
Abstract
The endotelin (ET) axis, that includes ET-1, ET-2, ET-3, and the ET receptors, ETA and ETB, plays an important physiological role, as modulator of vasomotor tone, tissue differentiation and development, cell proliferation, and hormone production. Recently, investigations into the role of the ET axis in mitogenesis, apoptosis inhibition, invasiveness, angiogenesis and bone remodeling have provided evidence of the importance of the ET-1 axis in cancer. Data suggest that ET-1 participates in the growth and progression of a variety of tumors such as prostatic, ovarian, renal, pulmonary, colorectal, cervical, breast carcinoma, Kaposi's sarcoma, brain tumors, melanoma, and bone metastases. ET-1 receptor antagonists beside providing ideal tools for dissecting the ET axis at molecular level have demonstrated their potential in developing novel therapeutic opportunity. The major relevance of ETA receptor in tumor development has led to an extensive search of highly selective antagonists. Atrasentan, one of such antagonists, is orally bioavailable, has suitable pharmacokinetic and toxicity profiles for clinical use. Preliminary data from clinical trials investigating atrasentan in patients with prostate cancer are encouraging. This large body of evidence demonstrates the antitumor activity of endothelin receptor antagonists and provides a rationale for the clinical evaluation of these molecules alone and in combination with cytotoxic drugs or molecular inhibitors leading to a new generation of anticancer therapies targeting endothelin receptors.
Similar content being viewed by others


Introduction
The endothelins, that includes three 21-aa peptides ET-1, ET-2 and ET-3, are potent vasoconstricting peptides, involved in the pathophysiology of different malignancies [1, 2]. ET-1 is a relevant growth factor in several tumor types including carcinoma of the prostate, ovary, colon, cervix, breast, kidney, lung, colon, central nervous system (CNS) tumors as well as melanoma, Kaposi's sarcoma (KS) and bone metastasis [3]. ETs and their receptors have been implicated in cancer progression through autocrine and paracrine pathways [4]. ET-1 participates to a wide range of cancer relevant process, such as cell proliferation, inhibition of apoptosis, matrix remodeling, bone deposition, and metastases. The demonstration of ET-1 as an important mediator in the progression of many tumors clearly identifies the ET axis as a potential therapeutic target. This has propelled the development of several potent and selective ET-1 receptor antagonists. These small molecules have contributed to our understanding of the physiopathological relevance of the ET axis and the beginning of translation of this information into clinical trials [5, 6].
Pathophysiology of endothelin
Synthesis
ET-1, ET-2 and ET-3, are characterized by a single α-helix and two disulfide bridges. The three peptides are encoded by distinct genes and are regulated at the level of mRNA transcription. The primary translation product of the ET-1 gene is the 212-aa prepro-ET-1, which is cleaved by an endopeptidase to form the 38-aa big-ET-1. The biologically active ET-1 is formed by endothelin-converting-enzyme (ECE), an enzyme with intracellular and membrane-bound isoforms [7]. The half-life of ET-1 in circulation is seven minutes [8]. Two pathways have been described for clearance of endothelin: ETB receptor-mediated uptake followed by lisosomal degradation [9, 10] and catabolism by extracellular neutral endopeptidase (NEP) [11, 12]. ET-1 production is stimulated by a variety of cytokines and growth factors, including IL-1β, TNF-α, TGF-β, PDGF, vasopressin, hypoxia, and shear stress. Inhibitory factors include nitric oxide, prostacyclin and atrial natriuretic peptide [6].
Receptors and signaling pathways
Endothelins exert their effects by binding to two distinct cell surface ET receptors, ETA and ETB. The ETB receptor (ETBR) binds the three peptide isotypes with equal affinity. In contrast, ETAR binds ET-1 with higher affinity than the other isoforms. Both receptors belong to the G protein-coupled receptor (GPCR) system and mediates biological responses from a variety of stimuli, including growth factors, vasoactive polypeptides, neurotransmitters, hormones, and phospholipids [1, 2].
ET-1 is produced by a variety of normal cells, including endothelial cells, vascular smooth muscle cells, and various epithelial tissues (eg, bronchial, endometrial, mammary, and prostatic) and is mitogenic for a variety of cell types including endothelial cells, vascular and bronchial smooth muscle cells, fibroblasts, keratinocytes, mesangial cells, osteoblasts, melanocytes, and endometrial stromal cells. This peptide, which is the most common circulating form of ETs, is produced also by many epithelial tumors where it acts as an autocrine or paracrine growth factor [4]. Ligand binding to the endothelin receptor results in activation of a pertussis toxin-insensitive G protein that stimulates phospholipase C activity and increases intracellular Ca2+ levels, activation of protein kinase C, mitogen activated protein kinase (MAPK) and p125 focal adhesion kinase (FAK) phosphorylation. Among downstream events after endothelin receptor activation, ET-1 causes EGF receptor transactivation, which is partly responsible for MAPK activation [13, 14].
Endothelin axis in tumor
ET-1 and tumor cell proliferation
ET-1 stimulates DNA synthesis and cell proliferation in various cells, including vascular smooth muscle, osteoblasts, glomerular mesangial cells, fibroblasts and melanocytes. ET-1 is also a mitogen for different cell types including prostate, cervical, ovary cancer cells. In primary cultures and established ovarian carcinoma cell lines, spontaneous growth was significantly inhibited in the presence of ETAR antagonist. ETBR antagonist lacked this activity demostrating that endogenous ET-1 acts as an autocrine modulator of ovarian carcinoma cell proliferation only through ETAR [15].
