
Abstract
Clinical drug trials are often conducted in selective patient populations, with relatively small numbers of patients, and a short duration of follow-up. Observational studies are therefore important for collecting additional information on adverse drug events (ADEs). Currently, there is no guidance regarding the methodology for measuring ADEs in such studies. Our aim was to evaluate whether the methodology used to assess non-serious ADEs in observational studies is adequate for detecting these ADEs, and for addressing limitations from clinical trials in patients with type 2 diabetes mellitus. We systematically searched MEDLINE and EMBASE for observational studies reporting non-serious ADEs (1999-2008). Methods to assess ADEs were classified as: 1) medical record review; 2) surveillance by health care professionals (HCP); 3) patient survey; 4) administrative data; 5) laboratory/clinical values; 6) not specified. We compared the range of ADEs identified, number and selection of patients included, and duration of follow-up. Out of 10,125 publications, 68 studies met our inclusion criteria. The most common methods were based on laboratory/clinical values (n = 25) and medical record review (n = 18). Solicited surveillance by HCP (n = 17) revealed the largest diversity of ADEs. Patient surveys (n = 15) focused mostly on hypoglycaemia and gastrointestinal ADEs, laboratory values based studies on hepatic and metabolic ADEs, and administrative database studies (n = 5) on cardiovascular ADEs. Four studies presented ADEs that were identified with the use of more than one method. The patient population was restricted to a lower risk population in 19% of the studies. Less than one third of the studies exceeded pre-approval regulatory requirements for sample size and duration of follow-up. We conclude that the current assessment of ADEs is hampered by the choice of methods. Many observational studies rely on methods that are inadequate for identifying all possible ADEs. Patient-reported outcomes and combinations of methods are underutilized. Furthermore, while observational studies often include unselective patient populations, many do not adequately address other limitations of pre-approval trials. This implies that these studies will not provide sufficient information about ADEs to clinicians and patients. Better protocols are needed on how to assess adverse drug events not only in clinical trials but also in observational studies.
Similar content being viewed by others


Introduction
Medication safety assessment during the pre-approval regulatory phase is known to have limitations. Pre-approval clinical trials are often conducted in selective patient populations, with relatively small numbers of patients, and a short duration of follow-up [1, 2]. Because of these limitations, several systems have been developed to monitor drug safety after marketing, including spontaneous reporting systems and risk management plans. Such safety assessment focuses primarily on detection of serious adverse drug events (ADEs) [3]. Little attention is given to the assessment of symptomatic or non-life-threatening ADEs, while the proportion of such ADEs is relatively common [4, 5]. Symptomatic ADEs may affect patients' quality of life and adherence to treatment, and thereby the risk-benefit ratio of a drug.
Post-marketing observational studies are considered important to get more information on ADEs occurring in patient populations actually using the drugs [2, 6, 7]. This additional value, however, will only be achieved when the methodology used in such studies allows for adequate capturing of non-serious ADEs in an unrestricted population. The use of different methods for assessing ADEs, such as spontaneous and solicited reporting, medical record review, and patient surveys, may lead to differences in observed ADEs [8, 9]. No guidance exists regarding the methods to be used for measuring ADEs in post-marketing studies [10–13].
Our aim was to evaluate the current methodology for assessing non-serious ADEs in observational studies, using oral antihyperglycemic drugs (OAD) as case. Research questions addressed are: (1) which methods of ADE assessment are used, (2) what is the range of non-serious ADEs captured for each method, (3) do the observational studies address known limitations of pre-approval trials regarding patient population and follow-up.
Methods
Search Strategy
We conducted a systematic search of MEDLINE and EMBASE for observational studies reporting on ADEs in patients with diabetes, and published between January 1 1999 and January 1 2009. We searched for papers using MeSH headings, subheadings and free-text terms related to the following domains: (1) "adverse events", and (2) "observational study design", and (3) "drug treatment" combined with "diabetes" (see Additional file 1 for detailed description of the search strategy). Using the boolean operator 'AND', only papers satisfying all three domains were included.
