
Abstract
Hypertrophic cardiomyopathy (HCM) is the most common genetic disease of the heart. HCM is characterized by a wide range of clinical expression, ranging from asymptomatic mutation carriers to sudden cardiac death as the first manifestation of the disease. Over 1000 mutations have been identified, classically in genes encoding sarcomeric proteins. Noninvasive imaging is central to the diagnosis of HCM and cardiovascular magnetic resonance (CMR) is increasingly used to characterize morphologic, functional and tissue abnormalities associated with HCM. The purpose of this review is to provide an overview of the clinical, pathological and imaging features relevant to understanding the diagnosis of HCM. The early and overt phenotypic expression of disease that may be identified by CMR is reviewed. Diastolic dysfunction may be an early marker of the disease, present in mutation carriers prior to the development of left ventricular hypertrophy (LVH). Late gadolinium enhancement by CMR is present in approximately 60% of HCM patients with LVH and may provide novel information regarding risk stratification in HCM. It is likely that integrating genetic advances with enhanced phenotypic characterization of HCM with novel CMR techniques will importantly improve our understanding of this complex disease.
Similar content being viewed by others

Introduction
Hypertrophic cardiomyopathy (HCM) is the most common heritable cardiovascular disorder, with a prevalence of 1:500 in the general population. It is also the most common cause of sudden cardiac death (SCD) in young individuals and young athletes [1, 2]. HCM is an autosomal dominant disease caused by mutations in genes encoding sarcomere proteins [1]. Autosomal recessive, X-linked, and mitochondrial (matrilinear) patterns of inheritance also occur [1, 3]. The penetrance of left ventricular hypertrophy (LVH) is highly age-dependent and incomplete [4]. Although the most common expression of HCM is during adolescence [5, 6], patients may present at any point in life. Clinical screening of first-degree relatives is recommended annually during adolescence (age 12-18) [7]. Adult relatives of HCM affected individuals beyond age 18 may be screened at 5 year intervals.
The annual mortality rate of HCM ranges from < 1% in the general community to 3-6% in tertiary referral centers [1, 8]. Current risk factor stratification for SCD considers multiple factors, including nonsustained ventricular tachycardia, syncope, exercise blood pressure response, family history of sudden death, high risk genetic mutations as considered in individual patients [7, 9, 10]. On the imaging level, risk factors for SCD in HCM include left ventricular (LV) wall thickness and left ventricular outflow tract (LVOT) obstruction (Table 1).
The diagnosis of HCM is based largely on imaging features. Noninvasive diagnosis is based on left ventricular (LV) wall thickness ≥ 15 mm at end-diastole (frequently involving the interventricular septum) or septal to lateral wall thickness ratio higher than 1.3 in a non-dilated LV in the absence of a loading condition sufficient to cause the observed abnormality [5, 11, 12]. However, Maron et al., [1] have stated that virtually any LV wall thickness, even when within normal limits, can be consistent with the presence of an HCM-causing mutant gene. Further, only one major CMR manuscript has focused on establishing normal values for LV wall thickness using current steady state free precession (SSFP) pulse sequences at 1.5 T (Dawson, Dawson et al. Circ Cardiovasc Imaging 2011 [13]) in 60 men and 60 women Caucasian subjects. In that study, wall thickness was evaluated only on short axis views.
Recently there has been greatly increased use of cardiac magnetic resonance (CMR) for the diagnosis of HCM because of its precise determination of myocardial anatomy and the depiction of myocardial fibrosis. The purpose of this review is to provide the physician with a review of clinical, pathologic and genetic abnormalities relevant to understanding the potential role of CMR in HCM.
Pathologic basis of HCM
The diagnosis of HCM was historically made by pathologists only at the untimely death of an individual [14]. Advances in cardiovascular imaging and genetics have facilitated improved diagnosis allowing for deployment of interventions such as intracardiac defibrillators that can favorably alter the natural history of the disease in high risk patients. HCM in living individuals is seen by pathologists on septal myectomy, endomyocardial biopsy, left ventricular core from assist device placement, and cardiac explant specimens. Rarely is this the primary diagnostic modality for HCM.
Grossly, the defining feature of an HCM heart is increased LV mass and thickened wall, especially the interventricular septum [15]. Grossly-evident scattered fibrosis may or may not be present. In some cases, there is a predilection for fibrosis at the junction of the septum to the anterior and posterior walls of LV. The thickened septum often bulges into LV, diminishing the LV end diastolic lumen space. When prominent this septal bulge may dynamically block left ventricular outflow. When this obstruction occurs, anterior displacement of the papillary muscles/mitral leaflets occurs and the anterior leaflet of the mitral valve rubs the septal wall upon opening resulting in endocardial thickening, noted as a pearly white alteration along the outflow tract. These abnormalities may lead to altered hydrodynamic forces resulting in systolic anterior motion (SAM) of the mitral valve and mitral leaflet-septal contact with obstructive physiology [16]. Eventually, the valve leaflets and chordae tendineae become thickened by fibrosis [14, 17].
The key histologic feature of HCM is myocyte and myofibrillar disarray [15, 16]. Myocyte disarray is non-linear or haphazard arrangement of the myocytes observed by light microscopy. Typically, "herringbone" or "pinwheel" configurations are noted [15]. Various papers have suggested a need for a minimal amount of myocyte disarray (usually 5 or 10%) to make the diagnosis of HCM [18, 19]. This is because rare myocyte disarray can occur in other diseases or even in normal hearts where two muscle bundles come together [15].
In addition to disarray, three other non-specific findings are generally noted in HCM. One is obvious myocyte hypertrophy with nuclear enlargement, angulated nuclear borders and hyperchromasia. A second is a marked increase in both interstitial and replacement fibrosis that may be patchy of diffuse, mainly in the septal region [20]. The third is dysplasia of small arteries, seen as medial and intimal smooth muscle cell proliferation with luminal narrowing [21]. Small vessels are encased by dense perivascular collagen and also contain increased collagen within the media. Reduced arteriolar density may lead to small-vessel intramural coronary artery disease (SICAD) as an early manifestation of HCM [22]. This dysplasia is more prevalent in areas of replacement fibrosis. This suggests reduction in blood flow contributes to regional myocyte loss as a substrate for ventricular arrhythmia and subsequently SCD [1, 21]. Taken together, mid-septal myocyte disarray with myocyte hypertrophy, fibrosis, dysplastic small coronary arteries, and endocardial thickening are the histopathologic hallmarks of HCM.
Morphologic variants identified by noninvasive imaging
Multiple morphological variants of HCM have been described that can be identified by cardiac magnetic resonance (CMR). These are detailed below:
Asymmetric HCM with sigmoid septal contour ("septal HCM")
This is the most common morphologic presentation HCM, accounting for about two thirds of the spectrum [23, 24]. Hypertrophy of the anteroseptal myocardium results in sigmoidal contour of the septum [15, 25, 26]. As indicated above, these abnormalities may lead to altered hydrodynamic forces resulting in SAM of the mitral valve and mitral leaflet-septal contact with obstructive physiology [16]. This results in subaortic obstruction and a concomitant mitral regurgitant jet (Figure 1 B, b) directed posteriorly into the left atrium (LA) due to incomplete leaflet apposition. Peak instantaneous outflow tract gradients can be estimated using phase-contrast CMR [25, 27]. Anterior and posterior mitral valve leaflet length in HCM are greater than in control subjects by CMR (26 ± 5 versus 19 ± 5 mm, P < 0.001; and 14 ± 4 versus 10 ± 3 mm, P < 0.001, respectively). This anatomic feature has been postulated to be responsible for LVOT obstruction [28].

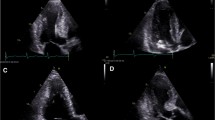
Left ventricular patterns in HCM, each drawing is accompanied by its corresponding image, (A, a) normal LV, (B, b) sigmoid septum showing SAM of mitral valve (white arrow), (C, c) reversed septal contour, note that there is no signs of LVOT, (D, d) mid ventricular hypertrophy, (E, e) Apical HCM, (F, f) symmetric HCM.