The mitogenic activity of ET-1 can be amplified by synergistic interactions with other growth factors including epidermal growth factor (EGF), basic fibroblast growth factor (bFGF), insulin, insulin-like growth factor (IGF), platelet-derived growth factor (PDGF), transforming growth factor (TGF), and interleukin-6 (IL-6) [16].
ET-1 and tumor neovascularization
ETs, which are mitogens for endothelial cells, vascular smooth muscle, fibroblasts, and pericytes, are also angiogenic factors. Endothelial cell mitogenesis is mediated by ETBR, while vascular smooth muscle cells and pericyte mitogenesis is mediated predominantly or solely by the ETAR. ET-1 modulates various stages of neovascularization, including endothelial cell proliferation, migration, invasion, protease production, tube formation and stimulates neovascularization in vivo [17]. Elevated expression of ET-1 and its cognate receptor is significantly associated with microvessel density (MVD) and vascular endothelial growth factor (VEGF) expression in ovarian carcinomas [18], suggesting that ET-1 and VEGF might have a complementary and coordinated role during neovascularization in this tumor. Thus, in ovarian carcinoma cell lines, ET-1 increases VEGF mRNA expression and induces VEGF levels in a time- and dose-dependent fashion, and does so to a greater extent during hypoxia. These activities were mediated through the ETAR, because the specific antagonist, BQ 123, reversed the stimulation of VEGF production [18].
The transcriptional upregulation of VEGF has been linked to a critical mediator of hypoxia signaling, the hypoxic inducible factor-1α (HIF-1α). ET-1 promotes VEGF production through HIF-1α and this mechanism might be responsible for increasing tumor angiogenesis. Degradation of HIF-1α was infact reduced in ET-1-treated ovarian carcinoma cells under both hypoxic and normoxic conditions. After ET-1 stimulation, HIF-1α protein levels increase in the cells, the HIF-1 transcription complex is formed and binds to the HRE binding site. Therefore, ET-1-induced HIF-1 accumulation activates all the signals necessary for a complete HIF-1 response [19]. The HIF-1α-mediated transcription of VEGF by ET-1 under normoxic conditions points to a general mechanism through which oncogenes and growth factors might upregulate VEGF, and could synergize with hypoxia during tumor growth. Addition of a specific ETAR antagonist blocked the ET-1-induced upregulation of VEGF expression and secretion as well as the ET-1-induced activation of HIF-1 transcription complex. In tumor cells, ET-1 might be upregulated by hypoxia and could promote angiogenesis by increasing VEGF production through an HIF-1α-dependent mechanism. Thus, under hypoxic conditions, ET-1 potentiates hypoxia stimulus by amplifying HIF-1α stability and VEGF production [25].
Prostaglandins (PG) and their rate-limiting enzymes cyclooxygenase (COX)-1 and -2 are involved in the onset and progression of a variety of malignancies [21]. Moreover high COX-1 and -2 have been reported in association with elevated levels of proangiogenic factors in ovarian cancer [22–25]. In ovarian carcinoma cells, ET-1 significantly increases the expression of COX-1 and -2, at mRNA and protein level, the COX-2 promoter activity and PGE2 production. These effects depend on the ETAR activation and involve multiple MAPK signal pathways, including p42/44 MAPK, p38 MAPK and the transactivation of EGFR.
There is increasing evidence that PGE2 contributes to tumor progression also by promoting angiogenesis and that this effect is mediated by VEGF. COX-2 and-1 inhibitors blocked ET-1-induced PGE2 and VEGF release demonstrating that both enzymes, although by a different extent, participate to PGE2 and VEGF production. These results indicate that impairing COX-1 and -2 and their downstream effect by targeting ETAR can be therapeutically advantageous [26] also in view that elevated COX-2 levels are associated with tumor progression and chemoresistance [24].
ET-1 and apoptosis
ET-1 is an antiapoptotic factor in different cell types, indicating that the peptide may also modulate cell survival pathways. In ovarian carcinoma cells, the addition of ET-1 markedly inhibited serum-withdrawal and paclitaxel-induced apoptosis in a concentration-dependent manner. Paclitaxel-induced apoptosis resulted in the phosphorylation of Bcl-2 that was suppressed by the addition of ET-1. Further analysis of the survival pathway demonstrated that ET-1 stimulated Akt activation dependent on PI3-K. Interestingly, the addition of a specific ETAR antagonist blocked the ET-1-induced resistance to paclitaxel-mediated apoptosis indicating that ET-1 contributes to trigger resistance to paclitaxel through ETAR binding via activation of anti-apoptotic signaling pathways such as Akt.
Specific ETAR antagonists may therefore provide an additional approach to the treatment of ovarian carcinoma in which ETAR blockade could result in the tumor growth inhibition by reducing tumor growth as well as by inducing apoptosis. Furthermore when combined with the conventional chemotherapy the ETAR antagonists would more effectively induce apoptosis by contributing to the reversal of paclitaxel resistance [27].
