Shigehara K, Hirasawa M, Tsujisaki M, Hinoda Y, Abe S: Soluble intercellular adhesion molecule-1 (ICAM-1) in sera and bronchoalveolar lavage (BAL) fluids of extrinsic allergic alveolitis. Clinical and Experimental Immunology. 1995, 102: 91-97.
Blann AD, Seigneur M, Steiner M, Miller JP, McCollum CN: Circulating ICAM-1 and VCAM-1 in peripheral artery disease and hypercholesterolaemia: relationship to the location of atherosclerotic disease, smoking, and in the prediction of adverse events. Thrombosis and Haemostasis. 1998, 79: 1080-1085.
Rifai N, Joubran R, Yu H, Asmi M, Jouma M: Inflammatory markers in men with angiographically documented coronary heart disease. Clinical Chemistry. 1999, 45: 1967-1973.
Barnett CC, Moore EE, Moore FA, Carl VS, Biffl WL: Soluble ICAM-1 (sICAM-1) provokes PMN elastase release. Journal of Surgical Research. 1996, 63: 6-10.
Barnett CC, Moore EE, Moore FA, Biffl WL, Partrick DA: Soluble intercellullar adhesion molecule-1 provokes polymorphonuclear leukocyte elastase release by CD18. Surgery. 1996, 120: 395-402.
Barnett CC, Moore EE, Mierau GW, Partrick DA, Biffl WL, Elzi DJ, Silliman CC: ICAM-1-CD18 interaction mediates neutrophil cytotoxicity through protease release. American Journal of Physiology. 1998, 274: C1634-1644.
Lukacs NW, Chensue SW, Strieter RM, Warmington K, Kunkel SL: Inflammatory granuloma formation is mediated by TNF-alpha-inducible intercellular adhesion molecule-1. Journal of Immunology. 1994, 152: 5883-5889.
McCabe SM, Riddle L, Nakamura GR, Prashad H, Mehta A, Berman PW, Jardieu P: sICAM-1 enhances cytokine production stimulated by alloantigen. Cellular Immunology. 1993, 150: 364-375.
Otto VI, Heinzel-Pleines UE, Gloor SM, Trentz O, Kossmann T, Morganti-Kossmann MC: sICAM-1 and TNF-alpha induce MIP-2 with distinct kinetics in astrocytes and brain microvascular endothelial cells. Journal of Neuroscience Research. 2000, 60: 733-742.
Schmal H, Czermak BJ, Lentsch AB, Bless NM, Beck-Schimmer B, Friedl HP, Ward PA: Soluble ICAM-1 activates lung macrophages and enhances lung injury. Journal of Immunology. 1998, 161: 3685-3693.
Cobb RR, Dubins JS, Warner J, Molony L: Functional expression of soluble ICAM-1 by baculovirus-infected Sf9 cells. Biochemical and Biophysical Research Communications. 1992, 185: 1022-1033.
Kusterer K, Bojunga J, Enghofer M, Heidenthal E, Usadel KH, Kolb H, Martin S: Soluble ICAM-1 reduces leukocyte adhesion to vascular endothelium in ischemia-reperfusion injury in mice. American Journal of Physiology. 1998, 275: G377-80.
Meyer DM, Dustin ML, Carron CP: Characterization of intercellular adhesion molecule-1 ectodomain (sICAM-1) as an inhibitor of lymphocyte function-associated molecule-1 interaction with ICAM-1. Journal of Immunology. 1995, 155: 3578-3584.
Ohno N, Ichikawa H, Coe L, Kvietys PR, Granger DN, Alexander JS: Soluble selectins and ICAM-1 modulate neutrophil-endothelial adhesion and diapedesis in vitro. Inflammation. 1997, 21: 313-324.
Rieckmann P, Michel U, Albrecht M, Bruck W, Wockel L, Felgenhauer K: Soluble forms of intercellular adhesion molecule-1 (ICAM-1) block lymphocyte attachment to cerebral endothelial cells. Journal of Neuroimmunology. 1995, 60: 9-15.
