
Abstract
Background
Gene targeting in vivo provides a potentially powerful method for gene analysis and gene therapy. In order to sensitively detect and accurately measure designed sequence changes, we have used a transgenic mouse system, MutaMouse, which has been developed for detection of mutation in vivo. It carries bacteriophage lambda genome with lacZ+ gene, whose change to lacZ-negative allele is detected after in vitro packaging into bacteriophage particles. We have also demonstrated that gene transfer with a replication-defective adenovirus vector can achieve efficient and accurate gene targeting in vitro.
Methods
An 8 kb long DNA corresponding to the bacteriophage lambda transgene with one of two lacZ-negative single-base-pair-substitution mutant allele was inserted into a replication-defective adenovirus vector. This recombinant adenovirus was injected to the transgenic mice via tail-vein. Twenty-four hours later, genomic DNA was extracted from the liver tissue and the lambda::lacZ were recovered by in vitro packaging. The lacZ-negative phage was detected as a plaque former on agar with phenyl-beta-D-galactoside.
Results
The mutant frequency of the lacZ-negative recombinant adenovirus injected mice was at the same level with the control mouse (~1/10000). Our further restriction analysis did not detect any designed recombinant.
Conclusion
The frequency of gene targeting in the mouse liver by these recombinant adenoviruses was shown to be less than 1/20000 in our assay. However, these results will aid the development of a sensitive, reliable and PCR-independent assay for gene targeting in vivo mediated by virus vectors and other means.
Similar content being viewed by others


Background
Gene targeting, which is the precise alteration of genomic information by homologous recombination, has provided a powerful means of genetic analysis in microorganisms and mammalian systems [1]. In mouse systems, embryonic stem-cell lines modified in vitro can be used to generate mice that are altered at the germ-line level. If the gene targeting of somatic cells is made possible by gene transfer in vivo, it will facilitate the analysis of gene function, and provide a means of gene therapy for genetic and other diseases [2].
There are two major inherent problems with the use of gene targeting in vivo. First, its low efficiency makes it difficult to detect and analyze. A sensitive and accurate measurement system is therefore needed to detect such low-frequency events. Although there have been several reports of gene targeting in the rat liver with specifically designed oligonucleotides [3, 4], their reproducibility remains controversial [5]. PCR-based detection methods might thus be inaccurate and prone to various artifacts. In order to detect and measure gene targeting in mice with sufficient sensitivity, we used a bacteriophage transgenic-mouse system, MutaMouse, which has been developed for the detection of mutagenesis in vivo (Figure 1) [6]. The MutaMouse carries tandem repeats of the bacteriophage lambda genome with the lacZ+ gene, in which the change to a lacZ-negative allele is detected after its in vitro packaging into viable bacteriophage particles.
The second major problem with gene targeting in vivo is that non-homologous recombination is much more frequent than homologous recombination in mammalian cells. Rare accurately modified cells are selected and purified in the case of embryonic stem cells that are treated in vitro. For gene targeting in vivo, imprecise modification would be detrimental for analytical uses and therapeutic purposes. Accurate gene modification has been achieved efficiently using replication-defective adenovirus vectors for gene delivery in vitro [7, 8]. Fujita and colleagues used a mammalian plasmid as a model target [7]. The gene targeting was frequent (~10-4 per cell) and analysis of the products revealed that homologous recombination was more frequent than non-homologous recombination. One possible reason for this high accuracy was protection of the viral DNA by the terminal protein, which is covalently attached to the ends of the viral DNA and to other viral proteins during its transfer to the nucleus and target DNA. Breaks in unprotected DNA would lead to non-homologous recombination.

Experimental steps to detect gene targeting in vivo. Gene targeting in vivo in liver cells was attempted after the delivery of donor DNA with an adenovirus vector. The gene with the required sequence change (lacZ-) on the lambda transgene in the mouse will be detected after its recovery in bacteriophage particles. Only lacZ-negative mutants can form plaques under the selective conditions.
The adenovirus is useful for gene delivery in vivo because it has a broad host-range, is easy to prepare to a high titer and only rarely integrates into the host genome by non-homologous recombination [9, 10]. To date, more than 170 clinical studies have used recombinant adenovirus vectors to express cDNA in humans [11]. Numerous adenovirus-infection experiments have been carried out with mice, and have established that the injection of adenovirus recombinants into the mouse tail-vein leads to the expression of their genes in approximately one-half of the liver cells [12, 13].
In the present study, we investigated gene targeting in the mouse liver using a replication-defective adenovirus vector and a transgenic mouse system (Figure 1). Although our initial attempts did not detect the predicted gene targeting (the frequency of the expected recombinants was less than 1/20,000 per lambda genome), the strategy and methods detailed here will aid the development of virus-mediated gene targeting in vivo.
Materials and methods
Bacteria, bacteriophages and plasmids
The bacteria, bacteriophages and plasmids used in this study are listed together with details of their construction in Additional file 1.
BIK12001 was used for the titration of bacteriophage lambda and the measurement of lacZ-negative bacteriophage lambda by phenyl beta-D-galactoside (p-gal) selection (see below). BIK1564 was used for the growth of all bacteriophage lambda strains in this study. BIK2206 was used for confirmation of the LacZ-negative phenotype of the bacteriophage selected with p-gal using 5-bromo-4-chloro-3-indlyl-beta-D-galactose (X-gal).