ET-1 and tumor invasion
As mentioned, high levels of ET-1 are detected in the majority of ascitic fluids of ovarian cancer patients and are significantly correlated with VEGF ascitic concentrations, suggesting that ET-1 may enhance the secretion of extracellular matrix-degrading proteinases and metastatization [18]. Thus, ET-1 acting through the ETAR consistently induced the activity of two families of metastasis-related proteinases, the matrix metalloproteinases (MMPs) and the urokinase type plasminogen activator system at several levels: mRNA transcription, zymogen secretion and pro-enzymes activation. ET-1 in fact activates MMP-2, MMP-9, MMP-3, MMP-7 and MMP-13. In addition to soluble MMPs, ET-1 enhanced the activation of MT1-MMP and the secretion of tissue inibitor of MMP (TIMP-1 and -2) increasing the net MMP/TIMP balance and the gelatinolytic capacity that results in rapid degradation of extracellular matrix (ECM). In the ovarian carcinoma cells, co-induction of uPA system, by the concomitant stimulation of production and secretion of uPA and uPAR, and MMPs by ET-1 resulted into the highest invasive potential of tumor cells through the Matrigel. Interestingly, the addition of BQ 123 blocked the ET-1-induced proteinase activation and tumor cell migration and invasion. Furthermore in these cells, ET-1 stimulated FAK and paxillin phosphorylation through ETAR binding [13] which directly correlated with tumor cell migration and invasion suggesting that ETAR antagonist can inhibit cell migration and possibly other FAK-associated processes which also contributes to invasion and metastasis in this tumor [28].
Following malignant transformation, stepwise changes in intercellular communications enable tumor cells to escape microenvironmental control from the normal surrounding tissue, thus allowing local invasiveness and metastatization. Human ovarian surface epithelial cells exhibit extensive gap junction intercellular communications (GJIC) and expression of different types of connexin (Cx), predominantly Cx43. Defects in intercellular communication, including reduced or inappropriate expression of Cx43 has emerged as key factors in ovarian carcinoma progression. In ovarian carcinoma cells, ET-1 via ETAR induces a transient and a dose-dependent reduction of GJIC (50–75%) and phosphorylates Cx43 through Src tyrosine kinase pathway indicating that ET-1 promotes cellular uncoupling at the level of connexin maturation and subsequent degradation [29]. The capacity of ET-1 to disrupt gap junctions could serve as a basis to further evaluate the cell-cell metabolic uncoupling and the cell detachment that occurs during tumor progression and adds further information on the overall relevance of ETAR in regulating the complex array of cell-cell or cell-matrix interactions promoting ovarian carcinoma growth [29].
Osteogenesis
Several observations implicate ET-1 in the osteoblastic response. Osteoblasts display a high density of ET receptors, and respond to ET-1 by increasing synthesis of both collagenous and noncollagenous proteins, including two osteoblast messengers such as osteopontin and osteocalcin [30]. Osteoblasts are also sensitive to the mitogenic and comitogenic activity of ET-1. Furthermore ET-1 inhibits osteoclasts activity and motility, upsetting the balance of osteoblasts and osteoclasts to favor new bone formation [31]. Alkaline phosphatase, a marker of new bone formation, is elevated in the presence of exogenous ET-1 [32]. ET-1 production is stimulated by IL-1β, TNF-α and TGF-β, which are normally secreted by immunocompetent cells, as well as osteoblasts and endothelial cells. This suggests that paracrine mediators secreted by osteoblasts and tumor cells are involved in a paracrine growth loop that results in the derangement of bone remodelling.
Pain
ET-1 is described as a new pain mediator implicated in the pathogenesis of pain states. Local cutaneous injection of ET-1 causes pain and excitation of nociceptors, both phenomena depending on ETAR. The peptide can trigger pain through ETAR of local nociceptors, and concurrently produces analgesia through ETBR by inducing the release of β-endorphin and the activation of opioid pool [33]. It is reasonable to speculate that the refractory pain of certain metastatic cancers is related to the direct nociceptive effects of ET-1. Thus antagonist of ETAR have been shown to ameliorate pain. This knowledge may lead to improved, targeted analgesia in patients with advanced cancer [34].
Targeting endothelin receptor as novel approach in cancer treatment
Ovarian carcinoma
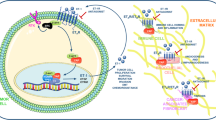
Ovarian cancer is the leading cause of gynecologic cancer-related death. ET-1/ETAR autocrine pathway is overexpressed in primary and metastatic ovarian carcinomas, compared with normal ovaries (15). In view of the fact that ETAR contributes to ovarian cancer progression by inducing cell proliferation, survival, angiogenesis and invasiveness, the ETAR has been proposed as a novel target for anticancer therapy. The recent identification of high-selective small molecules that inhibit ligand-induced activation of ETAR offers now the possibility of testing this therapeutic approach in a clinical setting. Among various ETAR antagonists, ABT-627 (atrasentan, Abbott Laboratories, Abbott Park, IL) is an orally bioavailable endothelin antagonist that potently (Ki = 34 pM) and selectively binds to the ETAR, blocking signal transduction pathways implicated in cancer cell proliferation and other host-dependent processes promoting cancer growth (Figure 1).

The ET-1/ETAR autocrine pathway is biologically relevant in tumor development and progression modulating cell proliferation, survival, angiogenesis, invasion. Targeted blockade of the ETAR using the potent and selective ETAR antagonist reduces tumor growth which concurs with the inhibition of the tumor-induced angiogenesis and invasiveness and with the induction of massive tumor cell apoptosis.
In ovarian cancer, ABT-627 inhibits in vitro cell proliferation, the ET-1 mediated protection against paclitaxel-induced apoptosis and the release of VEGF. A cooperative proapoptotic and VEGF inhibitory effect was observed when ABT-627 was used together with paclitaxel.