Altomonte M, Gloghini A, Bertola G, Gasparollo A, Carbone A, Ferrone S, Maio M: Differential expression of cell adhesion molecules CD54/CD11a and CD58/CD2 by human melanoma cells and functional role in their interaction with cytotoxic cells. Cancer Research. 1993, 53: 3343-3348.
Becker JC, Dummer R, Hartmann AA, Burg G, Schmidt RE: Shedding of ICAM-1 from human melanoma cell lines induced by IFN-gamma and tumor necrosis factor-alpha. Functional consequences on cell-mediated cytotoxicity. Journal of Immunology. 1991, 147: 4398-4401.
Becker JC, Dummer R, Schmidt RE, Burg G, Hartmann AA: Shedding of soluble intercellular adhesion molecule 1 (ICAM-1) from melanoma cells and the effect on cellular cytotoxicity. Immunitat und Infektion. 1992, 20: 62-63.
Becker JC, Termeer C, Schmidt RE, Brocker EB: Soluble intercellular adhesion molecule-1 inhibits MHC-restricted specific T cell/tumor interaction. Journal of Immunology. 1993, 151: 7224-7232.
Bossu P, Singer GG, Andres P, Ettinger R, Marshak-Rothstein A, Abbas AK: Mature CD4+ T lymphocytes from MRL/lpr mice are resistant to receptor-mediated tolerance and apoptosis. Journal of Immunology. 1993, 151: 7233-7239.
Kaihara A, Iwagaki H, Gouchi A, Hizuta A, Isozaki H, Takakura N, Tanaka N: Soluble intercellular adhesion molecule-1 and natural killer cell activity in gastric cancer patients. Research Communications in Molecular Pathology and Pharmacology. 1998, 100: 283-300.
Gho YS, Kleinman HK, Sosne G: Angiogenic activity of human soluble intercellular adhesion molecule-1. Cancer Research. 1999, 59: 5128-5132.
Wei W, Kim Y, Boudreau N: Association of smoking with serum and dietary levels of antioxidants in adults: NHANES III, 1988–1994. American Journal of Public Health. 2001, 91: 258-264.
Jilma B, Blann AD, Stohlawetz P, Eichler HG, Kautzky-Willer A, Wagner OF: Dexamethasone lowers circulating E-selectin and ICAM-1 in healthy men. Journal of Laboratory and Clinical Medicine. 2000, 135: 270-274.
Rothlein R, Mainolfi EA, Czajkowski M, Marlin SD: A form of circulating ICAM-1 in human serum. Journal of Immunology. 1991, 147: 3788-3793.
Seth R, Raymond FD, Makgoba MW: Circulating ICAM-1 isoforms: diagnostic prospects for inflammatory and immune disorders. Lancet. 1991, 338: 83-84.
Inuzuka H, Seita T, Okamoto K, Iida K, Ogawa Y, Iwasa S: A sensitive ELISA for the characterization of two forms of circulating intercellular adhesion molecule-1 in human plasma. Biological and Pharmaceutical Bulletin. 1995, 18: 1036-1040.
Wakatsuki T, Kimura K, Kimura F, Shinomiya N, Ohtsubo M, Ishizawa M, Yamamoto M: A distinct mRNA encoding a soluble form of ICAM-1 molecule expressed in human tissues. Cell Adhesion and Communication. 1995, 3: 283-292.
Champagne B, Tremblay P, Cantin A, St Pierre Y: Proteolytic cleavage of ICAM-1 by human neutrophil elastase. Journal of Immunology. 1998, 161: 6398-6405.
Miller J, Knorr R, Ferrone M, Houdei R, Carron CP, Dustin ML: Intercellular adhesion molecule-1 dimerization and its consequences for adhesion mediated by lymphocyte function associated-1. Journal of Experimental Medicine. 1995, 182: 1231-1241.
Reilly PL, Woska JR, Jeanfavre DD, McNally E, Rothlein R, Bormann BJ: The native structure of intercellular adhesion molecule-1 (ICAM-1) is a dimer. Correlation with binding to LFA-1. Journal of Immunology. 1995, 155: 529-532.
Young DG, Jackson AM, James K: Purification and characterisation of soluble intercellular molecule-1 and its effects on cell-mediated cytolysis of bladder tumour cells. Biochemical Society Transactions. 1997, 25: 365S.
