
Abstract
Background
Hepatitis C virus (HCV) causes liver fibrosis that may lead to liver cirrhosis or hepatocellular carcinoma (HCC), and may partially depend on infecting viral genotype. HCV genotype 3a is being more common in Asian population, especially Pakistan; the detail mechanism of infection still needs to be explored. In this study, we investigated and compared the gene expression profile between initial fibrosis stage and cirrhotic 3a genotype patients.
Methods
Gene expression profiling of human liver tissues was performed containing more than 22000 known genes. Using Oparray protocol, preparation and hybridization of slides was carried out and followed by scanning with GeneTAC integrator 4.0 software. Normalization of the data was obtained using MIDAS software and Significant Microarray Analysis (SAM) was performed to obtain differentially expressed candidate genes.
Results
Out of 22000 genes studied, 219 differentially regulated genes found with P ≤ 0.05 between both groups; 107 among those were up-regulated and 112 were down-regulated. These genes were classified into 31 categories according to their biological functions. The main categories included: apoptosis, immune response, cell signaling, kinase activity, lipid metabolism, protein metabolism, protein modulation, metabolism, vision, cell structure, cytoskeleton, nervous system, protein metabolism, protein modulation, signal transduction, transcriptional regulation and transport activity.
Conclusion
This is the first study on gene expression profiling in patients associated with genotype 3a using microarray analysis. These findings represent a broad portrait of genomic changes in early HCV associated fibrosis and cirrhosis. We hope that identified genes in this study will help in future to act as prognostic and diagnostic markers to differentiate fibrotic patients from cirrhotic ones.
Similar content being viewed by others

Background
Chronic hepatitis C is a major liver related health problem destroying liver architecture leading to cirrhosis and hepatocellular carcinoma. Almost 3% of the world population is infected with this deadly virus and in future, it is predicted that infection will rise to 3 fold of the present number [1–6]. HCV persist(s) beside the specific humoral responses and the mechanism of viral persistence and viral clearance is not fully understood. During HCV infection, initial fibrosis development is the method to overcome the damage caused by the virus. But the early events are the basis of disease outcome. Initial fibrosis is thought to be reversible, although many studies do not support this phenomenon. As extracellular matrix (ECM) tissues not only involve matrix production but also matrix degradation leading to ECM remodeling [7–9] Fibrosis is caused by excessive deposition of ECM by histological and molecular reshuffling of various components like collagens, glycoproteins, proteoglycans, matrix proteins and matrix bound growth factors. Fibrosis stage information not only indicates treatment response but also reflect/indicate cirrhosis development disaster [4, 10–16]. ECM metabolism is a balance between ECM deposition and removal influenced by cytokines and growth factors [17]. Genome-wide analysis of abnormal gene expression showed transcripts deregulation differences among normal, mild and severe fibrosis during HCC development with identification of novel serum markers for its early stage. Recent studies suggest that genetic markers may be able to define exact stage of liver fibrosis. For this purpose, limited but functional studies have proposed quite a few genetic markers with individual genes or group of genes [18, 19]. Advantage of genetic markers over liver biopsy is intrinsic and long-term while, liver biopsy represents only one time point [20]. Researchers found specific genes such as AZIN1, TLR4, CXCL9, CXCL10, CTGF, ITIH1, SERPINF2, TTR, PDGF, TGF-β1, collagens COL1-A1, TNFα, interleukin, ADAMTS, MMPs, TIMPs, LAMB1, LAMC1, Cadherin, CD44, ICAM1, ITGA, APO and CYP2C8 that showed deregulation during liver fibrosis and may be used to access liver fibrosis and cirrhosis [11–28]. Microarray is a powerful technique used for the identification of differentially expressed genes within control and experimental samples in different diseases and conditions like cancer development. Very few studies are available that use microarray for the identification of specific genes related to fibrosis [27, 28]. In a recent study, Caillot et al. used microarray technique and found a significant association of ITIH1, SERPINF2 and TTR gene expression and their related proteins with all fibrosis stages [28]. Expression of these genes and related proteins gradually decreased during the fibrosis development to its end stage cirrhosis. Mostly, HCV expression based studies using microarray are carried out with genotype 1 and 2. Very few studies exploring the role of HCV genotype 3a are done with limited set of genes using real Time PCR. Those do not represent complete picture of HCV and human gene interaction leading to disease progression [21–28]. In Pakistan, genotype 3a is the major contributor and has strong association with HCC. The aim of the present study was to examine gene expression profiles in the HCV associated liver disease progression. We have identified for the first time, those genes that are differentially regulated in initial fibrosis and advance stage liver cirrhosis 3a patients and identified potential targets that can be used as effective markers to differentiate between fibrotic and cirrhotic liver with genotype 3a. This data may also help to understand the disease stages between initial versus end stage cirrhosis, as there are limited studies concerning HCV genotype 3a disease progression.
Materials and methods
Patients
This study was conducted at Department of Pathology, Jinnah Hospital, Lahore, Mayo Hospital, Lahore and Liver Centre Faisalabad with collaboration of Applied and Functional Genomics Lab, National Centre of Excellence in Molecular Biology, University of the Punjab, Lahore, Pakistan. HCV RNA-positive patients were identified among HCV antibody (anti-HCV) positive patients. Patients who had received a previous course of INF or immunosuppressive therapy, or who had clinical evidence of HBV or HIV and any other type of liver cancer were excluded from the study. Patients who refused to have a liver biopsy or for whom it was contraindicated, i.e., because of a low platelet count, prolonged prothrombin time or decompensated cirrhosis were also excluded from the study. The liver biopsy procedure, its advantages and possible adverse effects were explained to the patients. Written informed consent for biopsy procedure was obtained from patients, also contained information about demographic data, possible transmission route of HCV infection, clinical, virological and biochemical data. The study was approved by institutional ethical committee.
Patients and liver biopsy
A group of patient was selected from previously described study with known fibrosis evaluation [29]. Two groups of samples consisted of early fibrosis (F1) and cirrhosis (F4) containing 9 samples each were made. Patient's characteristics are given in Table 1.
