
Abstract
Background
In humans, overproduction of apolipoprotein B (apoB) is positively associated with premature coronary artery diseases. To reduce the levels of apoB mRNA, we have designed an apoB mRNA-specific hammerhead ribozyme targeted at nucleotide sequences GUA6679 (RB15) mediated by adenovirus, which efficiently cleaves and decreases apoB mRNA by 80% in mouse liver and attenuates the hyperlipidemic condition. In the current study, we used an adeno-associated virus vector, serotype 2 (AAV2) and a self-complementary AAV2 vector (scAAV2) to demonstrate the effect of long-term tissue-specific gene expression of RB15 on the regulation apoB mRNA in vivo.
Methods
We constructed a hammerhead ribozyme RB15 driven by a liver-specific transthyretin (TTR) promoter using an AAV2 vector (rAAV2-TTR-RB15). HepG2 cells and hyperlipidemic mice deficient in both the low density lipoprotein receptor and the apoB mRNA editing enzyme genes (LDLR-/-Apobec1-/-; LDb) were transduced with rAAV2-TTR-RB15 and a control vector rAAV-TTR-RB15-mutant (inactive ribozyme). The effects of ribozyme RB15 on apoB metabolism and atherosclerosis development were determined in LDb mice at 5-month after transduction. A self-complementary AAV2 vector expressing ribozyme RB15 (scAAV2-TTR-RB15) was also engineered and used to transduce HepG2 cells. Studies were designed to compare the gene expression efficiency between rAAV2-TTR-RB15 and scAAV2-TTR-RB15.
Results
The effect of ribozyme RB15 RNA on reducing apoB mRNA levels in HepG2 cells was observed only on day-7 after rAAV2-TTR-RB15 transduction. And, at 5-month after rAAV2-TTR-RB15 treatment, the apoB mRNA levels in LDb mice were significantly decreased by 43%, compared to LDb mice treated with control vector rAAV2-TTR-RB15-mutant. Moreover, both the rAAV2-TTR-RB15 viral DNA and ribozyme RB15 RNA were still detectable in mice livers at 5-month after treatment. However, this rAAV2-TTR-RB15 vector mediated a prolonged but low level of ribozyme RB15 gene expression in the mice livers, which did not produce the therapeutic effects on alteration the lipid levels or the inhibition of atherosclerosis development. In contrast, the ribozyme RB15 RNA mediated by scAAV2-TTR-RB15 vector was expressed immediately at day-1 after transduction in HepG2 cells. The apoB mRNA levels were decreased 47% (p = 0.001), compared to the control vector scAAV2-TTR-RB15-mutant.
Conclusion
This study provided evidence that the rAAV2 single-strand vector mediated a prolonged but not efficient transduction in mouse liver. However, the scAAV2 double-strand vector mediated a rapid and efficient gene expression in liver cells. This strategy using scAAV2 vectors represents a better approach to express small molecules such as ribozyme.
Similar content being viewed by others

Background
Ribozymes are small RNA molecules with enzymatic RNA-cleaving activity [1]. Our laboratory [2, 3] has previously demonstrated that adenovirus mediated apolipoprotein B (apoB) mRNA-specific hammerhead ribozyme cleaved apoB mRNA efficiently. In the dyslipidemic mouse model, the effect of decreased apoB mRNA results in a marked decrease in plasma cholesterol, triglyceride and apo B levels [3] and these hypolipidemic effects persist to day-21. To assess the utility of this approach in treating hyperlipidemia, it is essential to explore other gene delivery vectors for prolonged gene expression. We sought to use adeno-associated viruses (AAV) for the longevity of transgene expression [4]. Furthermore, the AAV vector does not elicit T cells immune responses to the transgene product, because it is believed that AAV does not infect antigen-presenting cells [5]. These features suggest that AAV is a better vector for somatic gene transfer.
To decrease apoB mRNA expression, we have designed a hammerhead ribozyme targeted at GUA6679 of apoB mRNA (RB15) to cleave apoB mRNA [2, 3]. In this study, we constructed the apolipoprotein B mRNA-specific hammerhead ribozyme (RB15) driven by a liver-specific transthyretin (TTR) promoter [6] using the AAV2 serotype virus vector (rAAV2-TTR-RB15). We delivered rAAV2-TTR-RB15 to an atherosclerosis-susceptible mouse model, which is deficient in both the low density lipoprotein receptor (LDLR-/-) and the apoB mRNA editing enzyme (Apobec1-/-) genes [7–9], to evaluate the duration of gene expression by the AAV2 vector and the effect of apoB mRNA-specific hammerhead ribozymes on apoB mRNA gene expression. An improved strategy using self-complementary AAV2 vector shows rapid and efficient gene expression in HepG2 cells.
Methods
Construction and production of the AAV2 plasmid vectors
pAAV2-TTR-RB15 plasmid vectors
We chose transthyretin (TTR, kindly provided by Dr. Terry Van Dyke at the University of North Carolina, Chapel Hill, NC) as the liver-specific promoter. The enhancer/promoter region TTR has been shown to target transgene expression specifically to the liver [6, 10]. We have previously shown that hammerhead ribozyme targeted specifically at apoB mRNA sequences of GUA6679↓ (RB15) cleaves apoB mRNA efficiently [2, 3]. A point mutation of the conserved catalytic domain of ribozyme at nucleotide G5 → A (G5A) of RB15 completely abolished the catalytic activity (designated as RB15-mutant). RB15 or RB15-mutant driven by TTR promoter was cloned into plasmid pZAC2.0 (kindly provided by Dr. Alan Davis at Baylor College of Medicine, Houston, TX) flanking by the inverted terminal repeats of AAV2. A 1931-bp human genomic fragment of hypoxanthine guanine phosphorribosyltransferase (HPRT) as the stuffer sequences was inserted downstream of 5' ITR at the SspB1 site of pZAC2.0 to maintain the wild-type AAV genome size. The shuttle vectors were designated as pAAV2-TTR-RB15, pAAV2-TTR-RB15-mutant, which contains the viral genomic size of 3967-bp. The nucleotide sequences were confirmed by sequencing. As noted, this viral genomic size was suboptimal; the packaging limit of AAV size is 5 kb and a genome of 4.5 kb is the optimal size [11].