Study Selection
Observational studies, i.e. non-experimental studies where decisions regarding the prescription of drugs to each patient were made by their health care provider in every-day clinical practice, were included when they reported rates of non-serious ADEs in adult patients with type 2 diabetes mellitus treated with OAD. We excluded open-label extensions of clinical trials. Non-serious ADEs were defined as any unfavourable and unintended sign (including abnormal laboratory values) or symptom or disease that may present during treatment with a pharmaceutical product and which was not life-threatening, requiring hospitalization or resulted in significant disability or death.
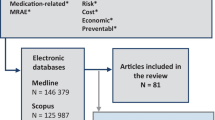
The first title and abstract screening was done by LH, excluding editorials, comments, notes, letters, randomized clinical trials (RCTs), case reports, and studies not including patients with diabetes or not including OAD (see also Figure 1 for exclusions). PD screened a 10% sample which showed that LH had not excluded any potentially relevant studies. Screening of the remaining abstracts and full-texts was done by two reviewers independently. We restricted our selection to studies published in English, German, French, Spanish or Dutch language.

Study flow diagram.
Data Extraction
Information was collected from the selected publications each by two reviewers (PD/LH, DD/LH or FHR/LH) using a standardized data extraction form. Data were extracted regarding methods used for assessing ADEs, the ADEs identified, inclusion and exclusion criteria of patient population, sample size, and duration of follow-up. In addition, we extracted data on study design and medications covered. Discrepancies in data extraction occurred in 3 cases regarding 'methods used for assessing ADEs', in 8 cases regarding 'sample size', and 9 cases regarding 'duration of follow-up'. These discrepancies were often the result of unclear descriptions in the publications, and were solved by consensus based on a joint re-evaluation of what was described in the publication.
Methods for ADE assessment
ADE assessment in observational studies can be based on review of existing practice-based data, such as medical records, laboratory reports, and administrative data, on surveillance by health care professionals (HCP) or on survey of patients [9, 10, 14]. Following this distinction, we defined the employed methods as: 1) medical record review, i.e. possible ADEs were collected from documentation or reports made by HCP in existing medical records; 2) solicited surveillance by HCP, i.e. requesting HCP to report possible ADEs either on Case Report Forms (prospective) or on socalled Prescription Event Monitoring forms (retrospective) [7]; 3) patient survey, including the use of open or closed patient questionnaires, checklists or diaries; 4) administrative data, making use of diagnostic codes related to possible ADEs in administrative or claims data; 5) laboratory or clinical values indicating ADEs, including results of laboratory measurements and physical examinations such as weight or blood pressure; 6) non-specified methods. Reported ADEs were categorized on anatomy or pathophysiology level according to Common Terminology Criteria for Adverse Events (CTCAE v3.0) classification [15].
Patient population
Based on the reported patient inclusion and exclusion criteria, we classified studies as: (A) restricting the patient population to lower risk patients, (B) restricting to higher risk patients, (C) applying restrictions needed to achieve reliable outcome assessment, e.g. by excluding patients with a condition or medication use at baseline which would confound the outcome, (D) no restrictions reported.
Sample size and duration of follow-up
We assessed the number of patients exposed to OAD, as well as the duration of their follow-up. For studies including more than one treatment group, we considered the sample size of the largest group exposed to OAD treatment. For studies including a diabetic subcohort, the overall number of exposed patients was considered as the sample size. Based on recommendations from regulatory agencies for safety assessment [11, 12, 16, 17], we categorized sample sizes into six levels: 1) < 100 patients; 2) 100 to 299 patients; 3) 300 to 599 patients; 4) 600 to 1499 patients; 5) 1500 to 5000 and 6) > 5000 patients. Duration of follow-up for cohort studies was classified into: 1) ≤6 months; 2) 7-12 months; 3) 13 to 24 months; 4) more than 2 years.