Treatment with ABT-627 produced a 65% tumor growth inhibition in well established HEY xenografts. This treatment which was generally well tolerated, with no detectable signs of acute or delayed toxicity was long-lasting and comparable to that achieved by paclitaxel. More marked and prolonged tumor growth inhibition (90% of controls) was obtained by combined treatment of ABT-627 with paclitaxel, with no toxicity and with four complete tumor regressions out of 10 treated animals. Immunohistochemical analysis of the xenografts revealed a marked reduction in the percentage of COX-2, VEGF and MMP-2 in treated mice. Tumor-induced vascularization, quantified as microvessel density (MVD) was directly proportional to the expression of VEGF. A significant increase in the percentage of TUNEL-positive cells was found [35]. The tumor growth inhibition induced by ABT-627, was also associated with a reduction of Cx43 phosphorylation and with an increase Cx43-based intercellular communication, suggesting that ETAR blockade may contribute to the control of ovarian carcinoma growth and progression also by preventing the loss of GJIC [29]. Almost complete inhibition of VEGF, MMP-2 expression, and tumor neovascularization, and an increase in apoptosis, were observed following combined treatment of ABT-627 with paclitaxel.
These findings demonstrating the antitumor, anti-angiogenic, apoptotic activities of ABT-627 in vivo provide a rationale for the clinical evaluation of this molecule, alone and in combination with other therapies, in patients with ovarian tumors and potentially in other epithelial tumors that overexpress functional ETAR.
Prostate carcinoma
The ET axis has recently been identified as contributing to the pathophysiology of prostate cancer [32, 36, 37]. In the normal prostate gland, ET-1 is produced by epithelial cells; the highest concentrations of ET-1 in the body are found in seminal fluid. In prostate cancer, key components of the ET-1 clearance pathway, ETB and neutral endopeptidease (NEP), are diminished, resulting in an increase in local ET-1 concentrations. Increased ETAR expression is also seen with advancing tumor stage and grade in both primary and metastatic prostate cancer. There are multiple pathways by which the ET-1/ETA axis may promote prostate cancer progression. In prostate cancer cell lines, ET-1 acting synergistically with other peptide growth factors is a mitogen and modulates apoptosis by affecting cell survival. Selective ETAR antagonists may block the proliferative effects of exogenous ET-1 in both prostate cancer cells and osteoblasts. In a phase I clinical trial, patients with advanced prostate cancer were treated with atrasentan. In this trial stabilization and decline of PSA occurred in 66% of patients. In a phase II study, atrasentan suppressed markers of biochemical and clinical prostate cancer progression in bone [38, 39]. These data substantiate the role of the ET-1/ETA axis as a growth and survival factor and as a therapeutic target in hormone-refractory prostate cancer. Atrasentan may inhibit tumor growth in bone both by direct effects on the tumor cells and by disrupting important bone/tumor interactions. In a randomized double-blind phase II study, 288 patients with hormone refractory prostate cancer were enrolled and were treated with atrasentan 2.5 mg and 10 mg administered orally once daily. In the evaluable patients (n = 244), atrasentan significantly delayed time to clinical and biochemical (PSA) progression. The most common side effects included rhinitis, peripheral edema, and headache. Atrasentan also maintained total and bone alkaline phosphatase concentrations at baseline values compared to the placebo-treated group. These findings suggest that atrasentan may inhibit progression of hormone refractory metastatic prostate cancer.
Osteoblastic bone metastases
Osteoblastic bone metastases occur in advanced prostate and breast cancers, but the mechanism underlaying new bone formation are unclear [40]. Many tumor-linked factors have been implicated in the onset and progression of skeletal metastases, including IGFI and II, TGFβ, PSA, uPA, FGF-1 and -2, bone morphogenic proteins, and ET-1. This latter peptide stimulates mitogenesis in osteoblasts and decreases osteoclastic bone resorption and osteoclast motility [40].
ET-1 is increased in the circulation of patients with prostate cancer with osteoblastic metastases, and is also produced by breast cancer cell lines that cause osteoblastic metastases. Treatment with the orally active ETAR antagonist atrasentan dramatically decreased bone metastases and tumor burden in mice inoculated with the human breast cancer cell line ZR-75-1 producing ET-1 [41]. The growing list of potential clinical applications for ETAR blockade should now include treatment and eventually prevention of osteoblastic bone metastases.
Cervical carcinoma
Human papillomavirus (HPV)-positive human cervical carcinoma cell lines overexpress ET-1 and ETAR mRNA and secrete ET-1 protein compared to HPV-negative cells. Binding studies show that HPV-infected cells express increased numbers of functional ETAR, and a specific antagonist of ETA inhibits ET-1-induced proliferation. An ETBR selective antagonist had no effect. These results indicate that the ET-1/ETAR axis could be targeted for antitumor therapy. Thus, atrasentan inhibited the growth of cervical carcinoma cell xenografts. Two cycles of treatment completely reverted tumor growth. As reported for ovarian cancer, this small molecule displayed additive effects when administrated in combination with the cytotoxic drug paclitaxel supporting its clinical use either in mono- or combination regimens [42, 43].
Breast carcinoma
ET-1 and its receptors, ETAR and ETBR, are overexpressed in breast carcinomas. Analysis of 176 breast carcinomas has shown that ET-1 was expressed in 43% whereas ETAR in 46% and ETBR in 53% of the cases. Elevated expression of ET-1 and its receptors was more common in breast carcinomas of patients with lower disease-free survival time and overall survival. In particular, a statistically significant correlation was observed between ETAR expression and reduced disease-free survival time [44]. ET-1, ETAR and ETBR expression was also associated with increased VEGF expression and higher vascularity [45]. Therefore analysis of the ET-axis and in particular of ETAR may improve the prediction of relapse and death and may identify patients who may profit from ETAR targeted adjuvant therapy.