The construction of the plasmids used in this study is detailed in additional file 1. The construction of pAdNY58 is also illustrated in Figure 2. The construction of pAdNY57 was as follows. The SmaI(1)-SacI fragment of LIA7 within the lacZ gene (Figure 2) was used to replace the shorter SmaI-SacI fragment of pUC18. The Glu461Gly mutation (Figure 3) was introduced into the resulting plasmid (pNY15) by site-directed mutagenesis using PCR [14] as follows. The PCR products generated with the primer pair LZG-U (5'-ACCGGCGATGAGCGAA-3') and LZG-MA (5'-GCCTGATCCATTCCCCAGCGACCA-3'), and the primer pair LZG-MS (5'-GGGAATGGATCAGGCCACGGCCGC-3') and LZG-D (5'-GGGCTGGTCTTCATCC-3'), were mixed and used as templates for the second round of PCR with the primer pair LZG-U and LZG-D. The MluI-BssHII fragment of the wild-type lacZ gene of pNY15 was replaced by the MluI-BssHII fragment of the PCR product. The targeted change in the resulting plasmid (pNY15G3.11) was confirmed by sequencing. pNY20 was produced by replacing the smaller SmaI-SacI fragment of pNY19 with the homologous SmaI-SacI fragment of pNY15G3.11, which carries the mutant sequence.

Construction of the recombinant adenovirus AdNY58. The bacteriophage lambda LIA7 was recovered from the MutaMouse by in vitro packaging. An SmaI-SacI fragment of LIA7 within its lacZ gene was inserted into pIK153. The Tyr105Stop mutation (Figure 3) was introduced into the resulting plasmid (pIK153LZS.6) using site-directed mutagenesis by PCR as follows. The PCR products generated with the primer pair LZT-U (5'-CGAAGAGGCCCGCAC-3') and LZT-MA (5'-TAATGGGCTAGGTTACGTTGGTGTAG-3'), and the primer pair LZT-MS (5'-TAACCTAGCCCATTACGGTCAATCC-3') and LZT-D (5'-GGCAACATGGAAATCGC-3') were mixed and used as templates for the second PCR with the primer pair LTZ-U and LZT-D. Replacement of an FspI-AatII fragment of pIK153LZS.6 by the FspI-AatII fragment of the resulting PCR product resulted in pIK153 T10.1. A BamHI-SmaI fragment covering the lacZ gene of LIA7 was inserted into the BamHI site of pIK153 (resulting in pNY19). pNY21 was made by replacing the smaller SmaI-SacI fragment of pNY19 with the homologous SmaI-SacI fragment of pIK153T10.1, which carries the mutant sequence. An XbaI-BglII fragment of pNY21 was used to replace the smaller XbaI-BamHI fragment of pHM5 (resulting in pNY58). pAdNY58 was made by replacement of the smaller I-CeuI-PI-SceI fragment of pAdHM4 with an I-CeuI-PI-SceI fragment of pNY58. The longer PacI fragment of pAdNY58 was transfected into 293 cells. The recombinant adenovirus AdNY58 was prepared and purified from the cell culture.
These two lacZ mutations were transferred back to lambda by homologous recombination in vivo [15] in order to generate LIA15 and LIA11, respectively. The recombinational transfer was carried out as follows. Cells of BIK12015 or BIK12018 were grown to OD600 = ~0.3 in LB (10 g bactotrypton, 5 g yeast extract and 10 g NaCl per liter) containing 20 μg/ml chloramphenicol, 0.2% maltose and 10 mM MgSO4. LIA7 was adsorbed onto the cells at a multiplicity of 1.0 at 37°C for 15 minutes. The mixture was shaken at 37°C until the OD600 dropped below 0.3. One drop of CHCl3 was added to the mixture, which was then shaken for 30 seconds. The mixture was centrifuged and the supernatant was recovered. The supernatant was assayed for BIK12001 on agar plates containing p-gal as detailed below. The plaques on the p-gal plates were isolated and analyzed for the designed sequence change by restriction of the PCR products (see Analysis of the mutant bacteriophage DNA).
Selection of lacZ-negative bacteriophage with p-gal
The lacZ-negative bacteriophage particles were detected using positive selection [15, 16]. BIK12001 cells were grown with shaking at 37°C to OD600 = 1.0 in LB containing ampicillin (50 μg/ml), kanamycin (20 μg/ml) and 0.2% maltose. The culture was centrifuged at 3,500 rpm for 15 minutes at 4°C. The pellets were dissolved into one-half the volume of LB containing 10 mM MgSO4. The bacteriophage was adsorbed onto these cells at room temperature for 20 minutes. To estimate the total number of bacteriophages, 2.5 ml molten 1/4 LB top agar (5 g LB broth base (Gibco BRL, Rockville, MD, USA), 6.4 g NaCl and 7.5 g Bactoagar per liter) was added to 0.25 ml of the mixture of cells and bacteriophages, and the entire content was poured onto a 1/4 LB plate (5 g LB broth base, 6.4 g NaCl and 15 g Bactoagar per liter). To estimate the number of lacZ-negative bacteriophages, 2 ml of the mixture of cells and bacteriophages, and 22 ml of molten 1/4 LB top agar containing 0.3% p-gal (Sigma Chemical Co., MO, USA), were mixed and poured onto four 1/4 LB plates. The plates were incubated at 37°C for 12 hours.