Data Analysis
Some publications reported on multiple studies with different patient populations and methods. We conducted analysis at this study level. We present the type, median number and interquartile range (IQR) of ADEs at category level reported for the six different methods of ADE assessment. Sample size and duration of follow-up are also compared for the different ADE assessment methods. We calculated the number of studies reaching regulatory recommendations for pre-approval safety assessment of drugs intended for long-term treatment of non-life-threatening conditions, i.e. 100 patients exposed for a minimum of 1 year or 300-600 patients for 6 months can be adequate to assess the pattern of ADEs over time [11, 12].
Results
The search resulted in 10,125 articles, out of which we selected 904 articles for full-text screening (Figure 1), resulting in 64 relevant articles reporting on 68 studies (see Additional file 2 for a description of the included studies).
Methods of ADE assessment
The most commonly employed methods for assessing ADEs were based on laboratory/clinical values (n = 25), medical record review (n = 18), and solicited surveillance by HCP (n = 17) (Table 1). Surveillance by HCP was conducted prospectively using Case Report Forms in 12 studies, and retrospectively in 5 Prescription Event Monitoring studies. Among the 15 studies which used patient survey methods, 10 studies used a closed questionnaire, including two validated questionnaires [18, 19], one used a checklist [20], one used a semi-structured interview guide where patients could report any perceived ADEs [21], and one used a 16-item content-validated questionnaire, containing closed and open-ended questions focusing among other issues on specific adverse events [22]. A patient diary was used in two studies [23, 24]. Administrative databases were used in 5 studies, and in 7 studies, the data collection method was not fully specified.
ADEs identified with different methods
The largest range of ADEs was identified with solicited surveillance by HCP, yielding a median of 4 ADE categories (Table 1). The range was even higher for retrospective surveillance (median 7, IQR 4-9) in comparison to prospective surveillance (median 3.5, IQR 2-6). Medical record review identified a median of 2 ADE categories (Table 1), covering many different areas (Table 2). Other specified methods assessed mostly 1 ADE category per study. Patient survey methods often focused on perceived hypoglycaemia or gastrointestinal ADEs (Table 2). Administrative databases were mainly used for cardiac ADEs, and laboratory/clinical values often included hepatic or metabolic problems or weight increase (Table 2). Four studies identified the same ADE, either hypoglycaemia or hepatic dysfunction, using more than one method, in particular a combination of laboratory values and other methods [25–28].
Patient population
In 28 studies (41%), there were no specific limitations regarding the patient population included. In two studies (3%), no inclusion or exclusion criteria were specified [29, 30]. Thirteen studies (19%) limited inclusion of patients to lower risk patients (category A) by including only patients with less severe diabetes [20, 26, 27, 31–33] or patients on monotherapy [19, 24, 27, 33–36], or OAD-naïve patients [27, 35] or by excluding high risk patients who failed previous therapy [37] or with multiple comorbidity [20, 38, 39]. Fifteen studies (22%) limited the inclusion to more complicated cases (category B), such as inadequately controlled by or not tolerating previous medication [40–45], receiving combination treatment [46–48] or insulin [21, 23, 45, 49] or treated with maximum dose of medication [50]. Furthermore, 18 studies (27%) excluded patients based on the presence at baseline of the outcome or a condition that could influence the outcome [18, 24, 25, 33, 37–39, 47, 51–55], non-availability of measurements and/or clinical visits [35, 37, 46, 47, 50, 54, 56, 57], inability to fill in questionnaires (category C) [18, 21, 46, 56].
Sample size and duration of follow-up
Studies using patient survey methods, medical record review, or laboratory data often included less than 300 patients (Figure 2). A sample size of equal or more than 1500 was achieved by all studies using administrative databases, and in many studies using solicited surveillance by HCP. Overall, the follow-up period did not exceed one year in 77% of the cohort studies. Longer follow-up periods were mostly seen in studies using administrative data or laboratory/clinical values. Evaluation of sample size and follow-up jointly showed that all 3 cohort studies using administrative data exceeded the requirements of the guidelines for pre-approval safety assessment, whereas this was the case in less than a quarter of the studies using any of the other specified methods (Table 3).