Asymmetric HCM with reversed septal contour
In this form, the septum hypertrophies as a reversed S-type curve (Figure 1 C, c) that does not cause LVOT obstruction [24, 29, 30]. LV mass may not be increased in patients with asymmetric HCM, especially with focal hypertrophy involving less than two LV segments [24, 31]. Subtle evidence of regional wall hypocontractility may be present despite a normal LV ejection fraction (EF). Systolic function declines as LVH becomes more severe [24, 30].
Asymmetric HCM is identified on short axis images characterized by a septal: free wall thickness ratio > 1.3 [15].
HCM with mid-ventricular obstruction (with or without a LV apical diverticulum)
In this variant, there is marked mid-ventricular hypertrophy (Figure 1 D, d) together with mid-cavity narrowing giving a 'dumb-bell' configuration of LV [27, 29]. In severe cases of narrowing, apical dilatation may occur. The apical dilatation is thought to result from the generation of increased systolic pressures within the cardiac apex due to mid-ventricular obstruction [15, 27].
In approximately 10% of patients, there is progression to a "burned out apex" with apical aneurysm formation and late gadolinium enhancement (LGE) after gadolinium administration [32]. This appearance is thought to result from ischemia resulting from reduced capillary density, hyperplasia of the arterial media, increased perivascular fibrosis and myocardial bridging [15, 33].
Apical HCM
In apical HCM, there is obliteration of LV cavity at the apex, giving a characteristic spade-like configuration of the cavity [34] seen on vertical long axis views (Figure 1 E, e). An apical wall thickness of > 15 mm or a ratio between apical and basal LV wall thicknesses ≥ 1.3-1.5 are characteristically present [35, 36]. This variant is more frequent in the Japanese population (~25% of HCM patients) compared to western countries (~2% of HCM patients) [24, 35].
CMR is particularly helpful in identifying apical HCM. In 10 patients with apical HCM by CMR (including one with LV wall thickness of 28 mm), echocardiograms were consistently negative [35]. Other comparisons of CMR to echocardiography for HCM diagnosis are shown in Table 2.
Symmetric (Concentric) HCM
In up to 42% of HCM cases, hypertrophy symmetrically involves the ventricular wall with no regional preferences (Figure 1 F, f). LV cavity dimensions are reduced in a concentric fashion [26, 29]. Symmetric LVH in the absence of hypertension or aortic stenosis may also be present in a variety of disorders, including amyloidosis, sarcoidosis and Fabry's disease. Hypertrophy in the athlete's heart is typically relatively modest, LV cavity size is normal to enlarged (as compared to the typically small LV cavity seen in HCM), and there is no evidence of diastolic dysfunction nor LGE. These features help to distinguish athlete's heart from symmetric HCM [8].
Focal HCM
Patients with HCM occasionally present with focal, mass-like thickening of LV. In these cases, CMR may help distinguish HCM from other cardiac masses using CMR tagging by spatial modulation of magnetization (SPAM)[37]. Tag lines show evidence of myocardial contractility in HCM, whereas neoplastic masses should not be contractile [38]. In addition, the signal intensities of tumor-related masses in the myocardium on spin echo CMR, first-pass perfusion and LGE-CMR are frequently different from normal myocardium [29]. The CMR characteristics of tumor-like HCM more closely resemble that of adjacent normal myocardium.
Right ventricular (RV) involvement in HCM
RV hypertrophy has been reported in approximately 18% of HCM patients [24, 29]. This typically involves the mid-to-apical portion of the RV. Severe involvement of the RV may result in RV outflow obstruction or reduced RV diastolic filling [15].
Genotype-phenotype correlations in HCM
Although several echocardiographic studies have evaluated the relationship between various morphologic subtypes and sarcomeric mutation [39, 40], such relationships are controversial and need further study. Sigmoidal HCM appears to be most common in the elderly and less likely to have an underlying sarcomere mutation.
In a cohort of 382 HCM patients, 73% of patients with sarcomeric HCM had reversed curve septum while only 10% had sigmoid septum. The incidence of SCD was found to be higher in specific MYH7 mutations [41]. Progression to heart failure was reported to be more commonly associated with mutations in MYH7 and TNNI3 than from other HCM mutations [42, 43]. One particular actin mutation (p.Glu101Lys) has been reported in at least 5 families with either apical hypertrophy or LV noncompaction [44, 45]. Approximately 3-5% of HCM patients may have multiple mutations (i.e., compound or digenic heterozygosity). These patients often have a more severe phenotype and increased incidence of SCD [46–49], suggesting a gene-dosage effect might contribute to disease severity. Most cases of compound heterozygosity involve MYBPC3 [46]. A recent report highlights the consequences of HCM resulting from 3 distinct mutations [49]. These investigators identified 4 such cases among 488 unrelated HCM probands (0.8%). Three of the 4 had severe disease that progressed to end-stage HCM by the fourth decade.
Increased left ventricular mass in HCM
LVH is widely regarded as a requirement for clinical diagnosis of HCM [30, 50, 51]. However, recent genotype-phenotype correlations have shown that virtually any wall thickness may be found in patients with a HCM gene mutation [24]. Greater CMR defined LV mass is associated with less favorable clinical outcome. This is attributed to the relationship between LVH and both the presence of LVOT obstruction and more advanced heart failure [52, 53]. Olivotto et al. [30] demonstrated that LV mass index is a more sensitive indicator for risk of death than peak wall thickness.
CMR provides better diagnostic accuracy than echocardiography in definition of the ventricular size, magnitude and distribution of hypertrophy, especially in identification of the anterolateral wall of LV [23, 35, 54]. Therefore, assessment of LV mass using CMR plays an important role in risk stratification of the disease.
Assessment of diastolic function
Diastolic dysfunction (DD) is an early functional marker of HCM. DD occurs secondary to abnormal dissociation of actin and myosin filaments during the active phase of relaxation in early diastolic filling [55, 56]. The assessment of diastolic function with CMR using myocardial tissue tagging has been previously reviewed [57]. Passive late diastole filling is also impaired due to increased interstitial fibrosis. Myocyte disarray may affect both ventricular relaxation and stiffness [55, 58]. Shirani et al., [33] suggested that the expanded and disorganized architecture of collagen matrix may contribute to DD. This results in decreased peak filling rate (PFR) and increased time to PFR [59, 60]. With diastole, prolongation of isovolumic relaxation is thought to be an early and sensitive marker of DD [61].
Tissue Doppler echocardiographic studies on genotyped subjects have demonstrated that sarcomere mutation carriers have diastolic abnormalities early in life, prior to the development of LVH. Impaired relaxation can be detected as decreased early myocardial relaxation velocities [62–64]. These studies indicate that diastolic abnormalities are an early, potentially, direct manifestation of the underlying sarcomere mutation, rather than simply a secondary consequence of altered myocardial compliance characteristics due to the hypertrophy, fibrosis and disarray that accompanies development of clinically overt disease.
Different CMR methods can be used to assess diastolic dysfunction. The first approach aims to parallel measures used by other imaging modalities [55, 56]. Ventricular time-volume curves can provide accurate assessment of global diastolic function. From these, PFR and time to PFR can be estimated [58]. PFR may be "normalized" relative to end-diastolic volume (PFR/EDV) and to stroke volume (PFR/SV). Unfortunately, PFR is only partially representative of compliance [56, 59]. Mitral inflow velocities (early filling "E" and atrial systolic filling" A") and pulmonary vein flow (systolic "S" and diastolic "D" velocities) derived from phase contrast CMR (PC-CMR) provide highly reproducible and accurate data. Velocity data can be used to calculate pressure gradients (ΔP) using the modified Bernoulli equation (ΔP = 4V2, where V = velocity) to classify DD [55]. Finally, myocardial motion velocity may be evaluated by PC-CMR. This approach can acquire images in multiple oblique planes, allowing the operator to choose in-plane or through-plane velocity acquisitions [55] with a high spatial and temporal resolution [65].