Construction of recombinant adenoviruses
pNY56 was constructed by replacing the shorter XbaI-BamHI fragment of pHM5 by the XbaI-BglII fragment of pNY19 (Figure 2). pAdHM4 includes the entire genome of the recombinant adenovirus vector. The plasmid pAdNY56 was constructed by replacing the shorter I-CeuI-PI-SceI fragment of pAdHM4 by an I-CeuI-PI-SceI fragment of pNY56. The PacI fragment of pAdNY56 was transfected into cells of cell-line 293, which allows replication of the replication-defective adenoviruses. The recombinant adenovirus AdNY56 was prepared and purified as described previously [18]. Similarly, AdNY57 was constructed from pNY20 via pNY57 (Additional file 1), and AdNY58 was constructed from pNY21 via pNY58 (Figure 2, Additional file 1).
Adenovirus infection
Female MutaMice (7 weeks old) were obtained from Covance Research Products Inc. (Denver, PA, USA). The MutaMice were maintained under specific pathogen-free conditions in the animal faculty of the Institute of Medical Science at the University of Tokyo, Japan. After the animals were anesthetized with Nembutal (Dainippon Pharmaceutical Co., Osaka, Japan), 3 × 109 plaque-forming units (PFU) of the recombinant adenovirus in 200 μl of PBS (137 mM NaCl, 8.10 mM Na2HPO4, 2.68 mM KCl, 1.47 mM KH2PO4, 0.9 mM CaCl2, 0.33 mM MgCl2) was injected into the tail-vein of each mouse using a 30-gauge needle. AdNY56 was injected into one mouse, AdNY57 was injected into two mice and AdNY58 was injected into two mice.
Isolation of genomic DNA, recovery of lambda bacteriophage and measurement of mutant frequency
Twenty-four hours after injection, the mice were sacrificed. A lobe of the liver of each animal was excised, frozen by submersion in liquid nitrogen and stored in a 1.5-ml plastic tube at -80°C. Genomic DNA was isolated from the liver tissue with phenol-chloroform and precipitated by ethanol/sodium as described in the manual for MutaMouse. Lambda bacteriophage particles were recovered from the isolated DNA by incubation with packaging extracts (Mutaplax, Epicentre, WI, USA). The lacZ-negative mutants were detected by p-gal selection as described above. Each plaque on the selective agar was recovered in 100 μl of SM buffer (50 mM Tris-HCl (pH 7.5), 10 mM MgSO4, 100 mM NaCl and 0.01% gelatin). In order to verify the lacZ-negative phenotype, each isolate was assayed on agar with X-gal using a spot assay as follows. BIK2206 was grown in LB containing ampicillin (50 μg/ml) and tetracycline (10 μg/ml). Twice-concentrated culture (1.25 ml) was mixed with 6 ml molten LB/MM agar (100 ml LB medium, 0.75 g Bactoagar, 10 mM MgSO4, 0.2% maltose and 0.35 mg/ml X-gal) and spread on agar. A 10-μl aliquot of each bacteriophage sample was spotted onto these cells. The plates were incubated overnight at 37°C. The mutant frequency was estimated by dividing the number of PFU on the selective plate (as verified with X-gal) by the number of total PFU on 1/4 LB agar.
Analysis of the mutant bacteriophage DNA
The lacZ-negative lambda bacteriophage DNA from the mice was analyzed using restriction enzymes following PCR. For the lacZ-negative lambda DNA from the AdNY57-treated mouse, PCR was carried out with the primer pair LG-1 (5'-TACCGGCGATGAGCGAAC-3') and LG-2 (5'-CTCCAGGTAGCGAAAGCC-3'). The 288-bp product was purified by ethanol/sodium precipitation, digested with TfiI (New England Biolabs, Beverly, MA, USA) (recognition site, 5'-G|AWTC-3' (W = A or T)) at 65°C and analyzed using agarose electrophoresis. The mutant sequence was resistant to TfiI, while the wild-type sequence was sensitive, yielding 204 and 84 bp fragments. The primer pair Lam-1 (5'-TACTGTCGTCGTCCCCTC-3') and Lam-2 (5'-CGCAGATGAAACGCCGAGT-3') was used for the lacZ-negative lambda DNA from the AdNY58-treated mouse. The 213-bp PCR product was digested with XspI (Takara Bio Inc., Shiga, Japan) (recognition site, 5'-C|TAG-3') at 37°C and analyzed using agarose electrophoresis. The wild-type sequence was resistant to XspI, while the mutant sequence was sensitive, yielding 146 and 67 bp fragments.
Results
Experimental design for the detection of gene targeting in vivo
Figure 1 illustrates our experimental design for the sensitive detection of gene targeting in vivo. The MutaMouse carries approximately 40 copies of bacteriophage lambda gt10lacZ on a chromosome [6, 19]. The single integration site is located in band C on chromosome 3 [20]. Our target sequence was the wild-type lacZ gene. The donor DNA was delivered to the liver cell nuclei by tail-vein injection of the recombinant adenovirus. Genomic DNA was isolated from the liver and its in vitro packaging allowed the recovery of the lambda genome in viable bacteriophage particles. A lacZ-negative mutant bacteriophage was selected as a plaque-former in an Escherichia coli mutant defective in the galE gene on an agar plate containing p-gal. This chemical is converted by the lacZ gene product (beta-galactosidase) into UDP-galactose, which accumulates in the absence of the GalE protein to induce cell death. The ratio of the mutant plaque-formers to the total plaque-formers was used to estimate the fraction of the mutated gene. The mutant gene was further analyzed using restriction enzymes.