Self-complementary recombinant AAV2 vector (scAAV2)
To construct scAAV2-TTR-RB15 and -RB15-mutant vectors, we deleted ~1479-bp of the stuffer sequences from pAAV2-TTR-RB15 and -RB15 mutant to generate a small size clone of 2462-bp. As described by McCarty et al [12], recombinant AAV DNA of less than half of wild-type AAV genome length can be packed as a dimer, and this double-stranded DNA viral vector would display a rapid onset of transgene expression.
Production and Purification of recombinant AAV2 (rAAV2)
We used the pDG helper plasmid (kindly provided by Dr. Jurgen Kleinschmidt from DKFZ, Heidelberg, Germany) [13], which contains AAV2 rep and cap genes plus the E2A and E4 sequences from the adenovirus to produce the recombinant AAV2 (rAAV2). One of the advantages of using pDG is that no replication-competent adenovirus has been detected. To produce rAAV2, the shuttle vector and the helper plasmid (pDG) were co-transfected into 293 cells using the calcium phosphate-mediated transfection method (Promega). After a 48-h incubation, the cells were harvested, pelleted down, and re-suspended in lysis buffer (150 mM NaCl, 50 mM Tris-HCl, pH 8.5). Initially, we used the Iodixanol density gradient centrifugation procedure as described by Zolotukhin et al. [14], followed by purification using Heparin-Agarose Type I column (Sigma). The rAAV2 was eluted with 5 ml 1 M NaCl in PBS-MK (1 × phosphate-buffered saline, 1 mM MgCl2, and 2.5 mM KCl). The first 2 ml elute was discarded and the virus was collected in the subsequent 3.5 ml of the elution buffer. The virus was concentrated and desalted by centrifugation through an Ultrafree Biomax filter unit (Millipore). Later on, we used the single-step heparin chromatography described by Auricchio et al. [15] to purify the rAAV2 and scAAV2 vectors.
Quantification of rAAV2 particles
The purified viral stocks (rAAV2 vectors) were titrated to determine the genome copies either by slot-blot hybridization [16] or by TaqMan real-time polymerase chain reaction (PCR) (Applied Biosystems). In real-time quantitative PCR, primers and probes were designed to recognize the stuffer sequence of the human HPRT gene. The primers and dual-labeled probes used and the final PCR working concentrations were as follows:
Forward primer: 5' GCCAGGATGGTCTCCATCTC (900 nM)
Reverse primer: 5' GTGGGCCAGGCGTAGTG (900 nM)
Probe: 5' FAM-CCTCATGATCTGCCTGCTTCGGC-TAMRA (100 nM).
FAM is fluorescein aminohexylamidite and TAMRA is tetramethylrhodamine.
The infection potencies of rAAV vectors were evaluated by the infectious center assay. The assay was carried out to infect C12 cells as described by Zolotukhin et al. [17].
The titers of viral genome particle number of scAAV2 were determined by quantitative DNA dot blot method. Each scAAV2 particle was calculated as containing two copies of parent AAV2 (non-modified single strand rAAV2-TTR-RB15).
Identification of rAAV2 dimer and monomer by alkaline agarose gel electrophoresis
Agarose gel was cast as 0.8% in H2O; the gel was then soaked in alkaline buffer solution (50 mM NaOH, 1 mM EDTA) for 30 min. The viral DNA was extracted from 10 µl of purified scAAV2-TTR-RB15 viral vector in 50 µl reactions containing 0.4 mg/ml proteinase K, 1% SDS, and 10 mM EDTA at 50°C for 1 h, followed by phenol/chloroform extraction. The DNA was precipitated with salt and ethanol. The viral DNA was dissolved in H2O and applied to the alkaline agarose gel. The gel was electrophoreses in alkaline buffer for 3 h at 30 volts. At the end of gel electrophoresis, the gel was transferred to Hybond N+ membrane (Amersham) overnight and hybridized with 32P-labeled RB15 probe.
Western blot analysis
Western blot analysis was performed to detect AAV2 capsid proteins (VP1, VP2, and VP3). Virus (1 × 1010 particles) was separated on 10% SDS polyacrylamide gel under standard conditions and AAV2 capsid proteins (VP 1, VP2, and VP3) were detected using monoclonal antibody against AAV2 capsid proteins (American Research Products, Belmont, MA).
HepG2 Cells
Detection of ribozyme RB15 RNA in cells by one-step RT-PCR
A human hepatoma cell line (HepG2 cells, 2 × 105 cells) was seeded onto each well of a six-well plate until the cells reached 80% confluence (~5 × 105 cells). The cells were infected with 1 × 1010 particles of rAAV-TTR-RB15 or rAAV-TTR-RB15 mutant. At days 3 and 7 after infection, RNA was extracted from cells using Trizol reagent (Invitrogen). The expression level of ribozyme RB15 RNA was determined using the one-step RT-PCR kit (Qiagen), followed by Southern blot analysis using the 32P-end-labeled oligonucleotide RB15. Briefly, 1 µg of total RNA was treated with a DNase reagent using the DNA-free kit (Ambion) to remove the contaminating DNA. RT-PCR was then performed in the presence of the forward primer RBF2 (5' AGATCCACAAGCTCCTGA) and reverse primer RBR1 (5' ATAAGCTGCAATAAACAAGT) to amplify RB15 RNA. At the same time, PCR only control experiment, using the same amount of the treated total RNA was carried out. After RT-PCR, a product of 126-bp was detected using 1.5% agarose-1000 gel electrophoresis (Invitrogen). The PCR product was transferred using 0.4 N NaOH, followed by hybridization with radiolabeled oligonucleotide RB15 to detect RB15 DNA.