The second CMR approach to assess diastolic function is to directly measure myocardial diastolic relaxation. LV strain rate and torsion recovery rate are assessed using myocardial tissue tagging [56]. Tagged CMR can assess regional myocardial mechanics at different time-points during the cardiac cycle [66]. Reduced early diastolic strain rates are present in hypertrophied segments [67]. CMR tagging shows decreased relaxation rate and diminished early diastolic filling velocity as a result of delayed and prolongation of diastolic untwisting [66].
Assessment and quantification of myocardial fibrosis
Myocardial fibrosis or scaring detected by CMR occurs in up to 33-86% of patients with HCM [30, 53, 68, 69]. LGE-CMR characteristics are not specific for HCM, but the regional location of diffuse LGE within the septum is very suggestive of HCM [16].Table 3 shows incidence of LGE in multiple studies focused on HCM. The weighted mean reported prevalence of LGE, in 1814 LVH positive HCM patients, is 65% (Table 3).
The prognostic significance of the presence of LGE in HCM to adverse outcome is high. The presence of LGE in HCM patients has been associated with sudden cardiac death, systolic dysfunction and nonsustained ventricular tachycardia [69–73]. Table 4 lists studies that have evaluated the relationship between presence of LGE and various outcome parameters. Although the extent (rather than the presence) of fibrosis also was a predictor of major arrhythmic events, the degree of fibrosis has not yet been a significant predictor of events in multivariate analysis [73], possible due to small number of study subjects with events. Pathologically, fibrotic tissue is thought to be associated with re-entrant ventricular arrhythmia as well as myocardial dysfunction. The significance of LGE appears high, and larger, multi-center longitudinal trials to assess its prognostic significance seem warranted at this time.
Multiple factors have been proposed in the etiology of myocardial fibrosis in HCM patients, although the true origin has not yet been determined. Moon et al., [74] found greater collagen deposition correlated directly with LGE of the myocardium. Ho et al., [75] studied the PICP: CITP ratio that reflects the balance between collagen synthesis and degradation. They found a significantly higher PICP: CITP ratio in subjects with overt HCM compared to mutation carriers (without LVH) and the control group. The authors hypothesized that in overt HCM, collagen synthesis exceeds degradation, resulting in frank myocardial fibrosis.
Alternatively, LVH and dynamic LVOT pressure gradients may result in potential pressure necrosis causing small-vessel intramural coronary artery disease (SICAD) [21]. Ischemia may result from microvascular disease; increased end diastolic pressure together with the increased demand of LVH might initiate the processes of myocyte death and replacement fibrosis as a repair process [76].
Stress perfusion CMR may be performed in same setting as a LGE CMR study [77, 78]. The severity of myocardial perfusion defects correlate with areas of maximal wall thickness and the presence of CMR defined fibrosis in patients with HCM [79]. These studies suggest that microvascular abnormalities precede and predispose to the development of myocardial fibrosis. When present, perfusion abnormalities may represent an early risk marker and a possible therapeutic target [80].
Patterns of LGE in HCM
LGE in HCM characteristically appears as small punctuate, patchy mid-wall hyper-enhancement [29, 71]. There is a predilection to occur at the anterior and posterior RV insertion points (Figure 2). These RV insertion points represent plexiform fibrosis containing the crossing-fibers of LV and RV [80–82], although this pattern is not specific for HCM (e.g. patients with right ventricular hypertrophy also may demonstrate LGE at these sites). The interventricular septum is commonly involved by LGE, particularly the anteroseptal mid to basal segments (Figure 2). These segments are also the most commonly thickened segments, especially in patients with asymmetric HCM [81, 82]. Other foci of LGE are described as noncoronary in distribution, tending to occur in the hypertrophied regions (Figure 3) [29, 71]. An exception to this is areas of burned out HCM where the LV wall is typically thinned and full thickness LGE is present [83].

Late gadolinium enhancement patterns involving the anterior and posterior RV insertion points, as shown; the interventricular septum is involved, particularly the anteroseptal basal segment (three arrow heads).

Patchy mid wall, variable sized foci of hyperenhancement in a non-coronary distribution, involving mainly the hypertrophied parts (arrow heads). Right ventricle is also hypertrophied (large arrows).
In a cohort of 202 HCM patients; Maron et al., [84] showed that LGE was most commonly located in both the ventricular septum and LV free wall. Less frequently, LGE was confined to the LV free wall, followed by the septal LGE. The least common sites were the RV insertion into the ventricular septum followed by the LV apex. In patients without LVH, it is unusual to find LGE by CMR. This suggests that LGE occurs after development of LVH [85].
The pattern of LGE may be useful in distinguishing HCM from Anderson-Fabry disease [86, 87]. Anderson-Fabry disease is an x-linked disorder of sphingolipid metabolism associated with LVH. In one series, 50% of patients had LGE [86]. The most common location of LGE was in the basal inferolateral wall (12/13 patients) in a non ischemic pattern. The basal inferolateral wall is an infrequent location of isolated LGE for HCM.
Functional implications of late gadolinium enhancement
The presence and extent of LGE in relation to impaired ejection fraction (EF) is unclear. Some investigators found no significant relationship [76], but others have shown percent of LV involvement by LGE inversely correlates with LV function [84]. LGE may be associated with increased myocardial stiffness and LV adverse remodeling leading to cavity dilatation and eventually systolic dysfunction [72]. A larger extent of LGE is associated with higher incidence of regional wall motion abnormalities [35, 74]. In the presence of LVOT obstruction, there was no significant relationship between extent of LGE and magnitude of LV outflow gradient at rest [76, 84].
Quantification of diffuse myocardial fibrosis
High-resolution T1 mapping is an emerging technique for detection and quantification of subtle myocardial fibrosis. Abnormal T1 longitudinal relaxation may be useful to quantify the pathologic state of the tissue [88, 89]. T1 times are not specific for individual tissues, but each tissue has a normal range that varies by field strength [88, 90]. In patients with heart failure, myocardial segments with diffuse fibrosis showed shorter T1 time compared to segments with no LGE [91]. This technique may have potential for evaluation of HCM patients.
Following gadolinium administration in patients with HCM, Amano et al. [92] confirmed that T1 times of hyperenhanced regions of myocardium were lower than remote regions in the same patients. However, the remote regions without visually detected hyperenhancement had altered T1 time compared to a normal control population. This study suggests that T1 mapping may have a role in quantitative characterization of myocardial tissue in HCM. Since the method may detect diffuse fibrosis that is not visually detected, T1 mapping may be applicable to subclinical HCM, discussed further below.
T2 weighted sequences in HCM
High T2 signal intensity has been reported in HCM patients, frequently corresponding to LGE regions in HCM cases [93]. High T2 signal related to edema may accompany focal ischemia due to SICAD [94, 95]. The spatial relation between high T2 signal intensity and LGE could be explained by the pathologic progression of fibrosis: this may start with acute ischemia or inflammation (high T2 signal), and ending with mature chronic fibrous tissue (no obvious T2 abnormalities) [93, 96].
Other CMR applications in HCM
Myocardial disarray can be quantified with diffusion CMR [97], either by measuring the myocardial diffusion anisotropy, which is directly correlated with myocardial disarray, or by measuring myofibril orientations [98]. Tseng, et al. [99] found significant reduction of diffusion anisotropy in the hypertrophied septum due to an increase in the longitudinally oriented fibers. Rippplinger et al. evaluated a transgenic rabbit model of human hypertrophic cardiomyopathy. They reported that myocardial fiber anisotropy was similar in the wild type compared to the transgenic animal models [100]. Limitations of diffusion CMR include limited spatial resolution, motion artifact and long imaging times.