RNA isolation, cDNA and aRNA preparation, and dye labeling for microarray experiments
RNA from liver biopsy samples were isolated using RNeasy mini elute kit (Qiagen, USA) and preparation of cDNA and aRNA was carried out using RNA ampulse amplification and labeling kit (Kreatech, USA), according to manufacturer. aRNA from HCV infected patients and normal subjects were labeled with Cy3 and Cy5, respectively. A detailed protocol describing each step from start to microarray hybridization can be downloaded from (http://www.operon.com/products/microarrays/OpArray%20Protocol.pdf).
Array hybridization and scanning
Biopsy samples were analyzed on cDNA microarrays (Oparray) containing > 22000 named genes with 37584 spots. Equal amount of Cy3 and Cy5 (55 pmol each) labeled targets were mixed with 45 μl of OpArray Hyb Buffer. Pre-washing, array hybridization and post-washing of microarray labeled slides were performed according to the manufacturer protocols at 42°C for 18 hours on fully automated workstation "GeneTAC ™ HybStation".
Microarray data analysis
GeneTAC ™ UC4 × 4 scanner was used for scanning slides at 10 μm resolution for both Cy3 and Cy5 channels. GeneTAC Integrator 4.0 software was initially used for main data output as "csv" format file containing all necessary information. This "csv" file was converted to "mev" format for normalization by using software "ExpressConverter" (http://www.tm4.org/utilities.html). MIDAS (Microarray Data Analysis System) software was downloaded (http://www.tm4.org/midas.html) and used for normalization of data. Fold induction was determined by using formula log2Cy5/Cy3. A rank-based permutation method SAM was used to identify significantly expressed genes among fibrosis stages (http://www-stat.stanford.edu/~tibs/SAM/). Gene expression patterns through k-means clustering were produced and viewed using freely available programs CLUSTER 3.0 (http://rana.lbl.gov/EisenSoftware.htm) and Tree View 1.45 (http://rana.lbl.gov/downloads/TreeView/), respectively. To identify biological themes among gene expression profiles, the Expression Analysis Systematic Explorer (EASE) was used (http://david.abcc.ncifcrf.gov/content.jsp?file=/ease/ease1.htm&type=1) [30]. The microarray data have been deposited to the GEO accession database (http://www.ncbi.nlm.nih.gov/geo) with accession number GSE33258.
Real-time reverse transcriptase (RT)-PCR analysis
Genes with known function and significantly up-regulated or down-regulated were analyzed by real-time RT-PCR with RNA used for microarray analysis. Total RNA was converted to cDNA using MmLV (Moloney murine leukemia virus). Selected and tested oligonucleotide primer pairs for their specificity were used for real time RT-PCR using ABI 7500 real time PCR system using syber green chemistry. Each experiment was run in triplicate including GAPDH as endogenous control (Table 2). Each gene was quantified relative to the calibrator. Applied Biosystem Sequence Detection Software and calculations were made by instrument using the equation 2-ΔΔCT.
Results
Patient's characteristics
Among 18 patients, equal number of patients belonged to F1 (9) and cirrhotic (9) group. Out of these, six best samples each with good RNA were used for microarray experiments. Normal liver biopsies were also obtained in triplicate. The serum viral load, bilirubin, albumin, and platelet count of cirrhotic patients were significantly low (P < 0.05), while, serum ALP and AST levels were high when compared to patients with F1 stage. There were no significant differences between serum ALT and Hb level in the patients with F1 or cirrhotic stage (Table 1).
Microarray analysis: expression behavior of significant genes
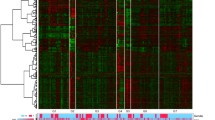
We found 219 differentially regulated genes in fibrosis versus cirrhotic groups (Figure 1). Among these, 107 genes were up-regulated (Figure 2) whereas, 112 genes were down-regulated (Figure 3). Significant genes with their symbols and functions are listed in Tables 3 and 4. Genes were classified into 31 categories according to their biological functions (Figure 4).

Significant host genes regulated by HCV infection. Clustering results for differentially expressed genes between HCV infected patients with initial fibrosis and cirrhosis. Clustering was performed by Cluster 3.0 software. The fold changes in mRNA expression are represented with green and red squares showing down- and up-regulation of genes in liver biopsy samples, respectively. Each vertical column represents an independent experiment, while color scale represents the fold change magnitude.

Heat map of up-regulated genes. Clustering results for differentially expressed genes between HCV infected patients with initial fibrosis and cirrhosis. Clustering was performed by Cluster 3.0 software. The fold changes in mRNA expression are represented with green and red squares showing down- and up-regulation of genes in liver biopsy samples, respectively. Each vertical column represents an independent experiment, while color scale represents the fold change magnitude.

Heat map of down-regulated genes in cirrhotic and non-cirrhotic sample. Clustering results for differentially expressed genes between HCV infected patients with initial fibrosis and cirrhosis. Clustering was performed by Cluster 3.0 software. The fold changes in mRNA expression are represented with green and red squares showing down- and up-regulation of genes in liver biopsy samples, respectively. Each vertical column represents an independent experiment, while color scale represents the fold change magnitude.

Distribution of genes according to their functions. Genes were grouped in 31 different categories.
Significantly synchronized genes with known biological functions
The differentially regulated genes were grouped according to their biological functions by EASE program that uses information from Entrez Gene (http://jura.wi.mit.edu/entrez_gene/) and KEGG database (http://www.genome.jp/kegg/kegg1.html). Our results showed variation in gene regulation in both early fibrosis and cirrhosis stages (Figure 1). Out of 107 up-regulated gens, 65 belonged to early fibrosis stage, whereas, 42 genes belonged to the cirrhotic stage. Genes related to immune response, cell signaling, kinase activity, lipid metabolism, metabolism, vision and transcriptional regulation were up-regulated in both early fibrosis and cirrhotic samples (Table 2). We found that most genes related to apoptosis, cell structure, cytoskeleton, nervous system protein metabolism, protein modulation, signal transduction, transcriptional regulation and transport were up-regulated in early fibrosis. Many uncharacterized genes were also found up-regulated in liver disease progression. We identified 112 genes (F1 = 92; F4 = 20) related to above mentioned pathways down-regulated when fibrosis lead to cirrhotic stage (Table 2 and Figure 2). Genes related to these pathways showed varied response and none of biological function was specifically related to any liver disease stage (Table 4 and Figure 3).