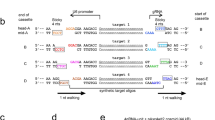
Replication-defective recombinant adenoviruses constructed by an in vitro-ligation method were used to deliver the donor DNA [18, 21]. Figure 3 shows the structure of the recombinant adenoviruses used in the present study (see Figure 2, Additional file 1, and Materials and methods for further details). An 8077-bp fragment of lambda gt10lacZ was inserted into the E1 deletion site of the mutant adenovirus [18, 21]. AdNY56 had wild-type lacZ, while AdNY57 and AdNY58 had a point mutation in lacZ (Figure 3B).

Design for gene targeting and its detection. (A) The donor carrying the mutant lacZ gene is inserted into an adenovirus vector. The lacZ mutation will be transferred to the lacZ gene of the lambda transgene in the mouse genome. (B) Expected sequence changes and their detection using restriction analysis.
AdNY57 was constructed so as to introduce a point mutation at the active site of LacZ. The target sequence was the 5' GAA that codes for Glu461, which is essential for the activity of LacZ [22, 23]. AdNY57 was expected to change its second base (that is, the 1437 th base) from A to G, thereby generating the Glu461Gly mutant, which shows a 76-fold decrease in activity [23]. The mutant and wild-type sequences can be distinguished using the restriction enzyme TfiI (Figure 3B).
AdNY58 was constructed so as to introduce a point mutation at the 5' TAT that codes for Tyr105. AdNY58 was expected to change its third base (that is, the 369th base) from T to G, thereby generating the Tyr105Stop mutant. The mutant and wild-type sequences can be distinguished using the restriction enzyme XspI (Figure 3B).
Control experiments
We demonstrated that lacZ mutants that were predicted to be generated by the recombinant adenovirus could be selected with p-gal as follows. Bacteriophage lambda strains carrying the mutations were produced by transferring each mutation on a plasmid back to lambda through homologous recombination in E. coli (as detailed in Materials and methods). The two bacteriophage strains, lambda gt10lacZ- Tyr105Stop (LIA11) and lambda gt10lacZ- Glu461Gly (LIA15), were then used in the p-gal selection. As shown in Table 1, lambda with wild-type lacZ showed a plaque-formation efficiency of less than 1/10,000 on the selective agar relative to that on the non-selective agar. By contrast, each of the mutant lambda strains showed similar or slightly decreased plaque-formation efficiency on the selective agar. We concluded that the expected targeted product with AdNY57 and AdNY58, if it was produced, should be selected and measured using the p-gal-selection procedure.
Delivery of donor DNA and measurement of mutant frequency
The recombinant adenovirus particles (3 × 109 PFU in 200 μl of PBS) were injected into the tail-vein of a MutaMouse. It is well established that the adenovirus genome accumulates in the liver cell nuclei after tail-vein injection [12, 13]. Most of the hepatocyte nuclei are expected to receive several copies of the adenovirus genome under these conditions (see Discussion). After 24 hours, the liver was excised from the MutaMouse, genomic DNA was isolated from the liver tissue and the lambda genome was recovered as a bacteriophage particle by in vitro packaging. The lacZ-negative phage was detected selectively on agar with p-gal. The plaques on these selective plates were isolated and the LacZ-negative phenotype was confirmed on agar plates containing X-gal. The mutant frequency was estimated as the fraction of the lacZ-negative phage (Table 2). The control mouse (animal number 0) received no injections.
The mutant frequencies of the AdNY56-injected and control mice were similar (Table 2, Experiment 1), and did not differ significantly from those reported previously using this method (see [15] and the references cited therein). No significant increase in the mutant form of the gene was induced by injection of the recombinant adenovirus: the mutant frequency of the AdNY57- and AdNY58-injected mice was similar to that of the control mouse, which was approximately 1/10,000 (Table 2).
All of the lacZ-negative bacteriophages were purified and their lacZ genes were analyzed using restriction-enzyme treatment of the PCR products (Figure 4). As shown in Figures 3B and 4A, the PCR product of the Glu461Gly mutant, as predicted from the AdNY57 injection, could not be cut with TfiI. By contrast, the wild-type and most of the other possible mutants could be cut with TfiI. In fact, all of the lacZ-negative bacteriophages from the AdNY57-injected mouse were cleavable with this restriction enzyme. As shown in Figure 3B and 4B, the PCR product of the Tyr105Stop mutant, as predicted from the AdNY58 injection, could be cut with XspI. By contrast, the wild-type and most of the other mutants could not be cut with XspI. None of the lacZ-negative bacteriophages from the AdNY58-injected mice were cleavable with this restriction enzyme.