CMR spectroscopy can be used to measure the ratio of PCr/ATP that reflects the energy status of the myocardial tissue. Decreased PCr/ATP was associated with the extent of hypertrophy and disturbed diastolic function in both symptomatic [101] and asymptomatic patients [102]. Shivu et al. showed PCr/ATP ratio was significantly reduced in HCM patients compared to controls [103]. Perhexiline, a modulator of substrate metabolism, was shown to significantly increase PCr/ATP ratios in HCM patients [104]. Current limitations of CMR spectroscopy including low spatial resolution, long scan time and special coils.
Role of CMR in early detection of negative phenotypes (pre-clinical HCM)
For the reason that SCD may be the first symptom of HCM [105], there is substantial interest in early identification of HCM mutation carriers who may be at risk for life-threatening events [72, 82]. Patients with pre-clinical HCM are negative for LVH, but are genetically positive for the disease. Because CMR accurately presents both anatomic and functional characteristics of HCM, its role in pre-clinical HCM is potentially relevant. To date, most imaging studies of pre-clinical HCM have been based on echocardiography.
Germans et al. [6] evaluated asymptomatic HCM carriers by CMR. They described LV crypts, best visualized at end-diastole, even when LV wall thickness was normal. However, Maron et al.[106] also described a deep LV crypt penetrating 12 mm into a hypertrophied basal posterior portion of ventricular septum of a patient with overt HCM. The significance of LV crypts in HCM remains unclear.
Diastolic function assessed by echocardiography has been reported to be abnormal in pre-clinical HCM [107]. This finding has not been studied by CMR. In addition, tissue characterization by MR spectroscopy or T1 mapping is yet to be explored in preclinical HCM.
Conclusions
CMR appears to be highly relevant in the clinical as well as research evaluation of patients with overt as well as pre-clinical HCM. Late enhancement after gadolinium administration allows tissue characterization of myocardial fibrosis. The method may potentially identify HCM patients at greatest risk for adverse cardiac events. CMR evaluation of HCM mutation carriers in an early stage of disease has yet to be extensively evaluated, but represents a promising method for exploring the inter-relationship between functional, morphologic and tissue abnormalities in HCM.
Abbreviations
- AS:
-
atrial systole
- BRISK:
-
block regional interpolation scheme of k-space
- CITP:
-
C-terminal telopeptide of type I collagen CMR cardiovascular magnetic resonance imaging
- CV:
-
cardiovascular
- CVA:
-
cardiovascular accident
- DD:
-
diastolic dysfunction
- LGE:
-
late gadolinium enhancement
- LGE CMR:
-
late gadolinium enhancement cardiovascular magnetic resonance
- E:
-
early filling
- EF:
-
ejection fraction
- HCM:
-
hypertrophic cardiomyopathy
- HF:
-
heart failure
- ICD:
-
implantable cardioverter-defibrillator
- LA:
-
left atrium
- LV:
-
left ventricle
- LVH:
-
left ventricular hypertrophy
- LVOT:
-
left ventricular outflow tract
- PC:
-
phase contrast
- PFR:
-
peak filling rate
- PICP:
-
C-terminal propeptide of type I procollagen
- RV:
-
right ventricle
- SAM:
-
systolic anterior motion
- SCD:
-
sudden cardiac death
- SERCA:
-
sarcoplasmic reticulum Ca2+ ATPase
- SICAD:
-
small-vessel intramural coronary artery disease
- VF:
-
ventricular fibrillation
- VT:
-
ventricular tachycardia.
References
Maron BJ: Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002, 287: 1308-1320. 10.1001/jama.287.10.1308.
O'Hanlon R, Pennell DJ: Cardiovascular magnetic resonance in the evaluation of hypertrophic and infiltrative cardiomyopathies. Heart Fail Clin. 2009, 5: 369-387. 10.1016/j.hfc.2009.02.003. vi
Judge DP, Johnson NM: Genetic evaluation of familial cardiomyopathy. J Cardiovasc Transl Res. 2008, 1: 144-154. 10.1007/s12265-008-9025-1.
Charron P, Carrier L, Dubourg O, Tesson F, Desnos M, Richard P, Bonne G, Guicheney P, Hainque B, Bouhour JB: Penetrance of familial hypertrophic cardiomyopathy. Genet Couns. 1997, 8: 107-114.
Maron BJ, Towbin JA, Thiene G, Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE, Young JB: Contemporary definitions and classification of the cardiomyopathies: an American Heart Association Scientific Statement from the Council on Clinical Cardiology, Heart Failure and Transplantation Committee; Quality of Care and Outcomes Research and Functional Genomics and Translational Biology Interdisciplinary Working Groups; and Council on Epidemiology and Prevention. Circulation. 2006, 113: 1807-1816. 10.1161/CIRCULATIONAHA.106.174287.
Germans T, Wilde AA, Dijkmans PA, Chai W, Kamp O, Pinto YM, van Rossum AC: Structural abnormalities of the inferoseptal left ventricular wall detected by cardiac magnetic resonance imaging in carriers of hypertrophic cardiomyopathy mutations. J Am Coll Cardiol. 2006, 48: 2518-2523. 10.1016/j.jacc.2006.08.036.
Maron BJ, McKenna WJ, Danielson GK, Kappenberger LJ, Kuhn HJ, Seidman CE, Shah PM, Spencer WH, Spirito P, Ten Cate FJ, Wigle ED: American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003, 42: 1687-1713. 10.1016/S0735-1097(03)00941-0.
Towbin JA: Hypertrophic cardiomyopathy. Pacing Clin Electrophysiol. 2009, 32 (Suppl 2): S23-31.
Elliott PM, Poloniecki J, Dickie S, Sharma S, Monserrat L, Varnava A, Mahon NG, McKenna WJ: Sudden death in hypertrophic cardiomyopathy: identification of high risk patients. J Am Coll Cardiol. 2000, 36: 2212-2218. 10.1016/S0735-1097(00)01003-2.
Spirito P, Seidman CE, McKenna WJ, Maron BJ: The management of hypertrophic cardiomyopathy. N Engl J Med. 1997, 336: 775-785. 10.1056/NEJM199703133361107.
Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kuhl U, Maisch B, McKenna WJ: Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008, 29: 270-276.
Rose AG: Evaluation of pathological criteria for diagnosis of hypertrophic cardiomyopathy. Histopathology. 1984, 8: 395-406. 10.1111/j.1365-2559.1984.tb02352.x.
Dawson DK, Maceira AM, Raj VJ, Graham C, Pennell DJ, Kilner PJ: Regional thicknesses and thickening of compacted and trabeculated myocardial layers of the normal left ventricle studied by cardiovascular magnetic resonance. Circ Cardiovasc Imaging. 2011, 4: 139-146. 10.1161/CIRCIMAGING.110.960229.
Teare D: Asymmetrical hypertrophy of the heart in young adults. Br Heart J. 1958, 20: 1-8. 10.1136/hrt.20.1.1.
Hughes SE: The pathology of hypertrophic cardiomyopathy. Histopathology. 2004, 44: 412-427. 10.1111/j.1365-2559.2004.01835.x.
Davies MJ, McKenna WJ: Hypertrophic cardiomyopathy-pathology and pathogenesis. Histopathology. 1995, 26: 493-500. 10.1111/j.1365-2559.1995.tb00267.x.
Klues HG, Maron BJ, Dollar AL, Roberts WC: Diversity of structural mitral valve alterations in hypertrophic cardiomyopathy. Circulation. 1992, 85: 1651-1660.
Maron BJ, Roberts WC: Quantitative analysis of cardiac muscle cell disorganization in the ventricular septum of patients with hypertrophic cardiomyopathy. Circulation. 1979, 59: 689-706.
Davies MJ: The current status of myocardial disarray in hypertrophic cardiomyopathy. Br Heart J. 1984, 51: 361-363. 10.1136/hrt.51.4.361.