Quantification of apoB mRNA levels in HepG2 cells by RNase Protection assay
The apoB mRNA levels in HepG2 cells after rAAV-TTR-RB15 infection were determined using RNase protection assay as described previously [2]. Briefly, the RNase protection assay was carried out with 10 µg of total RNA and 3 × 104 cpm of 32P-UTP-labeled antisense apoB RNA probe in 20 µl of hybridization buffer (RPA III kit, Ambion). After RNase digestion, the protected fragment of 640 nucleotides was analyzed with 5% polyacrylamide-urea gel electrophoresis and quantified using the FX phosphoimage system (Bio-RAD).
Animal experiments
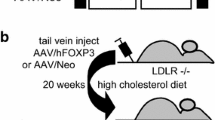
Mice deficient in both apoB mRNA editing enzyme (Apobec1-/-) and LDL receptor (LDLR-/-) were produced in our laboratory [8, 9] and were used for this study. This mouse model produces apoB100 only. It has markedly increased plasma total cholesterol and LDL cholesterol and develops atherosclerosis on a chow diet [7–9]. Recombinant adeno-associated virus, rAAV2-TTR-RB15 or rAAV2-TTR-RB15 mutant (as control) of 2 × 1011 virus particles was injected into the jugular vein of 2-months-old mice (n = 10 for each rAAV2 virus vector). These mice were maintained on laboratory chow (4% mouse/rat diet 7001; Harlan Teklad, Madison, WI). Samples of blood or tissue were obtained at the indicated time points after rAAV2 vectors injection. All animal experiments were conducted in accordance with the guidelines of the Animal Protocol Review Committee of the University of Texas Health Science Center at Houston (Houston, TX).
Analysis of DNA from tissues
Genomic DNA was prepared as described previously [18] from mouse liver and other organs including spleen, fat, colon, ileum, jejunum, duodenum, kidney, stomach, lung, and heart after rAAV2 transduction. rAAV2-TTR-RB15 or RB15-mutant genomes were determined in 1 µg of genomic DNA by PCR using forward primer RBF2 (5' AGATCCACAAGCTCCTGA) and reverse primer RBR1 (5' ATAAGCTGCAATAAACAAGT) to amplify RB15 DNA in the tissue. A product of 126-bp was detected using 1.5% agarose-1000 gel electrophoresis (Invitrogen). The PCR product was transferred using 0.4 N NaOH, followed by hybridization with radiolabeled oligonucleotide RB15 to detect RB15 DNA. Liver DNA from untreated mouse was used as negative control.
Detection of ribozyme RB15 RNA in tissues by one-step RT-PCR
Total RNA from livers and other organs of Apobec1-/-LDLR-/- mice was extracted using the Trizol system (Invitrogen). The expression levels of RB15 RNA in tissues after treatment were detected using the one-step RT-PCR method as described above.
Quantification of mouse apoB mRNA by real-time quantitative RT-PCR after rAAV2-TTR-RB15 treatment
The mice apoB mRNA levels after rAAV2 transductions were determined by real-time quantitative RT-PCR using ABI 7700 Sequence Detection System (Applied Biosystems). The sequence-specific primers and probes used for the mouse apoB mRNA and the endogenous control 18S ribosomal RNA were designed using Primer Express Software (Applied Biosystems). The nucleotide sequences were as follows:
Mouse apoB mRNA Forward primer 5' ATGTACTAATTGCCATAGATAGTGCCA,
Reverse primer 5' TCGCGTATGTCTCAAGTTGAGAG,
Probe: FAM-ATCAACTTCAATGAAAAA-MGBNFQ
Mouse 18S RNA Forward primer 5' TAACGAACGAGACTCTGGCAT,
Reverse primer 5' CGGACATCTAAGGGCATCACAG
Probe 5' FAM-TGGCTGAACGCCACTTGTCCCTCTAA-TAMRA.
The nucleotide sequences of each primer and probe were Blast searched against the Genebank database to confirm the uniqueness of each primer.
We used TaqMan One-Step RT-PCR Master Mix reagent (Applied Biosystems) to quantify RNA as described previously [8, 9]. The RNA standard curve for the specific gene was generated from T7-cDNA plasmid vector; serial dilutions of 103–109 molecules were employed in duplicate for the assay. Total RNA was treated with DNase (DNA-free kit; Ambion) to remove DNA contamination. The optimum RNA concentration for each gene was determined initially using real-time quantitative RT-PCR. Each RNA sample was then diluted accordingly. We used 150 ng and 50 pg of total RNA to quantify apoB mRNA and 18S RNA, respectively. Each of the RNA samples was normalized with an endogenous control of 18S ribosomal RNA. The copy numbers were calculated from the standard curve. The results are expressed as the ratio of specific mRNA/18S RNA.