Restriction analysis of the lacZ -negative gene from mice treated with a recombinant adenovirus. (A) AdNY57-injected mouse. The PCR product of the lambda bacteriophage DNA with primers that flank the target site is 288 bp long. The wild-type PCR product is cut with TfiI into 84 and 204 bp fragments, whereas the Glu461Ala mutant PCR product is not cut. Lane M: Marker DNA prepared by HinfI digestion of the plasmid pUC19; 1–12, lacZ-negative bacteriophages from animal number 2; lacZ+: Lambda bacteriophage recovered from control mouse; lacZ-Glu461Gly: lambda bacteriophage LIA15. (B) AdNY58-injected mouse. The PCR product of the lambda bacteriophage DNA with primers that flank the target site is 213 bp long. The Tyr105Stop mutant PCR product is cut with XspI into 146 and 67 bp fragments, whereas the wild-type product is not. Lane M: Marker DNA prepared by HinfI digestion of plasmid pUC19; 1–4, lacZ-negative bacteriophages from animal number 3; lacZ+: Lambda bacteriophage recovered from control mouse; lacZ-Tyr105Stop: lambda bacteriophage LIA11.
We did not detect the expected gene replacement in any of the isolates. Moreover, the gene-correction frequency by these adenovirus constructs was shown to be less than 1/20,000 in the present system.
Discussion
Here we attempted to perform gene targeting in a transgenic mouse system that allowed the sensitive detection of mutagenesis by various agents, such as those directly interacting with DNA in the liver and other organs [24, 25]. The limit of sensitivity in this system was 1/20,000 (see also [15]). This procedure might provide an alternative to the PCR-based assay for gene targeting in vivo, although our initial trials did not detect any of the expected recombinants.
In the present system, the sensitivity appeared to be limited by the high level of spontaneous mutagenesis in the target gene. The MutaMouse system was produced to detect mutagenesis at numerous sites within a gene, rather than to study gene targeting. Experimental designs involving the specific selection of homologous recombination events, such as those used in the previous work in vitro [7], would therefore be preferred.
Also, in the present system, a successful gene-targeting event would not be distinguishable in the phenotype of the mouse cell. In transgenic mice with a single copy of the mutant lacZ gene [26], correction to the wild-type gene would result in a direct positive readout in the mouse body (for example, through staining with dye). However, as the authors admit, it would be difficult to detect the targeting events with a high sensitivity. The presence of multiple copies of the target gene would improve the sensitivity because the lacZ+ allele is dominant over, and epistatic to, the lacZ- alleles with respect to the above phenotype. The MutaMouse carries multiple (approximately 40) copies of the target gene, which amount to 0.4% of the genome. This should be able to improve the sensitivity of detection of gene targeting, although the sensitivity is limited by spontaneous mutagenesis. In addition, the presence of tandem repeats might have other types of negative effect on gene targeting, as detailed below.
How efficient is adenovirus infection and delivery to the hepatocyte nucleus? Tail-vein injection is an established method for the delivery of adenovirus to liver cells. The average copy number of a replication-defective recombinant adenovirus genome per liver cell has been estimated as 14–28 copies using Southern hybridization after tail-vein injection of 5 × 109 PFU of the virus [12]. This corresponds to 40% of the injected adenovirus. Fluorescence in situ hybridization revealed that, after tail-vein injection of 2 × 109 PFU, all of the hepatocyte nuclei had 1–100 copies of a recombinant adenovirus genome, with an average of 20 copies [27]. After tail-vein injection of 2 × 108 PFU of a recombinant adenovirus with the lacZ expression cassette, 40% of the hepatocytes expressed beta-galactosidase [13]. We assumed that the majority of the liver cells received several copies of the adenovirus genome, at least sufficient for gene expression, after injecting 3 × 109 PFU in our experiment. (We cannot raise the titer any more because of the toxicity of the virus.) This type of information can be confirmed by Southern hybridization and fluorescence in situ hybridization.
The gene-targeting frequency with recombinant adenoviruses in vitro varies from ~10-7–10-4 per cell [7, 8, 28]. We did not detect any signal using recombinant adenovirus for gene delivery in the mouse liver. In order to achieve gene targeting in vivo using an adenovirus vector or any other means, it will be necessary to increase the frequency of gene targeting. So how can we achieve this goal?
The efficiency of gene targeting in vitro varies from one locus to another [29, 30]. Such locus-dependence might reflect drastic effects of the chromatin structure on the frequency of homologous recombination [30, 31]. Thus, the target transgene could be placed at a different locus that is known to be a hot spot in gene targeting in embryonic stem (ES) cells.
Repetitive sequences are methylated in the mouse genome [32]. Ikehata and colleagues suggested that the whole coding region of the MutaMouse lacZ transgene is methylated to a high degree at every CpG site [33]. One possible reason for this phenomenon is that the CpG content of the lacZ gene (9%) [34] is much higher than the average CpG content of the mouse genome (~1%) [35]. Methyl-CpG binding protein 2 (MeCP2) might bind to methylated CpG and somehow compact chromatin [36]. Furthermore, Manuelidis analyzed the structure of a mouse chromosome bearing a huge (~11 Mb) insert of a tandem-repeated transgene (~1,000 copies) [37]. This transgene was localized on an arm of chromosome 3 at a distance from the centromere. According to Manuelidis, the transgene is heterochromatic and highly condensed. Therefore, the MutaMouse transgene might be heterochromatic. The accessibility of nucleases to the heterochromatic structure is lower than that of euchromatin [38, 39]. Reducing the copy number of the transgene and/or using another transgene that is lower in CpG content might increase gene targeting, although the decrease in copy number might affect the sensitivity of detection. An important experiment that can be done is to test whether the coding region of the MutaMouse lacZ transgene is really heterochromatic, using, for example, CHIP assay with the antibody against the methylated histones and PCR primers on the lacZ genes.