Factor SM, Butany J, Sole MJ, Wigle ED, Williams WC, Rojkind M: Pathologic fibrosis and matrix connective tissue in the subaortic myocardium of patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 1991, 17: 1343-1351. 10.1016/S0735-1097(10)80145-7.
Maron BJ, Wolfson JK, Epstein SE, Roberts WC: Intramural ("small vessel") coronary artery disease in hypertrophic cardiomyopathy. J Am Coll Cardiol. 1986, 8: 545-557. 10.1016/S0735-1097(86)80181-4.
Takemura G, Takatsu Y, Fujiwara H: Luminal narrowing of coronary capillaries in human hypertrophic hearts: an ultrastructural morphometrical study using endomyocardial biopsy specimens. Heart. 1998, 79: 78-85.
Rickers C, Wilke NM, Jerosch-Herold M, Casey SA, Panse P, Panse N, Weil J, Zenovich AG, Maron BJ: Utility of cardiac magnetic resonance imaging in the diagnosis of hypertrophic cardiomyopathy. Circulation. 2005, 112: 855-861. 10.1161/CIRCULATIONAHA.104.507723.
Maron MS, Maron BJ, Harrigan C, Buros J, Gibson CM, Olivotto I, Biller L, Lesser JR, Udelson JE, Manning WJ, Appelbaum E: Hypertrophic cardiomyopathy phenotype revisited after 50 years with cardiovascular magnetic resonance. J Am Coll Cardiol. 2009, 54: 220-228. 10.1016/j.jacc.2009.05.006.
Elliott P, McKenna WJ: Hypertrophic cardiomyopathy. Lancet. 2004, 363: 1881-1891. 10.1016/S0140-6736(04)16358-7.
Cannavale A, Ordovas KG, Higgins CB: Magnetic Resonance Imaging of Hypertrophic Cardiomyopathy. J Thorac Imaging. 2010
Wigle ED: Cardiomyopathy: The diagnosis of hypertrophic cardiomyopathy. Heart. 2001, 86: 709-714. 10.1136/heart.86.6.709.
Maron MS, Olivotto I, Harrigan C, Appelbaum E, Gibson CM, Lesser JR, Haas TS, Udelson JE, Manning WJ, Maron BJ: Mitral valve abnormalities identified by cardiovascular magnetic resonance represent a primary phenotypic expression of hypertrophic cardiomyopathy. Circulation. 2011, 124: 40-47. 10.1161/CIRCULATIONAHA.110.985812.
Hansen MW, Merchant N: MRI of hypertrophic cardiomyopathy: part I, MRI appearances. AJR Am J Roentgenol. 2007, 189: 1335-1343. 10.2214/AJR.07.2286.
Olivotto I, Maron MS, Autore C, Lesser JR, Rega L, Casolo G, De Santis M, Quarta G, Nistri S, Cecchi F: Assessment and significance of left ventricular mass by cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2008, 52: 559-566. 10.1016/j.jacc.2008.04.047.
Seidman JG, Seidman C: The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell. 2001, 104: 557-567. 10.1016/S0092-8674(01)00242-2.
Spirito P, Maron BJ, Bonow RO, Epstein SE: Occurrence and significance of progressive left ventricular wall thinning and relative cavity dilatation in hypertrophic cardiomyopathy. Am J Cardiol. 1987, 60: 123-129. 10.1016/0002-9149(87)90998-2.
Shirani J, Pick R, Roberts WC, Maron BJ: Morphology and significance of the left ventricular collagen network in young patients with hypertrophic cardiomyopathy and sudden cardiac death. J Am Coll Cardiol. 2000, 35: 36-44. 10.1016/S0735-1097(99)00492-1.
Sperling RT, Parker JA, Manning WJ, Danias PG: Apical hypertrophic cardiomyopathy: clinical, electrocardiographic, scintigraphic, echocardiographic, and magnetic resonance imaging findings of a case. J Cardiovasc Magn Reson. 2002, 4: 291-295. 10.1081/JCMR-120003956.
Moon JC, Fisher NG, McKenna WJ, Pennell DJ: Detection of apical hypertrophic cardiomyopathy by cardiovascular magnetic resonance in patients with non-diagnostic echocardiography. Heart. 2004, 90: 645-649. 10.1136/hrt.2003.014969.
Suzuki J, Shimamoto R, Nishikawa J, Yamazaki T, Tsuji T, Nakamura F, Shin WS, Nakajima T, Toyo-Oka T, Ohotomo K: Morphological onset and early diagnosis in apical hypertrophic cardiomyopathy: a long term analysis with nuclear magnetic resonance imaging. J Am Coll Cardiol. 1999, 33: 146-151. 10.1016/S0735-1097(98)00527-0.
Osman NF, Kerwin WS, McVeigh ER, Prince JL: Cardiac motion tracking using CINE harmonic phase (HARP) magnetic resonance imaging. Magn Reson Med. 1999, 42: 1048-1060. 10.1002/(SICI)1522-2594(199912)42:6<1048::AID-MRM9>3.0.CO;2-M.
Bergey PD, Axel L: Focal hypertrophic cardiomyopathy simulating a mass: MR tagging for correct diagnosis. AJR Am J Roentgenol. 2000, 174: 242-244.
Lever HM, Karam RF, Currie PJ, Healy BP: Hypertrophic cardiomyopathy in the elderly. Distinctions from the young based on cardiac shape. Circulation. 1989, 79: 580-589. 10.1161/01.CIR.79.3.580.
Solomon SD, Wolff S, Watkins H, Ridker PM, Come P, McKenna WJ, Seidman CE, Lee RT: Left ventricular hypertrophy and morphology in familial hypertrophic cardiomyopathy associated with mutations of the beta-myosin heavy chain gene. J Am Coll Cardiol. 1993, 22: 498-505. 10.1016/0735-1097(93)90055-6.
Ackerman MJ, VanDriest SL, Ommen SR, Will ML, Nishimura RA, Tajik AJ, Gersh BJ: Prevalence and age-dependence of malignant mutations in the beta-myosin heavy chain and troponin T genes in hypertrophic cardiomyopathy: a comprehensive outpatient perspective. J Am Coll Cardiol. 2002, 39: 2042-2048. 10.1016/S0735-1097(02)01900-9.
Kokado H, Shimizu M, Yoshio H, Ino H, Okeie K, Emoto Y, Matsuyama T, Yamaguchi M, Yasuda T, Fujino N: Clinical features of hypertrophic cardiomyopathy caused by a Lys183 deletion mutation in the cardiac troponin I gene. Circulation. 2000, 102: 663-669.
Dhandapany PS, Sadayappan S, Xue Y, Powell GT, Rani DS, Nallari P, Rai TS, Khullar M, Soares P, Bahl A: A common MYBPC3 (cardiac myosin binding protein C) variant associated with cardiomyopathies in South Asia. Nat Genet. 2009, 41: 187-191. 10.1038/ng.309.
Arad M, Penas-Lado M, Monserrat L, Maron BJ, Sherrid M, Ho CY, Barr S, Karim A, Olson TM, Kamisago M: Gene mutations in apical hypertrophic cardiomyopathy. Circulation. 2005, 112: 2805-2811. 10.1161/CIRCULATIONAHA.105.547448.
Monserrat L, Hermida-Prieto M, Fernandez X, Rodriguez I, Dumont C, Cazon L, Cuesta MG, Gonzalez-Juanatey C, Peteiro J, Alvarez N: Mutation in the alpha-cardiac actin gene associated with apical hypertrophic cardiomyopathy, left ventricular non-compaction, and septal defects. Eur Heart J. 2007, 28: 1953-1961. 10.1093/eurheartj/ehm239.
Van Driest SL, Vasile VC, Ommen SR, Will ML, Tajik AJ, Gersh BJ, Ackerman MJ: Myosin binding protein C mutations and compound heterozygosity in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004, 44: 1903-1910. 10.1016/j.jacc.2004.07.045.