Kaposi's sarcoma
A different approach in targeting ET-1 receptor in cancer treatment is represented by Kaposi's sarcoma (KS) in which ET-1 acts as an autocrine growth factor through both ETAR and ETBR. Binding of ET-1 and ET-3 to both receptors increased the proliferation, migration and invasiveness of the KS derived cell line, KS IMM cells, by stimulating secretion and activation of multiple tumor proteases. Treatment of KS IMM xenografts with the small molecule ETA/ETB antagonist A182086 produced a tumor growth inhibition most likely related to antiproliferative effect on tumor cells and to the antiangiogenic effect on endothelial cells expressing ETBR. Thus ET-1 receptor antagonists may be effective for treatment of this malignancy because capable of interfering simultaneously with cell proliferation, invasiveness and angiogenesis [46, 47].
Melanoma
Transformed melanocytes express both ETAR and ETBR. Gene expression profiling [48] and immunophenotyping of human cutaneous melanoma [49] have recently identified ETBR as critical in the progression of this malignancy. Through the same receptor, ET-1 acts as antiapoptotic factor for melanoma cells and melanocytes. Thus, ETBR blockade by the ETBR peptide antagonist BQ788 resulted in growth inhibition and death of melanoma cells in vivo and in vitro [50]. While these early studies defined a relevant role of the ET-1/ETBR pathway in the biology of melanocytic tumors, the molecular events underlying this activity are becoming now identified. Early melanoma growth is the result of disrupted intercellular homeostatic regulation. Once this balance is lost and malignant transformation has occurred, microenviromental factors such as cell adherence to extracellular matrix, host-tumor interactions, degradation of matrix components, migration and invasion became essential for the tumor progression to the metastatic phenotype. Changes in cadherins, gap junctions and matrix metalloproteinases expression have emerged as key factors in melanoma progression. In this regards ET-1 and ET-3 by ETBR signalling induce the inactivation of the gap junctions through the phosphorylation of the Cx43, which are responsible for contact mediated regulatory control of keratinocytes. Additionally, activation of ETBR pathway by ET-1 and ET-3 contributes to disruption of normal host-tumor interactions by downregulating the expression of E-cadherin and associated β-catenin adhesion proteins. Significant ET-1-induced mRNA expression of the transcription factor Snail, which has been identified a potent inhibitor of E-cadherin expression in melanoma, closely correlates with downregulation of E-cadherin. ETs also causes a tyrosine phosphorylation of β-catenin which contributes to loss of E-cadherin function, with a concomitant upregulation of N-cadherin. This latter change can mediate homotypic adhesive interactions as well as heterotypic melanoma cell-cell interactions. Concurrently ETs increase αvβ3 and α2β1 integrin expression and matrix metalloproteinase (MMP)-2, -9, and membrane type-1-MMP activation. These effects were associated with ETBR-mediated enhancement of cell adhesion, migration and invasiveness. Due to the resistance of melanoma to current therapies, the identification of molecular mechanisms underlying local and metastatic growth is mandatory for the development of novel treatments. The small molecule A-192621, an orally biovailable non peptide ETBR antagonist, significantly inhibited melanoma growth in nude mice. In conclusion, because multiple molecular pathways, such as FAK and MAPK, elicited by ET-1 and ET-3 are triggered by the ETBR leading to the activation of all the molecular effectors involved in melanoma progression, including integrins, tumor proteases, cell-cell adhesion and communication molecules, blockade of this receptor by small molecules results into inhibition of melanoma growth in vitro and in vivo, thus offering an unprecedented opportunity of targeted therapy in this malignancy [51].
Conclusions
A large body of in vitro and in vivo studies indicate the ET axis as a potential novel therapeutic target for cancer. ET-1 is overexpressed in many malignancies, acting as an autocrine/paracrine growth factor. Engagement of the cognate receptor by ET-1 triggers tumor proliferation, VEGF-induced angiogenesis, metastatic potential, antiapoptotic effect and is synergistic with other growth factors.
Direct mechanistic evidence of the role of ETARs in various malignancies supports the concept that ETAR antagonists may significantly revert the malignant phenotype [3]. The antitumor activity of selective ETAR antagonists observed in preclinical studies has been strongly supported in early clinical trials. Among these, atrasentan deserves at the present particular attention for clinical testing, in view of its oral bioavailability, suitable pharmacokinetic, toxicity profile and clinical activity.
The role of the ET axis and the therapeutic relevance of ET-1 receptor antagonists in a range of malignancies requires future investigation that may lead to a new generation of molecularly targeted therapies for cancer (Table 1) [15, 32, 35, 40–47, 50–57].
Resistance to apoptosis is a principal mechanism whereby tumors are able to overcome cytotoxicity induced by chemotherapy. ETAR blockade sensitized tumor cells to the apoptotic potential of chemoterapeutic agent resulting in tumor regression. The cooperative antitumor effect of combination therapy in which ETAR antagonist, by increasing the commitment of tumor cells towards apoptosis, potentiates the therapeutic efficacy of conventional cytotoxic drugs, offers a rationale for its clinical evaluation in malignancies expressing ETAR [58].
A novel approach to cancer therapy emerges by the treatment with multiple selective inhibitors to different growth factor receptors or to key post receptor signaling pathways. Engagement of the endothelin receptor by ET-1 induces tumor-promoting effects that are mediated by different downstream effectors, such as EGFR, VEGF COX-2, AKT, MAPK that could became the preferentially escaping pathways utilized by tumor cells. In this context, concomitant blockade of multiple molecular targets by using inhibitor of EGFR, or COX-2, or others in combination with ETAR antagonist could represent an attractive therapeutic strategy to be explored in clinical settings.