Chromosome replication is known to stimulate homologous recombination. Partial hepatectomies in mice might stimulate liver cell proliferation and DNA replication, which in turn might stimulate recombination. Hara et al. (1999) reported that partial hepatectomies increased mutagenesis with N-ethyl-N-nitrosourea, which is a direct-acting DNA-ethylation agent, in the MutaMouse [40].
It might be easier to modify the donor DNA than the recipient DNA. One can generate recombinogenic damage on the donor DNA. Irradiating adenovirus particles with ultraviolet light of 1500 J/m2 resulted in an approximately three-fold increase in their mutual homologous recombination [41]. Recombinogenic cross-links are induced by some mutagens, such as psoralens, cisplatin (cis-diamminedichloroplatinum) and mitomycin C [42]. Such agents, both mutagenic and recombinogenic, might be suitable for gene targeting in vivo if they are shown to be active in mutagenesis in a transgenic-reporter mouse system. The effect of such recombinogenic damage might be much larger with replication-defective adenovirus recombinants than with replication-competent adenoviruses, because their replication-intermediates are responsible for their high recombination frequency [41, 43–45].
The gene-targeting frequency is strongly dependent on the length of homology; the frequency increases as the homology length increases up to 10 kb [46–48]. If the deviation from this rule above 10 kb is due to the shearing and/or degradation of longer DNA after electroporation in embryonic stem cells, donor DNAs that are protected by the DNA binding proteins in the adenovirus particle might show greater length dependence over a wider range of values. Adenoviral vectors with a larger capacity for inserts, which are known as high-capacity 'gutless' vectors [49–51] might therefore be suitable for use in this approach.
Conclusion
Here we attempted to perform gene targeting in a transgenic mouse system that allowed the sensitive detection of mutagenesis. The frequency of gene targeting in the mouse liver by these recombinant adenoviruses was shown to be less than 1/20000 with the sensitive and PCR-independent detection system.
Abbreviations
- PCR:
-
polymerase chain reaction
- PFU:
-
plaque-forming unit
- RFLP:
-
restriction fragment length polymorphism
- p-gal:
-
phenyl-beta-D-galactoside
- X-gal:
-
5-bromo-4-chloro-3-indlyl-beta-D-galactose
References
Capecchi MR: Altering the genome by homologous recombination. Science. 1989, 244: 1288-1292.
Yanez RJ, Porter AC: Therapeutic gene targeting. Gene Ther. 1998, 5: 149-159. 10.1038/sj.gt.3300601.
Kren BT, Bandyopadhyay P, Steer CJ: In vivo site-directed mutagenesis of the factor IX gene by chimeric RNA/DNA oligonucleotides. Nat Med. 1998, 4: 285-290. 10.1038/nm0398-285.
Kren BT, Parashar B, Bandyopadhyay P, Chowdhury NR, Chowdhury JR, Steer CJ: Correction of the UDP-glucuronosyltransferase gene defect in the gunn rat model of crigler-najjar syndrome type I with a chimeric oligonucleotide. Proc Natl Acad Sci USA. 1999, 96: 10349-10354. 10.1073/pnas.96.18.10349.
Taubes G: Gene therapy. The strange case of chimeraplasty. Science. 2002, 298: 2116-2120. 10.1126/science.298.5601.2116.
Gossen JA, de Leeuw WJ, Tan CH, Zwarthoff EC, Berends F, Lohman PH, Knook DL, Vijg J: Efficient rescue of integrated shuttle vectors from transgenic mice: a model for studying mutations in vivo. Proc Natl Acad Sci USA. 1989, 86: 7971-7975.
Fujita A, Sakagami K, Kanegae Y, Saito I, Kobayashi I: Gene targeting with a replication-defective adenovirus vector. J Virol. 1995, 69: 6180-6190.
Mitani K, Wakamiya M, Hasty P, Graham FL, Bradley A, Caskey CT: Gene targeting in mouse embryonic stem cells with an adenoviral vector. Somat Cell Mol Genet. 1995, 21: 221-231. 10.1007/BF02255777.
Doerfler W: The fate of the DNA of adenovirus type 12 in baby hamster kidney cells. Proc Natl Acad Sci USA. 1968, 60: 636-643.
Harui A, Suzuki S, Kochanek S, Mitani K: Frequency and stability of chromosomal integration of adenovirus vectors. J Virol. 1999, 73: 6141-6146.
Volpers C, Kochanek S: Adenoviral vectors for gene transfer and therapy. J Gene Med. 2004, 6 (Suppl 1): S164-171. 10.1002/jgm.496.
Vrancken Peeters MJ, Perkins AL, Kay MA: Method for multiple portal vein infusions in mice: quantitation of adenovirus-mediated hepatic gene transfer. Biotechniques. 1996, 20: 278-285.
Nakatani T, Kuriyama S, Tominaga K, Tsujimoto T, Mitoro A, Yamazaki M, Tsujinoue H, Yoshiji H, Nagao S, Fukui H: Assessment of efficiency and safety of adenovirus mediated gene transfer into normal and damaged murine livers. Gut. 2000, 47: 563-570. 10.1136/gut.47.4.563.
Cormack B: Mutagenesis by the polymerase chain reaction. Current protocols in moecular biology. Edited by: Ausubel MF, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, Struho K. 1991, John Wiley and Sons, Inc, 1: 8.5.1-8.5.9.