Ho CY, Lever HM, DeSanctis R, Farver CF, Seidman JG, Seidman CE: Homozygous mutation in cardiac troponin T: implications for hypertrophic cardiomyopathy. Circulation. 2000, 102: 1950-1955.
Olivotto I, Girolami F, Ackerman MJ, Nistri S, Bos JM, Zachara E, Ommen SR, Theis JL, Vaubel RA, Re F: Myofilament protein gene mutation screening and outcome of patients with hypertrophic cardiomyopathy. Mayo Clin Proc. 2008, 83: 630-638.
Girolami F, Ho CY, Semsarian C, Baldi M, Will ML, Baldini K, Torricelli F, Yeates L, Cecchi F, Ackerman MJ, Olivotto I: Clinical features and outcome of hypertrophic cardiomyopathy associated with triple sarcomere protein gene mutations. J Am Coll Cardiol. 2010, 55: 1444-1453. 10.1016/j.jacc.2009.11.062.
Wigle ED, Sasson Z, Henderson MA, Ruddy TD, Fulop J, Rakowski H, Williams WG: Hypertrophic cardiomyopathy. The importance of the site and the extent of hypertrophy. A review. Prog Cardiovasc Dis. 1985, 28: 1-83. 10.1016/0033-0620(85)90024-6.
Davies MJ: The cardiomyopathies: an overview. Heart. 2000, 83: 469-474. 10.1136/heart.83.4.469.
Maron BJ, Fananapazir L: Sudden cardiac death in hypertrophic cardiomyopathy. Circulation. 1992, 85: I57-63.
Spirito P, Bellone P, Harris KM, Bernabo P, Bruzzi P, Maron BJ: Magnitude of left ventricular hypertrophy and risk of sudden death in hypertrophic cardiomyopathy. N Engl J Med. 2000, 342: 1778-1785. 10.1056/NEJM200006153422403.
Lima JA, Desai MY: Cardiovascular magnetic resonance imaging: current and emerging applications. J Am Coll Cardiol. 2004, 44: 1164-1171. 10.1016/j.jacc.2004.06.033.
Rathi VK, Biederman RW: Expanding role of cardiovascular magnetic resonance in left and right ventricular diastolic function. Heart Fail Clin. 2009, 5: 421-435, vii. 10.1016/j.hfc.2009.02.005.
Lamb BPPHJ: Assessment of diastolic function by cardiac MRI. Cardiovascular magnetic resonance imaging. Edited by: CANNON CP. 2008, Totowa, NJ: Humana Press, 415-428.
Shehata ML, Cheng S, Osman NF, Bluemke DA, Lima JA: Myocardial tissue tagging with cardiovascular magnetic resonance. J Cardiovasc Magn Reson. 2009, 11: 55-10.1186/1532-429X-11-55.
Fujita N, Hartiala J, O'Sullivan M, Steiman D, Chatterjee K, Parmley WW, Higgins CB: Assessment of left ventricular diastolic function in dilated cardiomyopathy with cine magnetic resonance imaging: effect of an angiotensin converting enzyme inhibitor, benazepril. Am Heart J. 1993, 125: 171-178. 10.1016/0002-8703(93)90071-G.
De Marchi SF, Allemann Y, Seiler C: Relaxation in hypertrophic cardiomyopathy and hypertensive heart disease: relations between hypertrophy and diastolic function. Heart. 2000, 83: 678-684. 10.1136/heart.83.6.678.
Yamanari H, Kakishita M, Fujimoto Y, Hashimoto K, Kiyooka T, Katayama Y, Otsuka F, Emori T, Uchida S, Ohe T: Effect of regional myocardial perfusion abnormalities on regional myocardial early diastolic function in patients with hypertrophic cardiomyopathy. Heart Vessels. 1997, 12: 192-198. 10.1007/BF02767047.
Brutsaert DL, Sys SU: Relaxation and diastole of the heart. Physiol Rev. 1989, 69: 1228-1315.
Nagueh SF, Bachinski LL, Meyer D, Hill R, Zoghbi WA, Tam JW, Quinones MA, Roberts R, Marian AJ: Tissue Doppler imaging consistently detects myocardial abnormalities in patients with hypertrophic cardiomyopathy and provides a novel means for an early diagnosis before and independently of hypertrophy. Circulation. 2001, 104: 128-130.
Ho CY, Sweitzer NK, McDonough B, Maron BJ, Casey SA, Seidman JG, Seidman CE, Solomon SD: Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy. Circulation. 2002, 105: 2992-2997. 10.1161/01.CIR.0000019070.70491.6D.
Ho CY, Carlsen C, Thune JJ, Havndrup O, Bundgaard H, Farrohi F, Rivero J, Cirino AL, Andersen PS, Christiansen M: Echocardiographic strain imaging to assess early and late consequences of sarcomere mutations in hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2009, 2: 314-321. 10.1161/CIRCGENETICS.109.862128.
Kayser HW, van der Geest RJ, van der Wall EE, Duchateau C, de Roos A: Right ventricular function in patients after acute myocardial infarction assessed with phase contrast MR velocity mapping encoded in three directions. J Magn Reson Imaging. 2000, 11: 471-475. 10.1002/(SICI)1522-2586(200005)11:5<471::AID-JMRI1>3.0.CO;2-D.
Petrank YF, Dong SJ, Tyberg J, Sideman S, Beyar R: Regional differences in shape and load in normal and diseased hearts studied by three dimensional tagged magnetic resonance imaging. Int J Card Imaging. 1999, 15: 309-321. 10.1023/A:1006132709895.
Nagel E, Stuber M, Burkhard B, Fischer SE, Scheidegger MB, Boesiger P, Hess OM: Cardiac rotation and relaxation in patients with aortic valve stenosis. Eur Heart J. 2000, 21: 582-589. 10.1053/euhj.1999.1736.
Kwon DH, Smedira NG, Rodriguez ER, Tan C, Setser R, Thamilarasan M, Lytle BW, Lever HM, Desai MY: Cardiac magnetic resonance detection of myocardial scarring in hypertrophic cardiomyopathy: correlation with histopathology and prevalence of ventricular tachycardia. J Am Coll Cardiol. 2009, 54: 242-249. 10.1016/j.jacc.2009.04.026.
Bruder O, Wagner A, Jensen CJ, Schneider S, Ong P, Kispert EM, Nassenstein K, Schlosser T, Sabin GV, Sechtem U, Mahrholdt H: Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010, 56: 875-887. 10.1016/j.jacc.2010.05.007.
Adabag AS, Maron BJ, Appelbaum E, Harrigan CJ, Buros JL, Gibson CM, Lesser JR, Hanna CA, Udelson JE, Manning WJ, Maron MS: Occurrence and frequency of arrhythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. J Am Coll Cardiol. 2008, 51: 1369-1374. 10.1016/j.jacc.2007.11.071.
Choudhury L, Mahrholdt H, Wagner A, Choi KM, Elliott MD, Klocke FJ, Bonow RO, Judd RM, Kim RJ: Myocardial scarring in asymptomatic or mildly symptomatic patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2002, 40: 2156-2164. 10.1016/S0735-1097(02)02602-5.
Moon JC, McKenna WJ, McCrohon JA, Elliott PM, Smith GC, Pennell DJ: Toward clinical risk assessment in hypertrophic cardiomyopathy with gadolinium cardiovascular magnetic resonance. J Am Coll Cardiol. 2003, 41: 1561-1567. 10.1016/S0735-1097(03)00189-X.
O'Hanlon R, Grasso A, Roughton M, Moon JC, Clark S, Wage R, Webb J, Kulkarni M, Dawson D, Sulaibeekh L: Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010, 56: 867-874. 10.1016/j.jacc.2010.05.010.
Moon JC, Reed E, Sheppard MN, Elkington AG, Ho SY, Burke M, Petrou M, Pennell DJ: The histologic basis of late gadolinium enhancement cardiovascular magnetic resonance in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2004, 43: 2260-2264. 10.1016/j.jacc.2004.03.035.