References
Levin ER: Endothelins. N Engl J Med. 1995, 333: 356-363. 10.1056/NEJM199508103330607.
Masaki T: The endothelin family: an overview. J Cardiovasc Pharmacol. 2000, 35: S3-S5. 10.1097/00005344-200000002-00002.
Nelson J, Bagnato A, Battistini B, Nisen P: The endothelin axis: emerging role in cancer. Nat Rev Cancer. 2003, 3: 110-116. 10.1038/nrc990.
Bagnato A, Catt KJ: Endothelins as autocrine regulators of tumor cell growth. Trends Endocrinol Metab. 1998, 9: 378-383. 10.1016/S1043-2760(98)00094-0.
Opgenorth TJ: Endothelin receptor antagonism. Adv Pharmacol. 1995, 33: 1-65.
Remuzzi G, Perico N, Benigni A: New therapeutics that antagonize endothelin: promises and frustations. Nature Rev Drug Disc. 2002, 1: 986-1001. 10.1038/nrd962.
Xu D, Emoto N, Giaid A, Slaughter C, Kaw S, deWit D, Yanagisawa M: ECE-1: a membrane bound metalloprotease that catalyses the proteolytic activation of big endothelin-1. Cell. 1994, 78: 473-485. 10.1016/0092-8674(94)90425-1.
Rubin SA, Levin ER: The endocrinology of vasoactive peptides: synthesis to function. J Clin Endocr Metabol. 1994, 78: 6-10. 10.1210/jc.78.1.6.
Burkhardt M, Barton M, Shaw SG: Receptor-and non-receptor-mediated clearance of big-endothelin and endothelin-1: differential effects of acute and chronic ETA receptor blockade. J Hypertens. 2000, 18: 273-279. 10.1097/00004872-200018030-00006.
Bremnes T, Paasche JD, Mehlum A, Sandberg C, Mremnes B, Attramadal H: Regulation and intracellular trafficking pathways of the endothelin receptors. J Biol Chem. 2000, 275: 17596-17604. 10.1074/jbc.M000142200.
Vijayaraghavan J, Scicli AG, Carretero OA, Slaughter C, Moomaw C, Hersh LB: The hydrolysis of endothelins by neutral endopeptidase. J Biol Chem. 1990, 265: 14150-14155.
Battistini B, D'Orleans-Juste P, Sirois P: Endothelins: circulating plasma levels and presence in other biologic fluids. Lab Invest. 1993, 68: 600-628.
Bagnato A, Tecce R, Di Castro V, Catt KJ: Activation of mitogenic signaling by endothelin-1 in ovarian carcinoma cells. Cancer Res. 1997, 57: 1306-1311.
Vacca F, Bagnato A, Catt KJ, Tecce R: Transactivation of the epidermal growth factor receptor in endothelin-1-induced mitogenic signaling in human ovarian carcinoma cells. Cancer Res. 2000, 60: 5310-5317.
Bagnato A, Salani D, Di Castro V, Wu-Wong JR, Tecce R, Nicotra MR, Venuti A, Natali PG: Expression of endothelin 1 and endothelin A receptor in ovarian carcinoma: evidence for an autocrine role in tumor growth. Cancer Res. 1999, 59: 720-727.
Battistini B, Chailler P, D'Orleans-Juste P, Briere N, Sirois P: Growth regulatory properties of endothelins. Peptides. 1993, 14: 385-399. 10.1016/0196-9781(93)90057-N.
Salani D, Taraboletti G, Rosanò L, Di Castro V, Borsotti P, Giavazzi R, Bagnato A: Endothelin-1 induces an angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Am J Pathol. 2000, 157: 1703-1711.
Salani D, Di Castro V, Nicotra MR, Rosanò L, Tecce R, Venuti A, Natali PG, Bagnato A: Role of endothelin-1 in neovascularization of ovarian carcinoma. Am J Pathol. 2000, 157: 1537-1547.
Spinella F, Rosanò L, Di Castro V, Natali PG, Bagnato A: Endothelin-1 induces vascular endothelial growth factor by increasing hypoxia-inducible factor 1α in ovarian carcinoma cells. J Biol Chem. 2002, 277: 27850-27855. 10.1074/jbc.M202421200.
Bagnato A, Spinella F: Emerging role of endothelin-1 in tumor angiogenesis. Trend Endocrinol Metab. 2003, 14: 44-50. 10.1016/S1043-2760(02)00010-3.
Dannenberg AJ, Subbaramaiah K: Targeting cyclooxygenase-2 in human neoplasia: rationale and promise. Cancer Cell. 2003, 4: 431-436. 10.1016/S1535-6108(03)00310-6.
Denkert C, Kobel M, Pest S, Koch I, Berger S, Schwabe M, Siegert A, Reles A, Klosterhalfen B, Hauptmann S: Expression of cyclooxygenase-2 is an independent prognostic factor in human ovarian carcinoma. Am J Pathol. 2002, 160: 893-903.
Erkinheimo TL, Lassus H, Finne P, van Rees BP, Leminen A, Ylikorkala O, Haglund C, Butzow R, Ristimäki A: Elevated cyclooxygenase-2 expression is associated with altered expression of p53 and SMAD4, amplification of HER-2/neu, and poor outcome in serous ovarian carcinoma. Clin Cancer Res. 2004, 10: 538-545.