Independent validation of candidate genes using quantitative real-time RT-PCR
Total RNA extracted from infected liver biopsies was used for real time RT-PCR analysis to validate microarray data. Expression analysis of the genes involved in apoptosis, immune response and transcriptional regulation was performed. We randomly selected four genes, CASPASE9, FAM14B, OAS2 and TGFBR2 from our study. CASPASE9 is apoptosis related gene, FAM14B and OAS2 are immune responsive genes, whereas, TGFBR2 is multifunctional gene and found to be up-regulated in fibrosis.
Discussion
Liver fibrosis can progress to cirrhosis after an interval of 15-20 years in patients with HCV [31]. It is very important to identify such markers that can differentiate liver fibrosis from cirrhosis. Liver biopsy is a common tool for the detection of liver current situation but due to some limitations its use as diagnostic tool is denied. Microarray analysis is an emerging and novel approach to study gene expression in HCV associated fibrosis and cirrhosis. As liver gene expression in HCV patients is variable and it might be partially dependent on the corresponding genotype [32]. In this study, we specially focused on gene expression analysis in patients with genotype 3a that is most common in our region. We found that many genes associated with apoptosis, several cellular functions, immune response, metabolism including energy, liver, sulphur; protein metabolism, transcriptional regulation, signal transduction, transport, DNA replication were dys-regulated both in early fibrosis and cirrhosis. In some cases, gene expression tends to be increased from initial fibrosis to cirrhosis. Induction of gene expression associated with proapoptotic, proinflammatory and proliferative activities is in accordance with previous studies [18, 27, 33–35]. Although, we found some dysregulation of genes related to vision and nervous system first time.
Differential expression of apoptosis related genes in HCV associated initial fibrosis and cirrhosis
In this study, host genes involved in apoptosis (Figure 5) such as BCL212 and PDCD1 showed down-regulation in initial fibrosis and significant up-regulation in cirrhosis, whereas, expression levels for CASP9 and EMP1 genes were high at initial stage and were down-regulated in cirrhosis stage. Regulation of apoptotic inducer and program cell death genes, BCL212 and PDCD1 in cirrhosis is according to previous observations where pro-apoptotic gene signaling has been observed in infection with HCV [36, 37]. CASP9 is known as apoptosis initiator [38] and EMP1 is also found to induce apoptosis [39, 40]. Expression of caspases is higher in early and moderate HCV infection, and enhanced apoptosis occur through the intrinsic apoptotic pathway via mitochondria [41, 42].

Differential expression of apoptotic genes in HCV associated initial fibrosis and cirrhosis. Clustering results for differentially expressed genes between HCV infected patients with initial fibrosis and cirrhosis according to their functions. Clustering was performed by Cluster 3.0 software. Genes shown in red are up-regulated, while down-regulated genes are shown in green. Genes shown in black have no expression changes. Gene expression profiles were presented on a 2-fold change scale.
Cellular functions, cell cycle, signaling and cytoskeleton associated genes
Genes related to various cellular functions showed different expression patterns (Figure 6). The cytoskeleton (COMMD5, KRTAP19-5, LLGL1 and SNX17) related genes were down-regulated in cirrhosis (F4). Most cell structure related genes were up-regulated in initial fibrosis (HYLS1, MAP6D1 and TOR1AIP1) and genes related to cell adhesion, cell cycle and signaling showed differential expression in both initial fibrosis and cirrhosis. It has been observed that HCV RNA synthesis may require an intact cytoskeleton [43]; our data indicated that many genes related to cytoskeleton were regulated by HCV infection.

Parallel expression of genes associated with Cellular functions, cell cycle, signaling and cytoskeleton in F1 versus F4. Genes shown in red are up-regulated, while down-regulated genes are shown in green. Genes shown in black have no expression changes. Gene expression profiles were presented on a 2-fold change scale.
Genes associated with Immune response and cytokines
A number of genes related to immune response and cytokines were identified (Figure 7). ATG16L2, DEFB114, FAM14B, IFNA21, IL8RB and KIR2DL genes were up-regulated in cirrhosis, whereas, FCRL3, IFITM2 and OAS2 genes were up-regulated in initial fibrosis. Genes related to cytokine regulation, IL13RA2, PLEKHG6 and XCL2 were down-regulated in initial fibrosis except CRLF3 gene. Interleukin related gene expression has been found to be increased at pathology stage 3 and 4 and which is concurrent with the present study and is associated with metastatsis, cell proliferation or angiogenesis [37, 44]. An increased expression of immune responsive genes and cytokines as fibrosis progress is in agreement with previous evidence that liver inflammation may enhance with increase in infected hepatocytes [45]. FCRL3, a genetically conserved gene family encodes orphan cell surface receptors bearing high structural homology to classical Fc receptors, with multiple extracellular Ig domains and either ITAMs, ITIMs, or both in the intracellular domains. The natural ligands of these family members are still unknown but due to their signaling domains and expression on multiple immune cell types, these members likely modulate immune cell functions by affecting signaling pathways [46]. FCRL3 is expressed predominantly in B lymphocytes in lymph nodes and germinal centers [47–49].

Expression profiles of immune responsive and cytokines associated genes. Clustering results for differentially expressed genes between HCV infected patients with initial fibrosis and cirrhosis according to their functions. Clustering was performed by Cluster 3.0 software. Gene expression profiles were presented on a 2-fold change scale.
Previous studies revealed that IFITM2 and IFITM3 (two structurally related cell plasma membrane proteins) interrupt early steps entry and/or uncoating of the viral infection. Interferon-induced transmembrane (IFITM) genes are transcribed in most tissues with the exception of IFITM5 interferon inducible gene. IFITM genes are involved in early development, cell adhesion, and control of cell growth. Elevated gene expression triggered by past or chronic inflammation can prevent spreading of pathogens by limiting host cell proliferation. Low level of expression is sufficient to capture the growth of cells, whereas, the loss of expression causes tumor growth. This gene is termed as tumor suppressor. However, in many cancers it is observed that despite high level of IFITM, it represents tumor progression stage especially where the one of anti-proliferative interferon pathway is shut down. The role of ATG protein in membrane trafficking is mostly not clear. ATGL16 is thought to play role in autophagosome formation in association with RAB33B. It is also considered an active player in HCV replication and assembly [50, 51].