Ho CY, Lopez B, Coelho-Filho OR, Lakdawala NK, Cirino AL, Jarolim P, Kwong R, Gonzalez A, Colan SD, Seidman JG: Myocardial fibrosis as an early manifestation of hypertrophic cardiomyopathy. N Engl J Med. 2010, 363: 552-563. 10.1056/NEJMoa1002659.
Rudolph A, Abdel-Aty H, Bohl S, Boye P, Zagrosek A, Dietz R, Schulz-Menger J: Noninvasive detection of fibrosis applying contrast-enhanced cardiac magnetic resonance in different forms of left ventricular hypertrophy relation to remodeling. J Am Coll Cardiol. 2009, 53: 284-291. 10.1016/j.jacc.2008.08.064.
Sipola P, Lauerma K, Husso-Saastamoinen M, Kuikka JT, Vanninen E, Laitinen T, Manninen H, Niemi P, Peuhkurinen K, Jaaskelainen P: First-pass MR imaging in the assessment of perfusion impairment in patients with hypertrophic cardiomyopathy and the Asp175Asn mutation of the alpha-tropomyosin gene. Radiology. 2003, 226: 129-137. 10.1148/radiol.2261011874.
Knaapen P, van Dockum WG, Gotte MJ, Broeze KA, Kuijer JP, Zwanenburg JJ, Marcus JT, Kok WE, van Rossum AC, Lammertsma AA, Visser FC: Regional heterogeneity of resting perfusion in hypertrophic cardiomyopathy is related to delayed contrast enhancement but not to systolic function: a PET and MRI study. J Nucl Cardiol. 2006, 13: 660-667. 10.1016/j.nuclcard.2006.05.018.
Petersen SE, Jerosch-Herold M, Hudsmith LE, Robson MD, Francis JM, Doll HA, Selvanayagam JB, Neubauer S, Watkins H: Evidence for microvascular dysfunction in hypertrophic cardiomyopathy: new insights from multiparametric magnetic resonance imaging. Circulation. 2007, 115: 2418-2425. 10.1161/CIRCULATIONAHA.106.657023.
Moon JC: [What is late gadolinium enhancement in hypertrophic cardiomyopathy?]. Rev Esp Cardiol. 2007, 60: 1-4.
Amano Y, Takayama M, Takahama K, Kumazaki T: Delayed hyper-enhancement of myocardium in hypertrophic cardiomyopathy with asymmetrical septal hypertrophy: comparison with global and regional cardiac MR imaging appearances. J Magn Reson Imaging. 2004, 20: 595-600. 10.1002/jmri.20172.
Teraoka K, Hirano M, Ookubo H, Sasaki K, Katsuyama H, Amino M, Abe Y, Yamashina A: Delayed contrast enhancement of MRI in hypertrophic cardiomyopathy. Magn Reson Imaging. 2004, 22: 155-161. 10.1016/j.mri.2003.08.009.
Bogaert J, Goldstein M, Tannouri F, Golzarian J, Dymarkowski S: Original report. Late myocardial enhancement in hypertrophic cardiomyopathy with contrast-enhanced MR imaging. AJR Am J Roentgenol. 2003, 180: 981-985.
Maron MS, Appelbaum E, Harrigan CJ, Buros J, Gibson CM, Hanna C, Lesser JR, Udelson JE, Manning WJ, Maron BJ: Clinical profile and significance of delayed enhancement in hypertrophic cardiomyopathy. Circ Heart Fail. 2008, 1: 184-191. 10.1161/CIRCHEARTFAILURE.108.768119.
Moon JC, Mogensen J, Elliott PM, Smith GC, Elkington AG, Prasad SK, Pennell DJ, McKenna WJ: Myocardial late gadolinium enhancement cardiovascular magnetic resonance in hypertrophic cardiomyopathy caused by mutations in troponin I. Heart. 2005, 91: 1036-1040. 10.1136/hrt.2004.041384.
Moon JC, Sachdev B, Elkington AG, McKenna WJ, Mehta A, Pennell DJ, Leed PJ, Elliott PM: Gadolinium enhanced cardiovascular magnetic resonance in Anderson-Fabry disease. Evidence for a disease specific abnormality of the myocardial interstitium. Eur Heart J. 2003, 24: 2151-2155. 10.1016/j.ehj.2003.09.017.
Moon JC, Sheppard M, Reed E, Lee P, Elliott PM, Pennell DJ: The histological basis of late gadolinium enhancement cardiovascular magnetic resonance in a patient with Anderson-Fabry disease. J Cardiovasc Magn Reson. 2006, 8: 479-482. 10.1080/10976640600605002.
Kaldoudi E, Williams CR: Relaxation time measurements in NMR imaging. 1993
Damadian R: Tumor detection by nuclear magnetic resonance. Science. 1971, 171: 1151-1153. 10.1126/science.171.3976.1151.
Sparrow P, Messroghli DR, Reid S, Ridgway JP, Bainbridge G, Sivananthan MU: Myocardial T1 mapping for detection of left ventricular myocardial fibrosis in chronic aortic regurgitation: pilot study. AJR Am J Roentgenol. 2006, 187: W630-635. 10.2214/AJR.05.1264.
Iles L, Pfluger H, Phrommintikul A, Cherayath J, Aksit P, Gupta SN, Kaye DM, Taylor AJ: Evaluation of diffuse myocardial fibrosis in heart failure with cardiac magnetic resonance contrast-enhanced T1 mapping. J Am Coll Cardiol. 2008, 52: 1574-1580. 10.1016/j.jacc.2008.06.049.
Amano Y, Takayama M, Kumita S: Contrast-enhanced myocardial T1-weighted scout (Look-Locker) imaging for the detection of myocardial damages in hypertrophic cardiomyopathy. J Magn Reson Imaging. 2009, 30: 778-784. 10.1002/jmri.21921.
Abdel-Aty H, Cocker M, Strohm O, Filipchuk N, Friedrich MG: Abnormalities in T2-weighted cardiovascular magnetic resonance images of hypertrophic cardiomyopathy: regional distribution and relation to late gadolinium enhancement and severity of hypertrophy. J Magn Reson Imaging. 2008, 28: 242-245. 10.1002/jmri.21381.
Schwartzkopff B, Mundhenke M, Strauer BE: Alterations of the architecture of subendocardial arterioles in patients with hypertrophic cardiomyopathy and impaired coronary vasodilator reserve: a possible cause for myocardial ischemia. J Am Coll Cardiol. 1998, 31: 1089-1096. 10.1016/S0735-1097(98)00036-9.
Cecchi F, Olivotto I, Gistri R, Lorenzoni R, Chiriatti G, Camici PG: Coronary microvascular dysfunction and prognosis in hypertrophic cardiomyopathy. N Engl J Med. 2003, 349: 1027-1035. 10.1056/NEJMoa025050.
Abdel-Aty H, Zagrosek A, Schulz-Menger J, Taylor AJ, Messroghli D, Kumar A, Gross M, Dietz R, Friedrich MG: Delayed enhancement and T2-weighted cardiovascular magnetic resonance imaging differentiate acute from chronic myocardial infarction. Circulation. 2004, 109: 2411-2416. 10.1161/01.CIR.0000127428.10985.C6.
Sosnovik DE, Wang R, Dai G, Reese TG, Wedeen VJ: Diffusion MR tractography of the heart. J Cardiovasc Magn Reson. 2009, 11: 47-10.1186/1532-429X-11-47.
Dou J, Tseng WY, Reese TG, Wedeen VJ: Combined diffusion and strain MRI reveals structure and function of human myocardial laminar sheets in vivo. Magn Reson Med. 2003, 50: 107-113. 10.1002/mrm.10482.
Tseng WY, Dou J, Reese TG, Wedeen VJ: Imaging myocardial fiber disarray and intramural strain hypokinesis in hypertrophic cardiomyopathy with MRI. J Magn Reson Imaging. 2006, 23: 1-8. 10.1002/jmri.20473.