Quantification of atherosclerotic lesions
The mice were anesthetized, exsanguinated and the aorta was carefully excised with part of the heart still attached. Under the stereomicroscope (Leica MZ60), all the fat and adventitious tissues were removed. With the major branching vessels still attached, the aorta was opened longitudinally from the iliac bifurcation to the aortic arch, and all the branching vessels and the heart were then removed [8, 9]. The aorta was pinned flat on a white wax surface, fixed overnight in 10% (v/v) formalin and stained with freshly prepared, filtered Oil Red O solution [19]. The aorta was scanned using the Polaroid Sprint Scan 35 Plus with Geoscan Enabler, the image was captured using Adobe Photoshop 5.0, and the background was removed with the guidance of the stereomicroscope. The total areas of the aorta and the atherosclerotic plaques were quantified using SigmaScan Pro 4.0 imaging software (SPSS Science, Chicago, IL). The results were presented as the percentage of the aortic surface covered by lesions (mm2) divided by the total surface area of the aorta (mm2).
Other Assays
Plasma cholesterol and triglyceride levels were determined using commercial enzymatic assay kits (Sigma). Immunoblot analysis, as described previously [20], was used to detect mouse apoB. Mouse apoB-specific antiserum was kindly provided by Dr. Thomas Innerarity (Gladstone Institute, San Francisco, CA).
Statistical analysis
The results are expressed as means ± SD. Student t-test was used to evaluate differences between the two groups. The p < 0.05 was considered to be significantly different.
Results and Discussion
We have previously demonstrated that hammerhead ribozyme RB15 cleaves both human and mouse apoB mRNA efficiently [3]. In this study, we constructed a recombinant adeno-associated virus vector expressing hammerhead ribozyme RB15 driven by a liver-specific promoter TTR (rAAV2-TTR-RB15). This rAAV2-TTR-RB15 vector was used to demonstrate the effects of long-term gene expression of ribozyme RB15 in mice on the alteration of apoB mRNA levels and the progression of atherosclerosis.
Characterization of the purified rAAV2-TTR-RB15
The genome copies of the purified rAAV2 virus were measured by real-time quantitative PCR. The purity of each viral vector prepared using either an iodixanol density gradient centrifugation followed by heparin-agarose column or a single-step heparin chromatography was examined by 10% SDS/PAGE. A representative preparation of rAAV2 by a single-step heparin chromatography method was shown in Fig. 1A and 1B. The structural CAP proteins of the rAAV2 vector (VP1, VP2, and VP3) were detected by Coomassive blue staining (Fig. 1A). The Western blot analysis using a mouse monoclonal antibody against the AAV2 capsid proteins confirmed that VP1, VP2, and VP3 were AAV2 structural proteins (Fig. 1B).

The purified rAAV-TTR-RB15 vector. The purified rAAV-TTR-RB15 prepared by a single-step heparin chromatography was separated by 10% SDS-PAGE, stained with Coomassieve blue (A), and the AAV capsid proteins (VP1, VP2, and VP3) were detected with a monoclonal antibody (B). The molecular weights of the capsid proteins are shown.
Effect of adeno-associated virus-mediated Ribozyme RB15 (rAAV2-TTR-RB15) gene expression in HepG2 cells
To test if the recombinant rAAV2-TTR-RB15 vector would result in a decrease of apoB mRNA levels, we infected HepG2 cells with rAAV2-TTR-RB15 or rAAV2-TTR-RB15-mutant to confirm that the expressed RB15 was biologically active. Total RNA was extracted from cells at days 3 and 7 after infection, RB15 RNA expression was determined by RT-PCR, followed by Southern blot analysis. As shown in Fig 2A, RB15 RNA was detected by RT-PCR and Southern blot analysis at days 3 and 7 after infection. There was no detectable band in the corresponding samples analyzed by PCR only.

The expressed ribozyme RB15 in HepG2 cells. HepG2 cells (5 × 105 cells/well) were infected with rAAV-TTR-RB15 (1 × 1010 particles/well). (A). Total RNA was extracted from infected cells at days 3 (lane 2) and 7 (lane 3). Ribozyme RB15 RNA was detected using RT-PCR, followed by Southern blot analysis. Lane 1 is non-infected control RNA and lane 4 is H2O blank. For a negative control, the same experiment was performed also by PCR only. The DNA marker is 100-bp DNA ladder. The same experiment was carried out with the rAAV-TTR-RB15 mutant. The results are not shown here. (B). Total RNA (10 µg) was hybridized with 32P-UTP-labeled anti-apoB RNA and 32P-UTP-labeled anti-GAPDH RNA. The expression levels of apoB mRNA and GAPDH mRNA were determined by an RNase protection assay using an RPA III kit. After RNase digestion, the protected fragments (apoB RNA = 640 nt and GAPDH RNA = 316 nt) were generated and analyzed with 5% polyacrylamide gel electrophoresis. The concentrations of protected RNAs were determined by a Phosphoimager FX system (Bio-Rad). The levels of apoB RNA were normalized with GAPDH RNA and a representative experiment is shown here. The probes and protected fragments of apoB mRNA and GAPDH RNA are indicated.
We used the RNase protection assay to quantify apoB mRNA concentration after rAAV2 infection. As shown in Fig. 2B, a protected fragment of 640 nucleotides was detected in non-treated cells and cells treated with either the rAAV2-TTR-RB15 or the rAAV2-TTR-RB15-mutant. The levels of apoB mRNA was normalized with GAPDH transcripts. In comparison to non-treated cells, at day-3 after infection, apoB mRNA levels were decreased 17% (non-treated = 0.813 ± 0.08, n = 3; rAAV-TTR-RB15-treated = 0.672 ± 0.117, n = 3; p = 0.0849). By day-7 the apoB mRNA levels were significantly decreased 91.5% (rAAV-TTR-RB15-treated = 0.069 ± 0.0, n = 3; p = 0.0019). In contrast, the apoB mRNA levels in cells treated with rAAV2-TTR-RB15-mutant (control vector) did not vary significantly; there was an enhancement at day-3 after infection of RB15 mutant vector, when compared to non-treated cells (non-treated = 0.861 ± 0.055, rAAV-TTR-RB15-mutant at day-3 = 1.102 ± 0.174, n = 3, p = 0.0643; day-7 = 0.947 ± 0.053, n = 3, p = 0.0613). Therefore, the AAV2-mediated apoB mRNA-specific hammerhead ribozyme RB15 markedly reduced apoB mRNA transcripts in HepG2 cells only at day-7 after treatment.