Ino A, Yamamoto S, Kaneda Y, Kobayashi I: Somatic gene targeting with RNA/DNA chimeric oligonucleotides: an analysis with a sensitive reporter mouse system. J Gene Med. 2004, 6: 1272-1280. 10.1002/jgm.617.
Dean SW, Myhr B: Measurement of gene mutation in vivo using Muta Mouse and positive selection for lacZ- phage. Mutagenesis. 1994, 9: 183-185.
Gossen JA, Vijg J: A selective system for lacZ-phage using a galactose-sensitive E.coli host. Biotechniqnics. 1993, 14: 326-330.
Mizuguchi H, Kay MA: Efficient construction of a recombinant adenovirus vector by an improved in vitro ligation method. Hum Gene Ther. 1998, 9: 2577-2583. 10.1089/10430349850019418.
Blakey DH, Douglas GR, Huang KC, Winter HJ: Cytogenetic mapping of lambda gt10 lacZ sequences in the transgenic mouse strain 40.6 (Muta Mouse). Mutagenesis. 1995, 10: 145-148.
Swiger RR, Myhr B, Tucker JD: The LacZ transgene in MutaMouse maps to chromosome 3. Mutat Res. 1994, 325: 145-148. 10.1016/0165-7992(94)90077-9.
Mizuguchi H, Kay MA: A simple method for constructing E1- and E1/E4-deleted recombinant adenoviral vectors. Hum Gene Ther. 1999, 10: 2013-2017. 10.1089/10430349950017374.
Jacobson RH, Zhang XJ, DuBose RF, Matthews BW: Three-dimensional structure of beta-galactosidase from E. coli. Nature. 1994, 369: 761-766. 10.1038/369761a0.
Cupples CG, Miller JH: Effects of amino acid substitutions at the active site in Escherichia coli beta-galactosidase. Genetics. 1988, 120: 637-644.
Cosentino L, Heddle JA: A comparison of the effects of diverse mutagens at the lacZ transgene and Dlb-1 locus in vivo. Mutagenesis. 1999, 14: 113-119. 10.1093/mutage/14.1.113.
Krebs O, Favor J: Somatic and germ cell mutagenesis in lambda lacZ transgenic mice treated with acrylamide or ethylnitrosourea. Mutat Res. 1997, 388: 239-248.
Nickerson HD, Colledge WH: A LacZ-based transgenic mouse for detection of somatic gene repair events in vivo. Gene Ther. 2004, 11: 1351-1357. 10.1038/sj.gt.3302311.
Schowalter DB, Tubb JC, Liu M, Wilson CB, Kay MA: Heterologous expression of adenovirus E3-gp19K in an E1a-deleted adenovirus vector inhibits MHC I expression in vitro, but does not prolong transgene expression in vivo. Gene Ther. 1997, 4: 351-360. 10.1038/sj.gt.3300398.
Wang Q, Taylor MW: Correction of a deletion mutant by gene targeting with an adenovirus vector. Mol Cell Biol. 1993, 13: 918-927.
Bird D, Bradshaw R: Gene targeting is locus dependent in the filamentous fungus Aspergillus nidulans. Mol Gen Genet. 1997, 255: 219-225. 10.1007/s004380050492.
Petes TD: Meiotic recombination hot spots and cold spots. Nat Rev Genet. 2001, 2: 360-369. 10.1038/35072078.
Ohta K, Shibata T, Nicolas A: Changes in chromatin structure at recombination initiation sites during yeast meiosis. Embo J. 1994, 13: 5754-5763.
Lau S, Jardine K, McBurney MW: DNA methylation pattern of a tandemly repeated LacZ transgene indicates that most copies are silent. Dev Dyn. 1999, 215: 126-138. 10.1002/(SICI)1097-0177(199906)215:2<126::AID-DVDY5>3.0.CO;2-8.
Ikehata H, Takatsu M, Saito Y, Ono T: Distribution of spontaneous CpG-associated G:C --> A:T mutations in the lacZ gene of Muta mice: effects of CpG methylation, the sequence context of CpG sites, and severity of mutations on the activity of the lacZ gene product. Environ Mol Mutagen. 2000, 36: 301-311. 10.1002/1098-2280(2000)36:4<301::AID-EM6>3.0.CO;2-R.
Chevalier-Mariette C, Henry I, Montfort L, Capgras S, Forlani S, Muschler J, Nicolas JF: CpG content affects gene silencing in mice: evidence from novel transgenes. Genome Biol. 2003, 4: R53-10.1186/gb-2003-4-9-r53.
Takai D, Jones PA: Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci USA. 2002, 99: 3740-3745. 10.1073/pnas.052410099.
Bowen NJ, Palmer MB, Wade PA: Chromosomal regulation by MeCP2: structural and enzymatic considerations. Cell Mol Life Sci. 2004, 61: 2163-2167. 10.1007/s00018-004-4177-x.
Manuelidis L: Heterochromatic features of an 11-megabase transgene in brain cells. Proc Natl Acad Sci USA. 1991, 88: 1049-1053.
Vega-Palas MA, Venditti S, Di Mauro E: Heterochromatin organization of a natural yeast telomere. Changes of nucleosome distribution driven by the absence of Sir3p. J Biol Chem. 1998, 273: 9388-9392. 10.1074/jbc.273.16.9388.