Ripplinger CM, Li W, Hadley J, Chen J, Rothenberg F, Lombardi R, Wickline SA, Marian AJ, Efimov IR: Enhanced transmural fiber rotation and connexin 43 heterogeneity are associated with an increased upper limit of vulnerability in a transgenic rabbit model of human hypertrophic cardiomyopathy. Circ Res. 2007, 101: 1049-1057. 10.1161/CIRCRESAHA.107.161240.
de Roos A, Doornbos J, Luyten PR, Oosterwaal LJ, van der Wall EE, den Hollander JA: Cardiac metabolism in patients with dilated and hypertrophic cardiomyopathy: assessment with proton-decoupled P-31 MR spectroscopy. J Magn Reson Imaging. 1992, 2: 711-719. 10.1002/jmri.1880020616.
Jung WI, Sieverding L, Breuer J, Hoess T, Widmaier S, Schmidt O, Bunse M, van Erckelens F, Apitz J, Lutz O, Dietze GJ: 31P NMR spectroscopy detects metabolic abnormalities in asymptomatic patients with hypertrophic cardiomyopathy. Circulation. 1998, 97: 2536-2542.
Shivu GN, Abozguia K, Phan TT, Ahmed I, Henning A, Frenneaux M: (31)P magnetic resonance spectroscopy to measure in vivo cardiac energetics in normal myocardium and hypertrophic cardiomyopathy: Experiences at 3T. Eur J Radiol. 2010, 73: 255-259. 10.1016/j.ejrad.2008.10.018.
Abozguia K, Elliott P, McKenna W, Phan TT, Nallur-Shivu G, Ahmed I, Maher AR, Kaur K, Taylor J, Henning A: Metabolic modulator perhexiline corrects energy deficiency and improves exercise capacity in symptomatic hypertrophic cardiomyopathy. Circulation. 2010, 122: 1562-1569. 10.1161/CIRCULATIONAHA.109.934059.
Maron BJ: Sudden death in young athletes. N Engl J Med. 2003, 349: 1064-1075. 10.1056/NEJMra022783.
Maron BJ, Lindberg J, Lesser JR: Ventricular septal crypt in hypertrophic cardiomyopathy. Eur Heart J. 2010, 31: 1923-10.1093/eurheartj/ehq140.
Tadamura E, Yamamuro M, Kubo S, Kanao S, Saga T, Harada M, Ohba M, Hosokawa R, Kimura T, Kita T, Togashi K: Effectiveness of delayed enhanced MRI for identification of cardiac sarcoidosis: comparison with radionuclide imaging. AJR Am J Roentgenol. 2005, 185: 110-115.
Maron BJ, Haas TS, Lesser JR: Images in cardiovascular medicine. Diagnostic utility of cardiac magnetic resonance imaging in monozygotic twins with hypertrophic cardiomyopathy and identical pattern of left ventricular hypertrophy. Circulation. 2007, 115: e627-628. 10.1161/CIRCULATIONAHA.106.680512.
Pons-Llado G, Carreras F, Borras X, Palmer J, Llauger J, Bayes de Luna A: Comparison of morphologic assessment of hypertrophic cardiomyopathy by magnetic resonance versus echocardiographic imaging. Am J Cardiol. 1997, 79: 1651-1656. 10.1016/S0002-9149(97)00216-6.
Posma JL, Blanksma PK, van der Wall EE, Hamer HP, Mooyaart EL, Lie KI: Assessment of quantitative hypertrophy scores in hypertrophic cardiomyopathy: magnetic resonance imaging versus echocardiography. Am Heart J. 1996, 132: 1020-1027. 10.1016/S0002-8703(96)90016-2.
Rubinshtein R, Glockner JF, Ommen SR, Araoz PA, Ackerman MJ, Sorajja P, Bos JM, Tajik AJ, Valeti US, Nishimura RA, Gersh BJ: Characteristics and clinical significance of late gadolinium enhancement by contrast-enhanced magnetic resonance imaging in patients with hypertrophic cardiomyopathy. Circ Heart Fail. 3: 51-58.
Kwon DH, Setser RM, Popovic ZB, Thamilarasan M, Sola S, Schoenhagen P, Garcia MJ, Flamm SD, Lever HM, Desai MY: Association of myocardial fibrosis, electrocardiography and ventricular tachyarrhythmia in hypertrophic cardiomyopathy: a delayed contrast enhanced MRI study. Int J Cardiovasc Imaging. 2008, 24: 617-625. 10.1007/s10554-008-9292-6.
Paya E, Marin F, Gonzalez J, Gimeno JR, Feliu E, Romero A, Ruiz-Espejo F, Roldan V, Climent V, de la Morena G, Valdes M: Variables associated with contrast-enhanced cardiovascular magnetic resonance in hypertrophic cardiomyopathy: clinical implications. J Card Fail. 2008, 14: 414-419. 10.1016/j.cardfail.2008.02.006.
Melacini P, Corbetti F, Calore C, Pescatore V, Smaniotto G, Pavei A, Bobbo F, Cacciavillani L, Iliceto S: Cardiovascular magnetic resonance signs of ischemia in hypertrophic cardiomyopathy. Int J Cardiol. 2008, 128: 364-373. 10.1016/j.ijcard.2007.06.023.
Kim YJ, Choi BW, Hur J, Lee HJ, Seo JS, Kim TH, Choe KO, Ha JW: Delayed enhancement in hypertrophic cardiomyopathy: comparison with myocardial tagging MRI. J Magn Reson Imaging. 2008, 27: 1054-1060. 10.1002/jmri.21366.
Debl K, Djavidani B, Buchner S, Lipke C, Nitz W, Feuerbach S, Riegger G, Luchner A: Delayed hyperenhancement in magnetic resonance imaging of left ventricular hypertrophy caused by aortic stenosis and hypertrophic cardiomyopathy: visualisation of focal fibrosis. Heart. 2006, 92: 1447-1451. 10.1136/hrt.2005.079392.
Soler R, Rodriguez E, Monserrat L, Mendez C, Martinez C: Magnetic resonance imaging of delayed enhancement in hypertrophic cardiomyopathy: relationship with left ventricular perfusion and contractile function. J Comput Assist Tomogr. 2006, 30: 412-420. 10.1097/00004728-200605000-00011.
Bruder O, Wagner A, Jensen CJ, Schneider S, Ong P, Kispert EM, Nassenstein K, Schlosser T, Sabin GV, Sechtem U, Mahrholdt H: Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 56: 875-887.
Suk T, Edwards C, Hart H, Christiansen JP: Myocardial scar detected by contrast-enhanced cardiac magnetic resonance imaging is associated with ventricular tachycardia in hypertrophic cardiomyopathy patients. Heart Lung Circ. 2008, 17: 370-374. 10.1016/j.hlc.2008.03.080.
Acknowledgements
Dr. Radwa A. Noureldin is a fellow in the Imaging Sciences Training Program supported in part by the Radiology and Imaging Sciences Department, Clinical Center and Intramural Research Program at the National Institute of Biomedical Imaging and Bioengineering, NIH.
Funding sources
Radiology & Imaging Sciences Department, National Institutes of Health Clinical Center. Bethesda, MD. 20892. USA
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
RA carried out the review of literature, manuscript design and drafting, SL has been involved in drafting the manuscript, MN participated in manuscript design and revision, DJ participated in revision of data regarding genetics section, MH carried out the revision of data regarding pathology section, TA has been involved in revising the manuscript critically for important intellectual content, CH has been involved in revising the manuscript critically for important intellectual content, and DB carried out the final revision and approved the manuscript to be submitted.
All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Noureldin, R.A., Liu, S., Nacif, M.S. et al. The diagnosis of hypertrophic cardiomyopathy by cardiovascular magnetic resonance. J Cardiovasc Magn Reson 14, 17 (2012). https://doi.org/10.1186/1532-429X-14-17
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1532-429X-14-17