Natural killer cells are the important player of innate immune response. KIRDL gene expression is found to be high in chronic HCV patients [52]. We found the KIR2DL1 gene expression high in patients with cirrhosis as compared to initial fibrosis stage. OAS synthesized in response to IFN-alpha stimulation. In infected cells, OAS enzymatic activity is induced by double-stranded RNAs, such as the intermediates of replication of RNA viruses or folded single stranded RNAs. OAS catalyzes polymerization of adenosine triphosphate into oligoadenylate that, in turn, activates a cellular endoribonuclease, RNase L, at subnanomolar concentrations. RNase L degrades cellular and viral single-stranded RNAs. Thus, viral replication is inhibited as a result of protein synthesis inhibition in a totally non-virus specific way [53]. We found high expression of OAS2 gene in fibrotic samples as compared to the last stage cirrohsis. This may be a way to stop viral replication but as the disease steps forward, virus overcome the host immune response to replicate itself.
Genes associated with different metabolic processes
A number of genes associated with different metabolism (processes/pathways) like energy, kinases, lipid and sulphur metabolism were identified among significantly expressed arrays (Figure 8). Several studies observed that HCV induces alterations in lipid metabolism that can lead to oxidative stress [54, 55]. Consistent with these observations, we found six genes, ADA, CHKA, DEGS2, OSBPL2, PPAPDC3, and Q5R387; which are involved in lipid biosynthesis, tumor cell growth by phosphatidyl-ethanolamine biosynthesis, negative regulation of myoblast differentiation and hydrolyzation of phospholipids into fatty acids etc. This finding is in agreement with Diamond et al.; that host cell lipid metabolism may represent an area for future HCV antiviral therapies [56]. We found two genes FAM119B and FAM62B associated with sulphur metabolism which were up-regulated in cirrhotic samples. A number of genes related to energy mechanism such as PSMD4, PSMD11, ABHD2, ATAD2 and COX6A1 were up-regulated while, SYDE1 and Q5VTUB genes were down-regulated in cirrhotic samples. Two genes PDXK and PRKCB1 with kinase activity, and one gene, OGDHL linked to carbohydrate metabolism were also identified. Role of PRKCB1 (also known as PKC) in cell growth and differentiation control is known. It has been also found elevated in breast and pituitary tumors and malignant gliomas [57–59]. PKC was also found up regulated in hepatocellular carcinoma which can lead to hyper proliferation of the HCV infected tissues [60].

Genes associated with different metabolic processes. Clustering was performed by Cluster 3.0 software. Genes shown in red are up-regulated, while down-regulated genes are shown in green. Genes shown in black have no expression changes. Gene expression profiles were presented on a 2-fold change scale.
Genes associated with protein synthesis, modulation and metabolism
Many genes involved in protein synthesis, modulation and metabolism have increased or decreased expression in patients with HCV (Figure 9). Genes representing protein synthesis were down-regulated in initial fibrosis and showed significant increased expression in cirrhotic samples. Two genes associated with protein post-translational modifications (PTMs) were also identified that showed increased expression in cirrhosis. Some genes linked with protein metabolism like GON4L, OTUD7A, PHACTR4, Q96NT9 and WFDC13 showed low expression in initial fibrosis, while CSDE1, ENPP7, KIAA1147, KIAA2013 and KNG1 were up-regulated in early fibrosis. It was interesting to know that previous studies have not shown the regulation of PTMs and protein synthesis with respect to HCV, although other viruses such as HIV have shown these trends. However, our findings were in agreement with Blackham et al. who showed these types of regulations in HCV infected hepatocytes [61].

Genes associated with protein synthesis, modulation and metabolism. Genes shown in red are up-regulated, while down-regulated genes are shown in green. Genes shown in black have no expression changes.
Transcriptional regulation and signal transduction related genes
Several genes associated with transcriptional regulation and signal transductions were identified (Figure 10). Most genes were down-regulated both in HCV initial fibrosis and cirrhosis. However, ANKHD1, CRAMP1L, FOXK2, GTF2B, HMGN2, NR1I3, PAX8, RUNX2 and SUSD4 genes showed increased trend in cirrhotic samples. Xu et al. also reported up-regulation of liver enriched transcriptional factors in infected HCV tissues [62]. A comprehensive study is needed to address the exact role of these genes. Some genes associated with signal transduction like CACNB3, PCSK5, TMEM100 and VDAC3 were up-regulated in initial fibrosis. Up-regulation of signal transduction related genes in HCC due to HCV and HBV is previously reported [63, 64]. This can lead to the hypothesis that cirrhosis due to HCV genotype 3a may lead to HCC in future.

Expression of transcription and signal transduction related genes. Clustering was performed by Cluster 3.0 software.
Transport and ion channel transport related genes
A number of genes encoding cellular and ion transport functions were also recognized (Figure 11). AMICA1, HHLA3, KIF17, KIF1A and SLC10A5 showed significant high expression, while, CLPB, K1024, MUC6, SCGN and MT1E expression was down in cirrhotic arrays. Previous studies related to HCV infection and entry has shown that HCV replication needs regulations in cellular trafficking [65–67]. High expression of SLC10A5, also known as putative bile acid transporter gene, it may indicate dysregulation of liver as well as pancreas in patients infected with HCV. Up-regulation of kinesin family members KIF17 or KIF2B may upset inner segment and synaptic terminal and consequently results in cell death [68].

Regulation of transport and ion channel related genes by HCV. Clustering results for differentially expressed genes between HCV infected patients with initial fibrosis and cirrhosis according to their functions. Genes shown in red are up-regulated, while down-regulated genes are shown in green. Genes shown in black have no expression changes.