Ferrandina G, Lauriola L, Zannoni GF, Fagotti A, Fanfani F, Legge F, Maggiano N, Gessi M, Mancuso S, Ranelletti FO, Scambia G: Increased cyclooxygenase-2 expression is associated with chemotherapy resistance and outcome in ovarian cancer patients. Ann Oncol. 2002, 13: 1205-1211. 10.1093/annonc/mdf207.
Gupta RA, Tejada LV, Tong BJ, Das SK, Morrow JD, Dey SK, DuBois RN: Cyclooxygenase-1 is overexpressed and promotes angiogenic growth factor production in ovarian cancer. Cancer Res. 2003, 63: 906-911.
Spinella F, Rosanò L, Di Castro V, Nicotra MR, Natali PG, Bagnato A: Inhibition of cyclooxygenase-1 and -2 expression by targeting the endothelin A receptor in human ovarian carcinoma cells. Clin Cancer Res. 2004, 10 (14):
Del Bufalo D, Di Castro V, Biroccio A, Varmi M, Salani D, Rosanò L, Trisciuoglio D, Spinella S, Bagnato A: Endothelin-1 protects ovarian carcinoma cells against paclitaxel-induced apoptosis: requirement for Akt activation. Mol Pharmacol. 2002, 61: 524-532. 10.1124/mol.61.3.524.
Rosanò L, Varmi M, Salani D, Di Castro V, Spinella F, Natali PG, Bagnato A: Endothelin-1 induces tumor proteinase activation and invasiveness of ovarian carcinoma cells. Cancer Res. 2001, 61: 8340-8346.
Spinella F, Rosanò L, Di Castro V, Nicotra MR, Natali PG, Bagnato A: Endothelin-1 decreases gap-junctional intercellular communication by inducing phosphorylation of connexin 43 in human ovarian carcinoma cells. J Biol Chem. 2003, 278: 41294-41301. 10.1074/jbc.M304785200.
Shioide M, Noda M: Endothelin modulates osteopontin and osteocalcin messenger ribonucleic acid expression in rat osteoblastic osteosarcoma cells. J Cell Biochem. 1993, 53: 176-180.
Alam ASMT, Gallaghe A, Shankar V: Endothelin inhibits osteoclastic bone resorption by a direct effect on cell motility: implications for the vascular control of bone resorption. Endocrinology. 1992, 130: 3617-3624. 10.1210/en.130.6.3617.
Nelson JB, Hedican SP, George DJ, Reddi AH, Piantadosi S, Eisenberger MA, Simons JW: Identification of endothelin-1 in the pathophysiology of metastatic adenocarcinoma of the prostate. Nat Med. 1995, 1: 944-949.
Khodorova A, Navarro B, Jovaville LS, Murphy JE, Rice FL, Mazurkiewicr JE, Long-Woodward D, Stofel M, Strichartz GR, Yukhananov R, Davar G: Endothelin-B receptor activation triggers an endogenous analgesic cascade at sites of peripheral injury. Nat Med. 2003, 9: 1055-1061. 10.1038/nm885.
Davar G: Endothelin-1 and metastatic cancer pain. Pain Med. 2001, 2: 24-27. 10.1046/j.1526-4637.2001.002001024.x.
Rosanò L, Spinella F, Salani D, Di Castro V, Venuti A, Nicotra MR, Natali PG, Bagnato A: Therapeutic targeting of endothelin A receptor in human ovarian carcinoma. Cancer Res. 2003, 63: 2447-2453.
Nelson JB, Chan-Tack K, Hedican SP, Magnuson SR, Opgenorth TJ, Bova GS, Simons JW: Endothelin-1 production and decreased endothelin B receptor expression in advanced prostate cancer. Cancer Res. 1996, 56: 663-668.
Nelson JB, Lee WH, Nguyen SH, Jarrard DF, Brooks JD, Magnuson SR, Opgenorth TJ, Nelson WG, Bova GS: Methylation of the 5' CpG island of the endothelin B receptor gene is common in human prostate cancer. Cancer Res. 1997, 57: 35-37.
Carducci MA, Nelson JB, Bowling MK, Rogers T, Eisenberger MA, Sinibaldi V, Donehower R, Leahy TL, Carr RA, Isaacson JD, Janus TJ, Andre A, Hosmane BS, Padley RJ: Atrasentan, an endothelin-receptor antagonist for refractory adenocarcinomas: safety and pharmacokinetics. J Clin Oncol. 2002, 20: 2171-2218. 10.1200/JCO.2002.08.028.
Carducci MA, Padley RJ, Breul J, Vogelzang NJ, Zonnenberg BA, Daliani DD, Schulman CC, Nabulsi AA, Humerickhouse RA, Weinberg MA, Schmitt JL, Nelson JB: Effect of endothelin-A receptor blockade with atrasentan on tumor progression in men with hormone-refractory prostate cancer: a randomized, phase II, placebo-controlled trial. J Clin Oncol. 2003, 21: 679-689. 10.1200/JCO.2003.04.176.
Guise TA: Molecular mechanisms of osteolytic bone metastases. Cancer. 2000, 88: 2892-2898. 10.1002/1097-0142(20000615)88:12+<2892::AID-CNCR2>3.0.CO;2-Y.
Yin JJ, Mohammed KS, Kakonen SM, Harris S, Wu-Wong JR, Wessale JL, Padley RJ, Garrett IR, Chirgwin JM, Guise TA: A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastasis. Proc Natl Acad Sci USA. 2003, 100: 10954-10959. 10.1073/pnas.1830978100.