Carley W, Ligon G, Phan S, Dziuba J, Kelley K, Perry C, Gerritsen ME: Distinct ICAM-1 forms and expression pathways in synovial microvascular endothelial cells. Cellular and Molecular Biology. 1999, 45: 79-88.
Diamond MS, Staunton DE, Marlin SD, Springer TA: Binding of the integrin Mac-1 (CD11b/CD18) to the third immunoglobulin-like domain of ICAM-1 (CD54) and its regulation by glycosylation. Cell. 1991, 65: 961-971.
Fraser HS, Palmer RM, Wilson RF, Coward PY, Scott DA: Elevated systemic concentrations of soluble ICAM-1 are not reflected in the gingival crevicular fluid of smokers with periodontitis. Journal of Dental Research. 2001, 80: 1643-1647.
Scott DA, Coward PY, Wilson RF, Poston RN, Odell EW, Palmer RM: Serum concentration of total soluble CD44 is elevated in smokers. Biomarkers. 2000, 5: 240-254.
Guo YJ, Liu G, Wang X, Jin D, Wu M, Ma J, Sy MS: Potential use of soluble CD44 in serum as an indicator of tumour burden and metastasis in patients with gastric or colon cancer. Cancer Research. 1994, 65: 422-426.
Kopp R, Classen S, Wolf H, Gholam P, Possinger K, Wilmanns W: Predictive relevance of soluble CD44v6 serum levels for the responsiveness to second line hormone- or chemotherapy in patients with metastatic breast cancer. Anticancer Research. 2001, 21: 2995-3000.
Martin S, Jansen F, Bokelmann J, Kolb H: Soluble CD44 splice variants in metastasizing human breast cancer. International Journal of Cancer. 1997, 74: 443-445.
Ristamaki R, Joensuu H, Lappalainen K, Teerenhovi L, Jalkanen S: Elevated serum CD44 level is associated with unfavourable outcome in non-Hodgkin's lymphoma. Blood. 1997, 90: 4039-4045.
Ristamaki R, Joensuu H, Gron-Virta K, Salmi M, Jalkanen S: Origin and function of circulating CD44 in non-Hodgkin's lymphoma. Journal of Immunology. 1997, 158: 3000-3008.
Ristamaki R, Joensuu H, Jalkanen S: Serum CD44 in non-Hodgkin's Lymphoma. Leukemia and Lymphoma. 1999, 33: 433-440.
Sasaki K, Niitsu N: Elevated serum levels of soluble CD44 variant 6 are correlated with shorter survival in aggressive non-Hodgkin's lymphoma. European Journal of Haematology. 2000, 65: 195-202.
Yamane N, Tsujitani S, Makino M, Maeta M, Kaibara N: Soluble CD44 variant 6 as a prognostic indicator in patients with colorectal cancer. Oncology. 1999, 56: 232-238.
Haberhauer G, Kittl EM, Skoumal M, Hubl W, Wagner E, Bayer PM, Bauer K, Dunky A: Increased serum levels of soluble CD44-isoform v5 in rheumatic diseases are restricted to seropositive rheumatoid arthritis. Acta Medica Austriaca. 1997, 24: 23-25.
Kato S, Matsubara Y, Taniguchi Abe K, Yoshinaga R, Yamashiro S, Mukai H, Kadota J, Kawano S, Matsukura S: Evaluation of soluble CD44 in the BALF before and after treatment of DPB (diffuse panbronchitis) with macrolide antibiotics. Japanese Journal of Antibiotics. 1998, 51: S38-40.
Kittl EM, Haberhauer G, Ruckser R, Selleny S, Rech-Weichselbraun I, Hinterberger W, Bauer K: Serum levels of soluble CD44 variant isoforms are elevated in rheumatoid arthritis. Rheumatology International. 1997, 16: 181-186.
Bourguignon LYW, Gunja-Smith Z, Iida N, Zhu HB, Young LJT, Muller WJ, Cardiff RD: CD44v(3,8–10) is involved in cytoskeleton-mediated tumor cell migration and matrix metalloproteinase (MMP-9) association in metastatic breast cancer cells. Journal of Cellular Physiology. 1998, 176: 206-215.