Others significant genes
Irrespective of above mentioned genes; we have also found several genes related to DNA binding proteins, DNA replication, morphogenesis, reproduction and liver function (Figure 12). The expression of DNA binding protein and replication genes change from initial fibrosis to cirrhosis. The high expression in early fibrosis may underlie a repair mechanism, whereas, reduced gene expression in cirrhosis stage may indicate that virus has overcome the repair mechanism for its replication resulting in total deterioration of liver cells and structure. It is interesting to note that some genes associated with nervous system and vision pathways were also identified. A lot of uncharacterized genes were also recognized. The link of expression of vision related genes with HCV is not clear.

Differential expression of genes associated with DNA binding, DNA replication, liver function, nervous system, vision and uncharacterized. Clustering results for differentially expressed genes between HCV infected patients with initial fibrosis and cirrhosis according to their functions. Clustering was performed by Cluster 3.0 software. Genes shown in red are up-regulated, while down-regulated genes are shown in green. Genes shown in black have no expression changes. Gene expression profiles were presented on a 2-fold change scale.
Real time RT-PCR validation of results
Analysis with real time RT-PCR confirmed that the selected genes were significantly differentially expressed in initial fibrosis and cirrhotic samples (Figure 13). Although, we observed higher fold induction values with real time RT-PCR, however, the trend was same between both analysis indicating reproducible gene expression patterns. CASPASE9, OAS2 and TGFBR2 genes showed up-regulation, whereas, FAM14B gene expression was down-regulated in early fibrosis. These findings open a new spectrum of genetic markers to differentiate fibrosis from cirrhosis.

Validation of microarray data by RT-PCR. (A) Quantification of differential expression of randomly selected genes by real time RT-PCR. (B) Expression profile of selected genes from our microarray study.
A comprehensive review of literature revealed that very few studies related to HCV expression based studies leading to initial to final stage cirrhosis have been carried out in association to genotype. Walters et al. used J6/JFH (genotype 2a) infected Huh-7.5 cells for the expression analysis of host in response to virus at different time points of infection. They observed that TGF-beta signaling genes were up-regulated 72 hrs post infection, it induces ROS activity. Liver injury during chronic HCV infection is immune mediated [37]. Hagist et al. compared differentially expressed genes in patients with mild and severe iron depleted HCV genotype 1a liver samples with hereditary hemochromatosis. They found many ISG genes dysregulated in HCV infection and related to RNA processing and carcinogenesis [69]. We also found up-regulation of ISG genes in initial fibrosis stage as host defense system try to limit the viral pathogenesis. A study conducted by Blackham et al. in JFH1 infected huh-7 cells by microarray identified genes mainly apoptosis, proliferation, intracellular transport and cellular mechanism [61]. A few studies to explore the role of individual genes of HCV in pathogenesis have been studied in association to genotype. Shah et al. compared the expression of oxidative stress related genes in blood samples and found that the expression of COX-2, iNOs and VEGF was high in 3a in comparison to 1a [70]. We found the expression is high in initial fibrosis stage and down regulation at the advance stage of liver cirrhosis.
Conclusion
There are limited studies available dealing with gene expression profiling in cirrhotic and non-cirrhotic (initial fibrosis) patients infected with HCV. In this study, we have observed that HCV infection due to genotype 3a has widespread effects on host gene expression involved in apoptosis, metabolism, transport, transcriptional regulation and immune response. This gives comprehensive information about the pathogenesis caused by HCV genotype 3a leading from initial to end stage liver cirrhosis. Although, HCV genotype 3a showed same pathways activation caused by other genotypes, further studies are required to understand the mechanism by which different genotypes can affect various pathways. Meanwhile, we found that expression of these genes was significantly changed within initial and final stage of fibrosis. A study describing the progression of these genes in mild and severe fibrosis stages (F2 and F3) will be required for future perspectives.
Authors' information
WA and BI are research officers at CEMB, while SH (PhD Molecular Biology) is Principal Investigator at CEMB, University of the Punjab, Lahore.
References
Memon MI, Memon MA: Hepatitis C: an epidemiological review. J Viral Hepat. 2002, 9: 84-100. 10.1046/j.1365-2893.2002.00329.x.
WHO: Global distribution of hepatitis A, B and C, 2001. Weakly Epidimiological Records. 2002, 77: 41-48.
Marcellin P, Asselah T, Boyer N: Fibrosis and disease progression in hepatitis C. Hepatology. 2002, 36: S47-56.
Afdhal NH, Nunes D: Evaluation of liver fibrosis: a concise review. Am J Gastroenterol. 2004, 99: 1160-1174. 10.1111/j.1572-0241.2004.30110.x.
Dienstag JL, McHutchison JG: American Gastroenterological Association technical review on the management of hepatitis C. Gastroenterology. 2006, 130: 231-264. 10.1053/j.gastro.2005.11.010. quiz 214-237
Clark JM: The epidemiology of nonalcoholic fatty liver disease in adults. J Clin Gastroenterol. 2006, 40 (Suppl 1): S5-10.
Bataller Ramón, Brenner David: Liver fibrosis. J Clin Invest. 2005, 115 (2): 209-218.
Zois CD, Baltayiannis GH, Karayiannis P, Tsianos EV: Systematic review: hepatic fibrosis-regression with therapy. Aliment Pharmacol Ther. 2008, 28 (10): 1175-1187. 10.1111/j.1365-2036.2008.03840.x.
Friedman SL: Reversibility of hepatic fibrosis and cirrhosis--is it all hype?. Nat Clin Pract Gastroenterol Hepatol. 2007, 4 (5): 236-237.
Harbin WP, Robert NJ, Ferrucci JT: Diagnosis of cirrhosis based on regional changes in hepatic morphology: a radiological and pathological analysis. Radiology. 1980, 135: 273-283.
Gressner AM: The cell biology of liver fibrogenesis-an imbalance of proliferation, growth arrest and apoptosis of myofibroblasts. Cell Tissue Res. 1998, 292: 447-452. 10.1007/s004410051073.
Arthur MJ: Reversibility of liver fibrosis and cirrhosis following treatment for hepatitis C. Gastroenterology. 2002, 122: 1525-1528. 10.1053/gast.2002.33367.
Adinolfi LE, Gambardella M, Andreana A, Tripodi MF, Utili R, Ruggiero G: Steatosis accelerates the progression of liver damage of chronic hepatitis C patients and correlates with specific HCV genotype and visceral obesity. Hepatology. 2001, 33: 1358-1364. 10.1053/jhep.2001.24432.