Venuti A, Salani D, Manni V, Poggiali F, Bagnato A: Expression of endothelin-1 and endothelin A receptor in HPV-associated cervical carcinoma: new potential targets for anticancer therapy. FASEB J. 2000, 14: 2277-2283. 10.1096/fj.00-0024com.
Bagnato A, Cirilli A, Salani D, Simeone P, Muller A, Nicotra MR, Natali PG, Venuti A: Growth inhibition of cervix carcinoma cells in vivo by endothelin A receptor blockade. Cancer Res. 2002, 62: 6381-6384.
Wulfing P, Diallo R, Kersting C, Wulfing C, Poremba C, Rody A, Greb RR, Bocker W, Kiesel L: Expression of endothelin-1, endothelin-A, and endothelin-B receptor in human breast cancer and correlation with long-term follow-up. Clin Cancer Res. 2003, 9: 4125-4131.
Wulfing P, Kersting C, Tio J, Fischer RJ, Wulfing C, Poremba C, Diallo R, Bocker W, Kiesel L: Endothelin-1-, endothelin-A-, and endothelin-B-receptor expression is correlated with vascular endothelial growth factor expression and angiogenesis in breast cancer. Clin Cancer Res. 2004, 10: 2393-2400.
Bagnato A, Rosanò L, Di Castro V, Albini A, Salani D, Varmi M, Nicotra MR, Natali PG: Endothelin receptor blockade inhibits proliferation of Kaposi's sarcoma cells. Am J Pathol. 2001, 158: 841-847.
Rosanò L, Spinella F, Di Castro V, Nicotra MR, Albini A, Natali PG, Bagnato A: Endothelin receptor blockade inhibits molecular effectors of tumor invasion in Kaposi's sarcoma. Am J Pathol. 2003, 163: 753-762.
Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, Radmacher M, Simon R, Yakhini Z, Ben-Dor A, Sampas N, Dougherty E, Wang E, Marincola F, Gooden C, Lueders J, Glatfelter A, Pollock P, Carpten J, Gillanders E, Leja D, Dietrich K, Beaudry C, Berens M, Alberts D, Sondak V: Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature. 2000, 406: 536-540. 10.1038/35020115.
Demunter A, De Wolf-Peeters C, Degreef H, Stas M, van den Oord JJ: Expression of the endothelin-B receptor in pigment cell lesions of the skin. Evidence for its role as tumor progression marker in malignant melanoma. Virchows Arch. 2001, 438: 485-491. 10.1007/s004280000362.
Lahav R, Heffner G, Patterson PH: An endothelin receptor B antagonist inhibits growth and induces cell death in human melanoma cells in vitro and in vivo. Proc Natl Acad Sci USA. 1999, 96: 11496-11500. 10.1073/pnas.96.20.11496.
Bagnato A, Rosanò L, Spinella F, Di Castro V, Tecce R, Natali PG: Endothelin B receptor blockade inhibits dynamic of cell interactions and communications in melanoma cell progression. Cancer Res. 2003, 64: 1436-1443.
Thevarajah S, Udan MS, Zheng H, Pfluyg BR, Nelson JB: Endothelin axis expression in renal cell carcinoma. J Urol. 1999, 161: 137-143. 10.1097/00005392-199904010-00552.
Ahmed SI, Thompson J, Coulson JM, Woll PJ: Studies on the expression of endothelin, its receptor subtypes, and converting enzymes in lung cancer and in human bronchial epithelium. Am J Respir Cell Mol Biol. 2000, 22: 422-431.
Egidy G, Juillerat-Jeanneret L, Jeannin JF, Korth P, Bosman FT, Pinet F: Modulation of human colon tumor-stromal interactions by the endothelin system. Am J Pathol. 2000, 157: 1863-1874.
Eberl LP, Valdenaire O, Saintgiorgio V, Jeannin JF, Juillerat-Jeanneret L: Endothelin receptor blockade potentiates FasL-induced apoptosis in rat colon carcinoma cells. Int J Cancer. 2000, 86: 182-187. 10.1002/(SICI)1097-0215(20000415)86:2<182::AID-IJC6>3.0.CO;2-G.
Pagotto U, Arzberger T, Hopfner U, Sauer J, Renner U, Newton CJ, Lange M, Uhl E, Weindl A, Stalla GK: Expression and localization of endothelin-1 and endothelin receptors in human meningiomas. Evidence for a role in tumoral growth. J Clin Invest. 1995, 96: 2017-2025.
Harland SP, Kuc RE, Pickard JD, Davenport AP: Expression of endothelin A receptors in human gliomas and meningiomas, with high affinity for the selective antagonist PD156707. Neurosurgery. 1998, 43: 890-898.
Bagnato A, Natali PG: Targeting endothelin axis in cancer. In Molecular targeting and signal transduction. Edited by: Kumar R. 2004, Kluwer Academic Publishers, 293-314.
Acknowledgements
We thank L. Rosanò. F. Spinella, V. Di Castro and M.R. Nicotra for their contributions to studies on ovarian cancer cells, M.V. Sarcone for secretarial assistance and P. Nisen (Abbott Laboratories, Global Oncology Develepment, Abbott Park, IL) for kindly provided atrasentan. This work was supported by the Associazione Italiana Ricerca sul Cancro, Ministero della Salute and CNR-MIUR.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Bagnato, A., Natali, P.G. Endothelin receptors as novel targets in tumor therapy. J Transl Med 2, 16 (2004). https://doi.org/10.1186/1479-5876-2-16
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1479-5876-2-16