Granberg D, Wilander E, Oberg K, Skogseid B: Prognostic markers in patients with typical bronchial carcinoid tumors. Journal of Clinical Endocrinology and Metabolism. 2000, 85: 3425-3430.
Gunthert U, Hoffman M, Rudy W, Reber S, Zoller M, Haussmann I, Matzku S, Wenzel A, Ponta H, Herrlich P: A new variant of glycoprotein CD44 confers metastatic potential to rat carcinoma cells. Cell. 1991, 65: 13-24.
Kuo MY-P, Cheng S-J, Chen H-M, Kok S-H, Hahn L-J, Chiang C-P: Expresion of CD44s, CD44v5, CD44v6 and CD44v7–8 in betel chewing-associated oral premalignant lesions and squamous cell carcinomas in Taiwan. Journal of Oral Pathology and Medicine. 1998, 27: 428-433.
Nguyen VN, Mirejovsky T, Melinova L, Mandys V: CD44 and its v6 spliced variant in lung carcinomas: relation to NCAM, CEA, EMA and UP1 and prognostic significance. Neoplasma. 2000, 47: 400-408.
Pirinen R, Hirvikoski P, Bohm J, Kellokoski J, Moisio K, Viren M, Johansson R, Hollmen S, Kosma VM: Reduced expression of CD44v3 variant isoform is associated with unfavorable outcome in non-small cell lung carcinoma. Human Pathology. 2000, 3: 1088-1095.
Ross JS, Sheehan CE, Williams SS, Malfetano JH, Szyfelbein WM, Kallakury BV: Decreased CD44 standard form expression correlates with prognostic variables in ovarian carcinomas. American Journal of Clinical Pathology. 2001, 116: 122-128.
Wimmel A, Kogan E, Ramaswamy A, Schuermann M: Variant expression of CD44 in preneoplastic lesions of the lung. Cancer. 2001, 92: 1231-1236.
Khaldoyanidi S, Sikora L, Orlovskaya I, Matrosova V, Kozlov V, Sriramarao P: Correlation between nicotine-induced inhibition of hematopoiesis and decreased CD44 expression on bone marrow stromal cells. Blood. 2001, 98: 303-312.
Dalchau R, Kirkley J, Fabre JW: Monoclonal antibody to a human brain-granulocyte-T lymphocyte antigen probably homologous to the W 3/13 antigen of the rat. European Journal of Immunology. 1980, 10: 745-749.
Haynes BF, Hale LP, Patton KL, Martin ME, McCallum RM: Measurement of an adhesion molecule as an indicator of inflammatory disease activity. Up-regulation of the receptor for hyaluronate (CD44) in rheumatoid arthritis. Arthritis and Rheumatology. 1991, 34: 1434-1441.
Chiu RK, Carpenito C, Dougherty ST, Hayes GM, Dougherty GJ: Identification and characterization of CD44RC, a novel alternatively spliced soluble CD44 isoform that can potentiate the hyaluronan binding activity of cell surface CD44. Neoplasia. 1999, 1: 446-452.
Skelton TP, Zeng C, Nocks A, Stamenkovic I: Glycosylation provides both stimulatory and inhibitory effects on cell surface and soluble CD44 binding to hyaluronan. Journal of Cell Biology. 1998, 140: 431-446.
Yu Q, Toole BP, Stamenkovic I: Induction of apoptosis of metastatic mammary carcinoma cells in vivo by disruption of tumor cell surface CD44 function. Journal of Experimental Medicine. 1997, 186: 1985-1996.
Peterson RM, Yu Q, Stamenkovic I, Toole BP: Perturbation of hyaluronan interactions by soluble CD44 inhibits growth of murine mammary carcinoma cells in ascites. American Journal of Pathology. 2000, 156: 2159-2167.
Ahrens T, Sleeman JP, Schempp CM, Howells N, Hofmann M, Ponta H, Herrlich P, Simon JC: Soluble CD44 inhibits melanoma tumor growth by blocking cell surface CD44 binding to hyaluronic acid. Oncogene. 2001, 20: 3399-3408.