El-Serag HB: Hepatocellular carcinoma and hepatitis C in the United States. Hepatology. 2002, 36: S74-83.
Parkes J, Guha IN, Roderick P, Rosenberg W: Performance of serum marker panels for liver fibrosis in chronic hepatitis C. J Hepatol. 2006, 44: 462-474. 10.1016/j.jhep.2005.10.019.
Ziol M, Handra-Luca A, Kettaneh A, Christidis C, Mal F, Kazemi F, de Ledinghen V, Marcellin P, Dhumeaux D, Trinchet JC, Beaugrand M: Noninvasive assessment of liver fibrosis by measurement of stiffness in patients with chronic hepatitis C. Hepatology. 2005, 41: 48-54. 10.1002/hep.20506.
Ngo Y, Munteanu M, Messous D, Charlotte F, Imbert-Bismut F, Thabut D, Lebray P, Thibault V, Benhamou Y, Moussalli J, Ratziu V, Poynard T: A prospective analysis of the prognostic value of biomarkers (FibroTest) in patients with chronic hepatitis C. Clin Chem. 2006, 52: 1887-1896. 10.1373/clinchem.2006.070961.
Asselah T, Bieche I, Laurendeau I, Paradis V, Vidaud D, Degott C, Martinot M, Bedossa P, Valla D, Vidaud M, Marcellin P: Liver gene expression signature of mild fibrosis in patients with chronic hepatitis C. Gastroenterology. 2005, 129: 2064-2075. 10.1053/j.gastro.2005.09.010.
Smith MW, Walters KA, Korth MJ, Fitzgibbon M, Proll S, Thompson JC, Yeh MM, Shuhart MC, Furlong JC, Cox PP, Thomas DL, Philips JD, Kushner JP, fausto N, Carithers RL, Katze MG: Gene expression patterns that correlate with hepatitis C and early progression to fibrosis in liver transplant recipients. Gastroenterology. 2006, 130: 179-187. 10.1053/j.gastro.2005.08.015.
Huang H, Shiffman ML, Cheung RC, Layden TJ, Friedman S, Abar OT, Yee L, Chokkalingam AP, Schrodi SJ, Chan J, Catanese JJ, Leong DU, Ross D, Hu X, Monto A, McAllister LB, Border S, White T, Sninsky JJ, Wright TL: Identification of two gene variants associated with risk of advanced fibrosis in patients with chronic hepatitis C. Gastroenterology. 2006, 130: 1679-1687. 10.1053/j.gastro.2006.02.032.
Machida K, Cheng KT, Sung VM, Levine AM, Foung S, Lai MM: Hepatitis C virus induces toll-like receptor 4 expression, leading to enhanced production of beta interferon and interleukin-6. J Virol. 2006, 80: 866-874. 10.1128/JVI.80.2.866-874.2006.
Huang H, Shiffman ML, Friedman S, Venkatesh R, Bzowej N, Abar OT, Rowland CM, Catanese JJ, Leong DU, Sninsky JJ, Layden TJ, Wright TL, White T, Cheung RC: A 7 gene signature identifies the risk of developing cirrhosis in patients with chronic hepatitis C. Hepatology. 2007, 46: 297-306. 10.1002/hep.21695.
Zeremski M, Petrovic LM, Chiriboga L, Brown QB, Yee HT, Kinkhabwala M, Jacobson IM, Dimova R, Markatou M, Talal AH: Intrahepatic levels of CXCR3-associated chemokines correlate with liver inflammation and fibrosis in chronic hepatitis C. Hepatology. 2008, 48: 1440-1450. 10.1002/hep.22500.
Zeremski M, Dimova R, Brown Q, Jacobson IM, Markatou M, Talal AH: Peripheral CXCR3-associated chemokines as biomarkers of fibrosis in chronic hepatitis C virus infection. J Infect Dis. 2009, 200: 1774-1780. 10.1086/646614.
Sharma A, Chakraborti A, Das A, Dhiman RK, Chawla Y: Elevation of interleukin-18 in chronic hepatitis C: implications for hepatitis C virus pathogenesis. Immunology. 2009, 128: e514-522. 10.1111/j.1365-2567.2008.03021.x.
Gutierrez-Reyes G, Gutierrez-Ruiz MC, Kershenobich D: Liver fibrosis and chronic viral hepatitis. Arch Med Res. 2007, 38: 644-651. 10.1016/j.arcmed.2006.10.001.
Shao RX, Hoshida Y, Otsuka M, Kato N, Tateishi R, Teratani T, Shiina S, Taniguchi H, Moriyama M, Kawabe T, Omata M: Hepatic gene expression profiles associated with fibrosis progression and epatocarcinogenesis in hepatitis C patients. World J Gastroenterol. 2005, 11 (13): 1995-1999.
Caillot F, Hiron M, Goria O, Gueudin M, Francois A, Scotte M, Daveau M, Salier JP: Novel serum markers of fibrosis progression for the follow-up of hepatitis C virus-infected patients. Am J Pathol. 2009, 175: 46-53. 10.2353/ajpath.2009.080850.
Ahmad W, Ijaz B, Javed FT, Gull S, Kausar H, Sarwar MT, Asad S, Shahid I, Sumrin A, Khaliq S, Jahan S, Pervaiz A, Hassan S: A comparison of four fibrosis indexes in chronic HCV: Development of new fibrosis-cirrhosis index (FCI). BMC Gastroenterol. 2011, 11: 44-10.1186/1471-230X-11-44.
Hosack DA, Dennis G, Sherman BT, Lane HC, Lempicki RA: Identifying biological themes within lists of genes with EASE. Genome Biol. 2003, 4 (10): R70-10.1186/gb-2003-4-10-r70.
Pazienza V, Clément S, Pugnale P, Conzelmann S, Pascarella S, Mangia A, Negro F: Gene expression profile of Huh-7 cells expressing hepatitis c virus genotype 1b or 3a core proteins. Liver Int. 2009, 9 (5): 661-669.
Lau DT, Luxon BA, Xiao SY, Beard MR, Lemon SM: Intra-hepatic gene expression profiles and alpha-smooth muscle actin patterns in hepatitis C virus induced fibrosis. Hepatology. 2005, 42 (2): 273-281. 10.1002/hep.20767.