Hepatic uptake and expression of rAAV2-TTR-RB15 in mouse liver
Next, we injected rAAV2-TTR-RB15 or rAAV2-TTR-RB15-mutant (2 × 1011 particles) via the jugular vein into LDb mice (LDLR-/-Apobec1-/-) to examine the persistence of the RB15 gene expression and the specificity of the TTR promoter. Organs including liver, spleen, kidney, heart, lung, duodenum, jejunum, ileum, colon, fat, and muscle were collected at days 7 and 150 after virus injection. The DNA of the rAAV vector was detected mostly in the liver (95%) with trace amounts in the spleen (3.5%) and kidney (1%) at day-7 after injection. At day-150 after injection, vector DNA was still readily detectable in the liver with trace amounts in the kidney (Fig. 3A). In contrast, using RT-PCR method the ribozyme RNAs of RB15 and RB15 mutant were detected only in the mouse liver, but not in any other tissues that collected either at day-7 or at day-150 after injection (data not shown).

The effect of rAAV2-TTR-RB15-mediated gene expression in LDLR-/-Apobec1-/- mice. (A). The DNAs from various organs were extracted at days 7 and 150 after rAAV2-TTR-RB15 transduction. The AAV2-RB15 DNA was detected by amplification using PCR, followed by Southern blot analysis. The radioactive bands of RB15 are shown. (B). The expressed ribozymes RB15 and RB15 mutant RNAs in the livers of LDLR-/-Apobec1-/- mice treated with rAAV2-TTR-RB15 or RB15-mutant. The total liver RNA was extracted from each mouse at day-150 after rAAV-TTR-RB15 (2 × 1011 particles/animal, samples 1 – 5) or rAAV-TTR-RB15 mutant (2 × 1011 particles/animal, samples 6 – 10) transduction. Ribozymes RB15 or RB15 mutant RNAs were detected using RT-PCR, followed by Southern blot analysis. The radioactive bands of ribozymes RB15 and RB15 mutant are shown. The same samples were also subjected to PCR only, followed by Southern blot analysis. There were no detectable bands in these samples. (C). The effect of ribozymes RB15 and RB15 treatment on mouse apoB mRNA levels. Mouse apoB mRNA and endogenous 18S RNA from each mouse liver at day-150 after rAAV-TTR-RB15 or RB15 mutant treatment were determined by real-time quantitative RT-PCR. The results are expressed as the ratio of mouse apoB mRNA (mApoB mRNA) to 18S RNA. The results are shown as means ± standard deviation of ten animals. Each assay was performed in duplicate at two separate times. The results were analyzed by Student t-test.
Fig. 3B shows the results of ribozyme RB15 RNAs analyzed by RT-PCR in the livers of mice treated with rAAV2-TTR-RB15 (numbers 1 to 5) and rAAV2-TTR-RB15 mutant (numbers 6 to 10) at day-150 after injection. Control experiments using the same RNA samples analyzed by PCR only did not detect any RB15 RNA (Fig. 3B). Therefore, our results demonstrate that the gene expression of ribozyme RB15 persisted to day-150 (5 months) after virus injection and ribozyme RB15 was specifically expressed in the liver.
We, then, used real-time quantitative RT-PCR to examined the effect of rAAV2-TTR-RB15 and rAAV2-TTR-RB15-mutant on the levels of mouse apoB mRNA at day-150 after transduction. As shown in Fig. 3C, mouse apoB mRNA was significantly decreased 43% after RB15 treatment (p = 0.0262), compared to RB15-mutant treated group.
However, unlike the results observed in our previous experiments using adenovirus-mediated RB15, the persistent but low levels expression of RB15 ribozyme RNA did not have effects on the levels of plasma cholesterol (day-0 to day-150; 357 ± 42 to 435 ± 35 mg/dl) or triglycerides (day-0 to day-150; 237 ± 137 to 253 ± 15 mg/dl). The mouse plasma apoB levels also remained relatively the same throughout the study. We quantified the extent of atherosclerotic lesions on each animal after treatment. There was no difference in the severity of atherosclerotic lesions when comparing rAAV2-TTR-RB15-treated animals to rAAV2-TTR-RB15-mutant-control animals (rAAV2-TTR-RB15-treated = 11.9 ± 4.02 % lesions, rAAV2-TTR-RB15-mutant-treated = 11.6 ± 2.10 % lesions).
Taken together, rAAV2-mediated ribozyme gene expression in LDb (LDLR-/-Apobec1-/-) mice expressed active RB15 RNA, which decreased apoB mRNA by 43%. Nevertheless, this reduction levels of apoB mRNA did not yield a result of lowering the levels of apoB proteins or plasma cholesterol or triglyceride. Our previous works of expressing ribozyme RB15 mediated by an adenovirus vector in Apobec1-/-ERhB+/+ mice decrease apoB mRNA levels by 80%, which produce a markedly reduction of apoB protein of 62% and cholesterol and triglyceride of 42% and 51%, respectively [3]. The different observations demonstrated in our studies between using an adenovirus vector and an adeno-associated vector were a great concern.