Ye F, Signer ER: RIGS (repeat-induced gene silencing) in Arabidopsis is transcriptional and alters chromatin configuration. Proc Natl Acad Sci USA. 1996, 93: 10881-10886. 10.1073/pnas.93.20.10881.
Hara T, Sui H, Kawa Kami K, Shimada Y, Shibuya T: Partial hepatectomy strongly increased the mutagenicity of N-ethyl-N-nitrosourea in Mutamouse liver. Environ Mol Mutagen. 1999, 34: 121-123. 10.1002/(SICI)1098-2280(1999)34:2/3<121::AID-EM10>3.0.CO;2-R.
Rainbow AJ, Castillo JE: Homologous recombination of adenovirus DNA in mammalian cells: enhanced recombination following UV-irradiation of the virus. Mutat Res. 1992, 274: 201-210.
Wang YY, Maher VM, Liskay RM, McCormick JJ: Carcinogens can induce homologous recombination between duplicated chromosomal sequences in mouse L cells. Mol Cell Biol. 1988, 8: 196-202.
Flint SJ, Berget SM, Sharp PA: Characterization of single-stranded viral DNA sequences present during replication of adenovirus types 2 and 5. Cell. 1976, 9: 559-571. 10.1016/0092-8674(76)90038-6.
Epstein LH, Young CS: Adenovirus homologous recombination does not require expression of the immediate-early E1a gene. J Virol. 1991, 65: 4475-4479.
Young CS, Cachianes G, Munz P, Silverstein S: Replication and recombination in adenovirus-infected cells are temporally and functionally related. J Virol. 1984, 51: 571-577.
Deng C, Capecchi MR: Reexamination of gene targeting frequency as a function of the extent of homology between the targeting vector and the target locus. Mol Cell Biol. 1992, 12: 3365-3371.
Scheerer JB, Adair GM: Homology dependence of targeted recombination at the Chinese hamster APRT locus. Mol Cell Biol. 1994, 14: 6663-6673.
Fujitani Y, Yamamoto K, Kobayashi I: Dependence of frequency of homologous recombination on the homology length. Genetics. 1995, 140: 797-809.
Wang L, Hernandez-Alcoceba R, Shankar V, Zabala M, Kochanek S, Sangro B, Kramer MG, Prieto J, Qian C: Prolonged and inducible transgene expression in the liver using gutless adenovirus: a potential therapy for liver cancer. Gastroenterology. 2004, 126: 278-289. 10.1053/j.gastro.2003.10.075.
Kreppel F, Kochanek S: Long-term transgene expression in proliferating cells mediated by episomally maintained high-capacity adenovirus vectors. J Virol. 2004, 78: 9-22. 10.1128/JVI.78.1.9-22.2004.
Stilwell JL, McCarty DM, Negishi A, Superfine R, Samulski RJ: Development and characterization of novel empty adenovirus capsids and their impact on cellular gene expression. J Virol. 2003, 77: 12881-12885. 10.1128/JVI.77.23.12881-12885.2003.
Casadaban MJ, Cohen SN: Analysis of gene control signals by DNA fusion and cloning in Escherichia coli. J Mol Biol. 1980, 138: 179-207. 10.1016/0022-2836(80)90283-1.
Takahashi NK, Kusano K, Yokochi T, Kitamura Y, Yoshikura H, Kobayashi I: Genetic analysis of double-strand break repair in Escherichia coli. J Bacteriol. 1993, 175: 5176-5185.
Huynh T, Young R, Davis R: Constructing and Screening cDNA Libraries in λgt10 and λgt11. DNA cloning. Volume I a practical approach. Edited by: Glover D. 1984, Washinton DC: IRL PRESS, 49-78.
Naito Y, Naito T, Kobayashi I: Selfish restriction modification genes: resistance of a resident R/M plasmid to displacement by an incompatible plasmid mediated by host killing. Biol Chem. 1998, 379: 429-436.
Yanisch-Perron C, Vieira J, Messing J: Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene. 1985, 33: 103-119. 10.1016/0378-1119(85)90120-9.
Acknowledgements
Ms. Kuniko Iwasaki and Dr. Ryuichi Miura from the Laboratory Animal Research Center of the Institute of Medical Science, Japan, guided us in our manipulation of the mice. Dr. Noriko Takahashi from our laboratory helped with the maintenance of the mice. Dr. Yoichiro Iwakura of the Institute of Medical Science provided critical comments on an early version of the manuscript. This work was supported by grants from the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan (No.0828102: General Mechanisms of DNA Recombination Repair. 1996–1999) and the Japan Owners Association (JOA) (1999–2002) as arranged by the Japan Society for Gene Therapy.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
AI carried out the injection of the recombinant adenovirus and the analysis of the mouse DNA. YN and HM constructed the recombinant adenovirus. NH injected the recombinant adenovirus to the mouse. YN constructed the experimental design as well as cloning of the part of lambda DNA from the MutaMouse genomic DNA. IK provided the original experimental idea and coordinated the experimental design. All authors read and approved the final manuscript.
Electronic supplementary material
12965_2005_28_MOESM1_ESM.DOC
Additional file 1: Bacterial strains, plasmids, bacteriophage strains and recombinant adenovirus constructs. (DOC 59 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ino, A., Naito, Y., Mizuguchi, H. et al. A trial of somatic gene targeting in vivo with an adenovirus vector. Genet Vaccines Ther 3, 8 (2005). https://doi.org/10.1186/1479-0556-3-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1479-0556-3-8