Shackel NA, McGuinness PH, Abbott CA, Gorrell MD, McCaughan GW: Insights into the pathobiology of hepatitis C virus-associated cirrhosis: Analysis of intrahepatic differential gene expression. Am J Pathol. 2002, 160 (2): 641-654. 10.1016/S0002-9440(10)64884-5.
Smith MW, Yue ZN, Korth MJ, Do HA, Boix L, Fausto N, Bruix J, Carithers RL, Katze MG: Hepatitis C virus and liver disease: global transcriptional profiling and identification of potential markers. Hepatology. 2003, 38 (6): 1458-1467.
Lan L, Gorke , Rau SJ, Zeisel MB, Hildt E, Himmelsbach K, Carvajal-Yepes M, Huber R, Wakita T, Schmitt-Graeff A, Royer C, Blum HE, Fischer R, Baumert TF: Hepatitis C virus infection sensitizes human hepatocytes to TRAIL-induced apoptosis in a caspase 9-dependent manner. J Immunol. 2008, 181: 4926-4935.
Kubo F, Ueno S, Hiwatashi K, Sakoda M, Kawaida K, Nuruki K, Aikou T: Interleukin 8 in human hepatocellular carcinoma correlates with cancer cell invasion of vessels but not with tumor angiogenesis. Ann Surg Oncol. 2005, 12: 800-807. 10.1245/ASO.2005.07.015.
Walters KA, Syder AJ, Lederer SL, Diamond DL, Paeper B, Rice CM, Katze MG: Genomic analysis reveals a potential role for cell cycle perturbation in HCV-mediated apoptosis of cultured hepatocytes. PLoS Pathog. 2009, 5: e1000269-10.1371/journal.ppat.1000269.
Milleron RS, Bratton SB: Heat shock induces apoptosis independently of any known initiator caspase-activating complex. J Biol Chem. 2006, 281 (25): 16991-7000. 10.1074/jbc.M512754200.
Fischer R, Baumert T, Blum HE: Hepatitis C virus infection and apoptosis. World J Gastroenterol. 2007, 13 (36): 4865-4872.
Wang HT, Kong JP, Ding F, Wang XQ, Wang MR, Liu LX, Wu M, Liu ZH: Analysis of gene expression profile induced by EMP-1 in esophageal cancer cells using cDNA Microarray. World J Gastroenterol. 2003, 9 (3): 392-398.
Calabrese F, Pontisso P, Pettenazzo E, Benvegnu L, Vario A, Chemello L, Alberti A, Valente M: Liver cell apoptosis in chronic hepatitis C correlates with histological but not biochemical activity or serum HCV-RNA levels. Hepatology. 2000, 31: 1153-1159. 10.1053/he.2000.7123.
Malhi H, Guicciardi ME, Gores GJ: Hepatocyte death: a clear and present danger. Physiol Rev. 2010, 90 (3): 1165-94. 10.1152/physrev.00061.2009.
Ciccaglione RA, Marcantonio C, Tritarelli E, Tataseo P, Ferraris A, Bruni R, Dallapiccola B, Gerosolimo G, Costantino A, Rapicetta M: Microarray analysis identifies a common set of cellular genes modulated by different HCV replicon clones. BMC Genomics. 2008, 9: 309-10.1186/1471-2164-9-309.
Behrens C, Feng L, Kadara H, Kim HJ, Lee JJ, Mehran R, Hong WK, Lotan R, Wistuba II: Expression of interleukin-1 receptor-associated kinase-1 in non-small cell lung carcinoma and preneoplastic lesions. Clin Cancer Res. 2010, 16 (1): 34-44. 10.1158/1078-0432.CCR-09-0650.
Davis RS: Fc receptor-like molecules. Annu Rev Immunol. 2007, 25: 525-560. 10.1146/annurev.immunol.25.022106.141541.
Kochi Y, Yamada R, Suzuki A, Harley JB, Shirasawa S, Sawada T, Bae SC, Tokuhiro S, Chang X, Sekine A, Takahashi A, Tsunoda T, ohnishi Y, Kaufman KM, Kang CP, Kang C, Otsubo S, Yamura W, Mimori A, Koike T, Nakamura Y, Saazuki T, Yamamoto K: A functional variant in FCRL3, encoding Fc receptor-like 3, is associated with rheumatoid arthritis and several autoimmunities. Nat Genet. 2005, 37: 478-485. 10.1038/ng1540.
Miller I, Hatzivassiliou G, Cattoretti G, Mendelsohn C, Dalla-Favera R: IRTAs: a new family of immunoglobulinlike receptors differentially expressed in B cells. Blood. 2002, 99: 2662-2669. 10.1182/blood.V99.8.2662.
Polson AG, Zheng B, Elkins K, Chang W, Du C, Dowd P, Yen L, Tan C, Hongo JA, Koeppen K, Ebens A: Expression pattern of the human FcRH/IRTA receptors in normal tissue and in B-chronic lymphocytic leukemia. Int Immunol. 2006, 18: 1363-1373. 10.1093/intimm/dxl069.
Itoh T, Fujita N, Kanno E, Yamamoto A, Yoshimori T, Fukuda M: Golgi-resident small GTPase Rab33B interacts with Atg16L and modulates autophagosome formation. Mol Biol Cell. 2008, 19: 2916-2925. 10.1091/mbc.E07-12-1231.
Tanida I, Fukasawa M, Ueno T, Kominami E, Wakita T, Hanada K: Knockdown of autophagy-related gene decreases the production of infectious hepatitis C virus particles. Autophagy. 2009, 5: 937-945. 10.4161/auto.5.7.9243.
Dring MM, Morrison HM, McSharry PB, Guinan JK, Hagan R, Irish Research Consortium, Farrelly OC, Gardiner CM: Innate immune genes synergize to predict increased risk of chronic disease in hepatitis C virus infection. Proc Natl Acad Sci USA. 2011, 108: 5736-5741. 10.1073/pnas.1016358108.