AAV2 has been shown to have limit ability to transduce hepatocytes [21]. This low transduction ability of AAV2 to hepatocytes may be one of the main reasons that the expressed active RB15 RNA was limited to cleave apoB mRNA to a small percentage of cells. We have shown that apoB mRNA is constitutively expressed in hepatocytes [22] and the region of ribozyme RB15 target site GUA6679 is AU-rich, which was predicted to have a kinetic advantage for success in cleaving the transcript [2]. Thus, it is possible that the highly efficient RB15 catalytic RNA cleaved apoB mRNA in a limited number of transduced cells to yield a 43% reduction of apoB mRNA levels, but without a significant influence on the levels of apoB proteins or the lipids. Furthermore, LDb (LDLR-/-Apobec1-/-) mice have very high levels of cholesterol and triglyceride and the mice secrete cholesterol and triglyceride-rich LDL [9], therefore, we think a very efficient AAV gene transduction vector is probably needed to accomplish the goal of regulating the apoB production.
Improved AAV-mediated gene expression strategies
Ferrari et al. [23] have suggested that second-strand synthesis is a rate-limiting step for transduction of therapeutic genes by the AAV vectors. It was shown that the conversion of the input single-stranded DNA vector into a double-stranded DNA template for transcription is a slow and inefficient process in most non-dividing cells including hepatocytes. To enhance the transduction in vivo, McCarty et al [12] have shown that generation of a self-complementary AAV (scAAV) would bypass the rate-limiting step of second-strand synthesis. Ribozyme RB15 has a size of 61-bp in length, which is an ideal molecule for this approach. Accordingly, our laboratory has constructed the scAAV2 vectors expressing active ribozyme RB15 and inactive ribozyme RB15-mutant (Fig. 4A, scAAV2-TTR-RB15 or -RB15-mutant).

The expression of self-complementary AAV2-TTR-RB15 (scAAV2-TTR-RB15) vector in HepG2 cells. (A). The drawings depict the predicted conformations of monomer and dimer of TTR-RB15 virions. (B). Alkaline agarose gel electrophoresis (0.8%) of scAAV2-TTR-RB15 viral DNA, followed by Southern blot analysis, hybridized with 32P-labeled RB15 probe. The positions of monomer and dimmer are shown. (C). Comparison of the ribozyme RB15 RNA levels between cells treated with non-modified parent rAAV2-TTR-RB15 vectors and cells treated with scAAV2-TTR-RB15 vectors at days 1 and 3 after treatment. Total RNA was extracted from HepG2 cells treated with either non-modified parent AAV2-TTR-RB15 or scAAV2-TTR-RB15 vectors. The levels of RB15 RNA were determined using one-step RT-PCR and analyzed by 2% agarose gel electrophoresis. The same samples were also determined by PCR only. The position of RB15 RNA is shown. (D). Comparison of apoB mRNA levels in HepG2 cells at day-3 after treatment with non-modified parent rAAV2-TTR-RB15 vectors or with modified scAAV2-TTR-RB15 vectors. ApoB mRNA was determined by real-time quantitative RT-PCR. The apoB mRNA levels were normalized with 18S RNA. The results are expressed as % of cells treated with inactive RB15-mutant. The results are shown as means ± standard deviation of triple experiments. Experiment of cells infected with adenovirus-RB15-mutant, adenovirus-RB15 (positive control) are performed at the same time and used as a positive comparison purpose. Cells treated with parent-rAAV2-TTR-RB15-mutant, parent rAAV2-TTR-RB15, scAAV2-TTR-RB15-mutant, and scAAV2-TTR-RB15 are shown. The p values analyzed by student t-test are shown. ns = not significant
As shown in Fig. 4B, ~40% of AAV vectors produced were scAAV2-TTR-RB15 vector containing dimeric DNA genomes of 4856 nucleotides as analyzed by alkaline agarose gel electrophoresis. We used these scAAV2-TTR-RB15 and -RB15 mutant vectors to infect HepG2 cells, on days 1 and 3 after infection; scAAV-TTR-RB15 vector expressed much more ribozyme RB15 RNA, > 3-fold more ribozyme RNA produced compared to the non-modified parent RB15 vector (Fig. 4C) as determined by semi-quantitative RT-PCR. Moreover, on day-3 after infection, the apoB mRNA levels (determined by real-time RT-PCR, Fig. 4D) were significantly decreased 47% in scAAV-TTR-RB15 treated cells (p = 0.001), when compared to cells treated with inactive scAAV-TTR-RB15-mutant. In contrast, the apoB mRNA levels remained the same on cells treated with non-modified parent rAAV2-TTR-RB15 vector. Therefore, our results show that scAAV vector bypassed the conversion of single-stranded DNA to double-stranded DNA process, expressed ribozyme RB15 RNA faster and more efficiently.
Recently, two groups [24, 25] using double strand scAAV2 vector expressed GFP in mouse brain and liver. They showed that scAAV2 increased transgene expression faster, more efficiently, and last longer, when compared with non-modified single strand AAV2. Wang et al [25] showed that scAAV in liver remained as the circular low molecular weight episomal DNA, which gave the steady-state levels of transgene expression. From our experience shown on this study of using single-stranded rAAV2-TTR-RB15 vector in mice, we knew it is necessary to generate an optimal vector to obtain successful results in vivo. Wang et al. [25] and McCarty et al. [24] have shown that a modified scAAV2 vector by removing the D-sequence in the terminal resolution site of AAV2-5' ITR region [24, 25] yield high percentage of double-strand DNA and produce rapid and highly efficient transduction in mice. Furthermore, AAV2/8 vector has been shown to be more effective in transducing hepatocytes [26–28]. Thus, our laboratory are in the process of improving the scAAV vector by using the modified scAAV vector that produce mainly double-stranded DNA and package it in an AAV2/8 vector. These improvements should generate a much better AAV vector to express ribozyme RB15 in the liver in mice. We expect this improved AAV vector would provide a much better outcome than we have demonstrated using non-modified AAV2 and adenovirus vectors. The markedly reduction of apoB mRNA levels would lead to the inhibition of the development of atherosclerotic lesions.