Katoh S, McCarthy JB, Kincade PW: Characterization of soluble CD44 in the circulation of mice. Levels are affected by immune activity and tumor growth. Journal of Immunology. 1994, 153: 3440-3449.
Sy MS, Guo YJ, Stamenkovic I: Inhibition of tumor growth in vivo with a soluble CD44-immunoglobulin fusion protein. Journal of Experimental Medicine. 1992, 176: 623-627.
Zawadzki V, Perschl A, Rosel M, Hekele A, Zoller M: Blockade of metastasis formation by CD44-receptor globulin. International Journal of Cancer. 1998, 75: 919-924.
Thomas L, Byers HR, Vink J, Stamenkovic I: CD44H regulates tumor cell migration on hyaluronate-coated substrate. Journal of Cell Biology. 1992, 118: 971-977.
Peter K, Nawroth P, Conradt C, Nordt T, Weiss T, Boehme M, Wunsch A, Allenberg J, Kubler W, Bode C: Circulating vascular cell adhesion molecule-1 correlates with the extent of human atherosclerosis in contrast to circulating intercellular adhesion molecule-1, E-selectin, P-selectin, and thrombomodulin. Arteriosclerosis Thrombosis and Vascular Biology. 1997, 17: 505-512.
Osterud B, Elvevoll EO, Brox J, Anderssen T, Eliassen LT, Halvorsen H, Hogmo P, Kvernmo H, Lia K, Lund T, Olsen JO, Olsen RL, Engstad CS, Vognild E: Haemostatic parameters related to lipids and adhesion molecules. Blood Coagulation and Fibrinolysis. 1999, 10: 465-470.
Blann A, Morris J, McCollum C: Soluble L-selectin in peripheral arterial disease: relationship with soluble E-selectin and soluble P-selectin. Atherosclerosis. 1996, 126: 227-231.
Blann AD, Goode GK, Miller JP, McCollum CN: Soluble P-selectin in hyperlipidaemia with and without symptomatic vascular disease: relationship with von Willebrand factor. Blood Coagulation and Fibrinolysis. 1997, 8: 200-204.
Blann AD, Amiral J, McCollum CN: Prognostic value of increased soluble thrombomodulin and increased soluble E-selectin in ischaemic heart disease. European Journal of Haematology. 1997, 59: 115-120.
Circulating endothelial cell/leucocyte adhesion molecules in ischaemic heart disease. British Journal of Haematology. 1996, 95: 263-265.
Blann AD, Noteboom WM, Rosendaal FR: Increased soluble P-selectin levels following deep venous thrombosis: cause or effect?. British Journal of Haematology. 2000, 108: 191-193.
Blann AD, Faragher EB, McCollum CN: Increased soluble P-selectin following myocardial infarction: a new marker for the progression of atherosclerosis. Blood Coagulation and Fibrinolysis. 1997, 8: 383-390.
Blann AD, Seigneur M, Boisseau MR, Taberner DA, McCollum CN: Soluble P selectin in peripheral vascular disease: relationship to the location and extent of atherosclerotic disease and its risk factors. Blood Coagulation and Fibrinolysis. 1996, 7: 789-793.
Blann A, Morris J, McCollum C: Soluble L-selectin in peripheral arterial disease: relationship with soluble E-selectin and soluble P-selectin. Atherosclerosis. 1996, 126: 227-231.
Kitamura T, Tamada Y, Kato M, Yokochi T, Ikeya T: Soluble E-selectin as a marker of disease activity in pustulosis palmaris et plantaris. Acta Dermatologica Venereologica. 1999, 79: 462-464.
Acknowledgements
The authors would like to thank Dr. Andrew D. Blann of the Haemostasis, Thrombosis and Vascular Biology Unit, University Department of Medicine, City Hospital, Birmingham, UK, for his valuable input in the preparation of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Scott, D., Palmer, R. The influence of tobacco smoking on adhesion molecule profiles. Tob. Induced Dis. 1, 7 (2003). https://doi.org/10.1186/1617-9625-1-1-7
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1186/1617-9625-1-1-7