Hovanessian AG: Interferon-induced and double-stranded RNA-activated enzymes: a specific protein kinase and 29,59-oligoadenylate synthetases. J Interferon Res. 1991, 11: 199-205. 10.1089/jir.1991.11.199.
Pawlotsky JM: Hepatitis C virus resistance to antiviral therapy. Hepatology. 2000, 32 (5): 889-896. 10.1053/jhep.2000.19150.
Syed GH, Amako Y, Siddiqui A: Hepatitis C virus hijacks host lipid metabolism. Trends Endocrinol Metab. 2010, 21 (1): 33-40. 10.1016/j.tem.2009.07.005.
Simula MP, De Re V: Hepatitis C virus-induced oxidative stress and mitochondrial dysfunction: a focus on recent advances in proteomics. Proteomics Clin Appl. 2010, 4 (10-11): 782-793. 10.1002/prca.201000049.
Diamond DL, Syder AJ, Jacobs JM, Sorensen CM, Walters KA, Proll SC, McDermott JE, Gritsenko MA, Zhang Q, Zhao R, Metz TO, Camp DG, Waters KM, Smith RD, Rice CM, Katze MG: Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010, 6 (1): e1000719-10.1371/journal.ppat.1000719.
Alvaro V, Touraine P, Raisman Vozari R, Bai-Grenier F, Birman P, Joubert D: Protein kinase C activity and expression in normal and adenomatous human pituitaries. Int J Cancer. 1992, 50: 724-730. 10.1002/ijc.2910500510.
Craxi A, Pasqua P, Giannuoli G, Di Stefano R, Simonetti RG, Pagliaro L: Tissue markers of hepatitis B virus infection in hepatocellular carcinoma and cirrhosis. Hepatogastroenterology. 1984, 31: 55-59.
O'Brian C, Vogel VG, Singletary SE, a Ward NE: Elevated protein kinase C expression in human breast tumor biopsies relative to normal breast tissue. Cancer Res. 1989, 49: 3215-3217.
Tsai JH, Hsieh YS, Kuo SJ, Chen ST, Yu SY, Huang CY, Chang AC, Wang YW, Tsai MT, Liu JY: Alteration in the expression of protein kinase C isoforms in human hepatocellular carcinoma. Cancer Lett. 2000, 161: 171-175. 10.1016/S0304-3835(00)00597-8.
Blackham S, Baillie A, Al-habibi F, Remlinger K, You S, Hamatake R, mcGarvey MJ: Gene expression profiling indicates the roles of host oxidative stress, apoptosis, lipid metabolism, and intracellular transport genes in the replication of hepatitis C virus. J Virol. 2010, 84: 5404-5414. 10.1128/JVI.02529-09.
Xu L, Hui L, Wang S, Gong J, Jin Y, Wang Y, Ji Y, Wu X, Han Z, Hu G: Expression profiling suggested a regulatory role of liver-enriched transcription factors in human hepatocellular carcinoma. Cancer Res. 2001, 61: 3176-3181.
Xu XR, Huang J, Xu ZG, Qian BZ, Zhu ZD, Yan Q, Cai T, Zhang X, Xiao HS, Qu J, Liu F, Huang QH, Cheng ZH, Li N G, Du JJ, Hu W, Shen KT, Lu G, Fu G, Zhong M, Xu SH, Gu WY, Huang W, Zhao XT, Hu GX, Gu JR, Chen Z, Han ZG: Insight into hepatocellular carcinogenesis at transcriptome level by comparing gene expression profiles of hepatocellular carcinoma with those of corresponding noncancerous liver. Proc Natl Acad Sci USA. 2001, 98: 15089-15094. 10.1073/pnas.241522398.
Okabe H, Satoh S, Kato T, Kitahara O, Yanagawa R, Yamaoka Y, Tsunoda T, Furukawa Y, Nakamura Y: Genome-wide analysis of gene expression in human hepatocellular carcinomas using cDNA microarray: identification of genes involved in viral carcinogenesis and tumor progression. Cancer Res. 2001, 61: 2129-2137.
Bost AG, Venable D, Liu L, Heinz BA: Cytoskeletal requirements for hepatitis C virus (HCV) RNA synthesis in the HCV replicon cell culture system. J Virol. 2003, 77: 4401-4408. 10.1128/JVI.77.7.4401-4408.2003.
Hamamoto I, Nishimura Y, Okamoto T, Aizaki H, Liu M, Mori Y, Abe T, Suzuki T, Lai MM, Miyamura T, Moriishi K, Matsuura Y: Human VAP-B is involved in hepatitis C virus replication through interaction with NS5A and NS5B. J Virol. 2005, 79: 13473-13482. 10.1128/JVI.79.21.13473-13482.2005.
Lai CK, Jeng KS, Machida K, Lai MM: Association of hepatitis C virus replication complexes with microtubules and actin filaments is dependent on the interaction of NS3 and NS5A. J Virol. 2008, 82: 8838-8848. 10.1128/JVI.00398-08.
Insinna C, Humby M, Sedmak T, Wolfrum U, Beshase JC: Different roles for KIF17 and kinesin II in photoreceptor development and maintenance. Dev Dyn. 2009, 238: 2211-2222. 10.1002/dvdy.21956.
Hagist S, Sültmann H, Millonig G, Hebling U, Kieslich D, Kuner R, Balaguer S, Seitz HK, Poustka A, Mueller S: In vitro-targeted gene identification in patients with hepatitis C using a genome-wide microarray technology. Hepatology. 2009, 49 (2): 378-86. 10.1002/hep.22677.
Jahan S, Khaliq S, Ijaz B, Ahmad W, Hassan S: Role of HCV Core gene of genotype 1a and 3a and host gene Cox-2 in HCV-induced pathogenesis. Virol J. 2011, 8: 155-10.1186/1743-422X-8-155.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
WA and BI contributed equally to this work. They analyzed the data and wrote paper. All work was performed under supervision of SH. We all authors read and approved the final manuscript.
Waqar Ahmad, Bushra Ijaz contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ahmad, W., Ijaz, B. & Hassan, S. Gene expression profiling of HCV genotype 3a initial liver fibrosis and cirrhosis patients using microarray. J Transl Med 10, 41 (2012). https://doi.org/10.1186/1479-5876-10-41
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1479-5876-10-41