The recent discovery of using short interfering RNA (siRNA) species to silence gene expression has demonstrated its great potential as a therapeutic-based gene silencing. Both ribozyme and siRNA are RNA-based technologies. Ribozymes are catalytic RNAs, which cleaves the target sequences efficiently. siRNAs are short RNAs of 21–23 nucleotides in length and are incorporated into a nuclease complex, the RNA-induced silencing complex (RISC), which then targets and cleaves the mRNA. The factors influencing efficient gene target of ribozyme and siRNA are similar, including position of the binding site, secondary and tertiary structures in mRNA, base pairing of RNA, cleavage efficiency to the mRNA, and turnover of mRNA after cleavage. Ribozyme technology has been shown to have some difficulties to apply universally, although apoB mRNA-specific hammerhead ribozyme cleaves apoB mRNA efficiently. The applicability of siRNA in mammals is probably limited also, since the introduction of dsRNA longer than 30 nucleotides induces a sequence-nonspecific interferon response [29] and the induction of gene activation of siRNA appears to be transient. However, using synthetic 21 nucleotides targeted specific to gene Fas in mice down-regulated Fas mRNA and prevented liver injury [30]. Thus, RNAi provides the opportunity to be an exciting new therapeutic approach. Allude to all; the development of a suitable long-term gene transfer vector will benefit to deliver all of these molecules to cells to regulate gene expression.
Abbreviations
- rAAV2:
-
recombinant adeno-associated virus vector sera type 2
- TTR:
-
transthyretin
- apoB:
-
apolipoprotein B
- Apobec1:
-
apoB mRNA editing enzyme
- LDLR:
-
low density lipoprotein receptor
- LDb mice:
-
(Apobec1-/-LDLR-/-) mice
- scAAV:
-
self-complementary adeno-associated virus vector
References
Symons RH: Small catalytic RNAs. Annu Rev Biochem. 1992, 61: 641-671. 10.1146/annurev.bi.61.070192.003233.
Wang JP, Enjoji M, Tiebel M, Ochsner S, Chan L, Teng BB: Hammerhead ribozyme cleavage of apolipoprotein B mRNA generates a truncated protein. J Biol Chem. 1999, 274: 24161-24170. 10.1074/jbc.274.34.24161.
Enjoji M, Wang F, Nakamuta M, Chan L, Teng BB: Hammerhead ribozyme as a therapeutic agent for hyperlipidemia: production of truncated apolipoprotein B and hypolipidemic effects in a dyslipidemia murine model. Hum Gene Ther. 2000, 11: 2415-2430. 10.1089/104303400750038516.
Rabinowitz JE, Samulski J: Adeno-associated virus expression systems for gene transfer. Curr Opin Biotechnol. 1998, 9: 470-475. 10.1016/S0958-1669(98)80031-1.
Jooss K, Yang Y, Fisher KJ, Wilson JM: Transduction of dendritic cells by DNA viral vectors directs the immune response to transgene products in muscle fibers. J Virol. 1998, 72: 4212-4223.
Yan C, Costa RH, Darnell J. E., Jr., Chen JD, Van Dyke TA: Distinct positive and negative elements control the limited hepatocyte and choroid plexus expression of transthyretin in transgenic mice. Embo J. 1990, 9: 869-878.
Powell-Braxton L, Veniant M, Latvala RD, Hirano KI, Won WB, Ross J, Dybdal N, Zlot CH, Young SG, Davidson NO: A mouse model of human familial hypercholesterolemia: markedly elevated low density lipoprotein cholesterol levels and severe atherosclerosis on a low-fat chow diet. Nat Med. 1998, 4: 934-938.
Dutta R, Singh U, Li TB, Fornage M, Teng BB: Hepatic gene expression profiling reveals perturbed calcium signaling in a mouse model lacking both LDL receptor and Apobec1 genes. Atherosclerosis. 2003, 169: 51-62. 10.1016/S0021-9150(03)00133-3.
Singh U, Zhong S, Xiong M, Li TB, Sniderman A, Teng BB: Increased plasma non-esterified fatty acids and platelet-activating factor acetylhydrolase are associated with susceptibility to atherosclerosis in mice. Clin Sci (Lond). 2004, 106: 421-432. 10.1042/CS20030375.
Wang Y, DeMayo FJ, Tsai SY, O'Malley BW: Ligand-inducible and liver-specific target gene expression in transgenic mice. Nat Biotechnol. 1997, 15: 239-243.
Dong JY, Fan PD, Frizzell RA: Quantitative analysis of the packaging capacity of recombinant adeno-associated virus. Hum Gene Ther. 1996, 7: 2101-2112.
McCarty DM, Monahan PE, Samulski RJ: Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001, 8: 1248-1254. 10.1038/sj.gt.3301514.
Grimm D, Kern A, Rittner K, Kleinschmidt JA: Novel tools for production and purification of recombinant adenoassociated virus vectors. Hum Gene Ther. 1998, 9: 2745-2760.
Conway JE, Rhys CM, Zolotukhin I, Zolotukhin S, Muzyczka N, Hayward GS, Byrne BJ: High-titer recombinant adeno-associated virus production utilizing a recombinant herpes simplex virus type I vector expressing AAV-2 Rep and Cap. Gene Ther. 1999, 6: 986-993. 10.1038/sj.gt.3300937.
Auricchio A, Hildinger M, O'Connor E, Gao GP, Wilson JM: Isolation of highly infectious and pure adeno-associated virus type 2 vectors with a single-step gravity-flow column. Hum Gene Ther. 2001, 12: 71-76. 10.1089/104303401450988.
Clark KR, Voulgaropoulou F, Fraley DM, Johnson PR: Cell lines for the production of recombinant adeno-associated virus. Hum Gene Ther. 1995, 6: 1329-1341.
Zolotukhin S, Byrne BJ, Mason E, Zolotukhin I, Potter M, Chesnut K, Summerford C, Samulski RJ, Muzyczka N: Recombinant adeno-associated virus purification using novel methods improves infectious titer and yield. Gene Ther. 1999, 6: 973-985. 10.1038/sj.gt.3300938.
Bell GI, Karam JH, Rutter WJ: Polymorphic DNA region adjacent to the 5' end of the human insulin gene. Proc Natl Acad Sci U S A. 1981, 78: 5759-5763.
Tangirala RK, Rubin EM, Palinski W: Quantitation of atherosclerosis in murine models: correlation between lesions in the aortic origin and in the entire aorta, and differences in the extent of lesions between sexes in LDL receptor-deficient and apolipoprotein E-deficient mice. J Lipid Res. 1995, 36: 2320-2328.
Teng B, Blumenthal S, Forte T, Navaratnam N, Scott J, Gotto A. M., Jr., Chan L: Adenovirus-mediated gene transfer of rat apolipoprotein B mRNA-editing protein in mice virtually eliminates apolipoprotein B-100 and normal low density lipoprotein production. J Biol Chem. 1994, 269: 29395-29404.
Nakai H, Thomas CE, Storm TA, Fuess S, Powell S, Wright JF, Kay MA: A limited number of transducible hepatocytes restricts a wide-range linear vector dose response in recombinant adeno-associated virus-mediated liver transduction. J Virol. 2002, 76: 11343-11349. 10.1128/JVI.76.22.11343-11349.2002.
Pullinger CR, North JD, Teng BB, Rifici VA, Ronhild de Brito AE, Scott J: The apolipoprotein B gene is constitutively expressed in HepG2 cells: regulation of secretion by oleic acid, albumin, and insulin, and measurement of the mRNA half-life. J Lipid Res. 1989, 30: 1065-1077.
Ferrari FK, Samulski T, Shenk T, Samulski RJ: Second-strand synthesis is a rate-limiting step for efficient transduction by recombinant adeno-associated virus vectors. J Virol. 1996, 70: 3227-3234.
McCarty DM, Fu H, Monahan PE, Toulson CE, Naik P, Samulski RJ: Adeno-associated virus terminal repeat (TR) mutant generates self-complementary vectors to overcome the rate-limiting step to transduction in vivo. Gene Ther. 2003, 10: 2112-2118. 10.1038/sj.gt.3302134.
Wang Z, Ma HI, Li J, Sun L, Zhang J, Xiao X: Rapid and highly efficient transduction by double-stranded adeno-associated virus vectors in vitro and in vivo. Gene Ther. 2003, 10: 2105-2111. 10.1038/sj.gt.3302133.
Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM: Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc Natl Acad Sci U S A. 2002, 99: 11854-11859. 10.1073/pnas.182412299.
Sarkar R, Tetreault R, Gao G, Wang L, Bell P, Chandler R, Wilson JM, Kazazian H. H., Jr.: Total correction of hemophilia A mice with canine FVIII using an AAV 8 serotype. Blood. 2004, 103: 1253-1260. 10.1182/blood-2003-08-2954.
Thomas CE, Storm TA, Huang Z, Kay MA: Rapid uncoating of vector genomes is the key to efficient liver transduction with pseudotyped adeno-associated virus vectors. J Virol. 2004, 78: 3110-3122. 10.1128/JVI.78.6.3110-3122.2004.
Elbashir SM, Harborth J, Lendeckel W, Yalcin A, Weber K, Tuschl T: Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature. 2001, 411: 494-498. 10.1038/35078107.
Song E, Lee SK, Wang J, Ince N, Ouyang N, Min J, Chen J, Shankar P, Lieberman J: RNA interference targeting Fas protects mice from fulminant hepatitis. Nat Med. 2003, 9: 347-351. 10.1038/nm828.
Acknowledgements
This research was supported by National Institutes of Health (NIH) grant HL-53441 and Texas Advanced Technology Program. We thank Lawrence Chan (Baylor College of Medicine, Houston, TX) for providing Apobec1-/- mice, Terry Van Dyke (University of North Carolina, Chapel Hill, NC) for providing transthyretin (TTR) promoter, Alan Davis (Baylor College of Medicine, Houston, TX) for providing pZAC2.0 plasmid, Jurgen Kleinschmidt (DKFZ, Heidelberg, Germany) for providing pDG plasmid, and Thomas Innerarity (Gladstone Institute, San Francisco, CA) for providing anti-mouse apoB antiserum.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing Interests
None declared.
Authors' Contributions
SZ and SS cloned the plasmid vectors and performed the experimental studies presented in this paper. BBT developed and designed the experiments and assisted in analysis of the results. All authors have read and approved this manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Zhong, S., Sun, S. & Teng, BB. The recombinant adeno-associated virus vector (rAAV2)-mediated apolipoprotein B mRNA-specific hammerhead ribozyme: a self-complementary AAV2 vector improves the gene expression. Genet Vaccines Ther 2, 5 (2004). https://doi.org/10.1186/1479-0556-2-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1479-0556-2-5