Sample size included in studies using different assessment methods.
Discussion
Commonly used methods for assessing non-serious ADEs in patients with diabetes were based laboratory or clinical values, medical record review or solicited surveillance by HCP. The latter method identified the broadest range of ADE categories. Patient survey methods were used in 22% of the studies, and often focused on a limited range of ADEs, such as hypoglycaemia or gastrointestinal ADEs. The patient population was restricted to a lower risk population in a fifth of the studies. Less than one-third of studies exceeded pre-approval requirements regarding sample size and duration of follow-up.
Solicited surveillance by health care providers, using either prospective or retrospective data collection, revealed the largest diversity of ADEs, indicating that doctors register more events on such forms than in routine medical records. This is in line with previous findings that medical record review, although broadly used for assessing ADEs, results in incomplete findings [11, 58]. Since there is no systematic documentation of ADEs in medical records, partly due to limitations of the documentation systems [59, 60], review of such records lacks a standardized and reliable method to search for ADEs [61]. For non-serious, symptomatic ADEs the incomplete documentation of adverse events in medical records is even more the case when such ADEs do not warrant immediate action [1, 62]. Prescription Event Monitoring studies, which make use of an open question to report all events that occurred during drug use on special forms, or prospective studies using prespecified Case Report Forms may solve this problem.
Patient reports can be of great value for ADE assessment because of the differences between reports from patients and health care providers [4, 63–66]. Patients are a helpful source for the identification of many symptomatic ADEs, such as dizziness, malaise, fatigue, sexual function disorders, and pain [67–69]. Surprisingly, we found that patient survey methods were used in a relatively small number of studies, and moreover, often limited in their focus. Although comprehensive questionnaires have been developed to assess patient-perceived ADEs [70, 71], such questionnaires were not used in observational studies for diabetes treatment.
Laboratory values may have a limited value for assessing non-serious ADEs, since mainly hepatic and metabolic problems were identified by these methods. This is in contrast with previous estimates that more than half of the ADEs can be detected by biochemical tests [72]. Administrative databases are also limited regarding the types of ADEs that can be identified. Such databases can be useful for assessing ADEs leading to hospitalization but have less value for assessing non-serious ADEs. Diagnostic administrative coding is likely to be both incomplete and unspecific for detecting non-serious ADEs [73], because these ADEs do not always call for a documented action from the health care provider [1, 62]. Currently, European Medicines Agency regulators work on strengthening this source of information by establishing a European Network of Centres for Pharmacovigilance and Pharmacoepidemiology [74].
Combining methods for ADE assessment could address some limitations seen with all methods leading to under- or overreporting. ADEs which are likely to be underreported because of improper registration or coding in medical records might be complemented by laboratory values [73]. The same applies to doctor and patient reports that may complement each other [75]. In our review, however, only a four studies identified the same ADE using a combination of methods.
Observational post-marketing studies can provide additional information on ADEs when sufficient numbers of patients are being followed in daily practice, including those with higher risks, more comorbidity, concomitant drugs, and longer disease duration. The majority of studies in our review included such patient populations, thus adding valuable information on ADEs in patient groups underrepresented in pre-approval trials. The number of patients included and the duration of follow-up, however, showed similar limitations as pre-registration trials, and the majority of studies did not go beyond the pre-approval recommendations for safety assessment of diabetes medication. Because of workload, long follow-up for large numbers of patients can be problematic in studies where the patients or the health care providers need to provide the information. It is less problematic when data can be collected from existing databases.
Our study has some limitations. It has previously been recognized that searching the literature for studies reporting on drug safety is difficult [76, 77], and also indexing of observational studies may not be as robust as of RCTs. We therefore used a broad search strategy to identify possibly relevant studies. Second, the results are based on studies conducted in diabetes patients using OADs. For other therapeutic areas and other drugs, results may be different. Third, we used the CTCAE v3.0 classification to define ranges of ADEs identified by different methods. Although the CTCAE categories are quite similar to the primary system organ classes in the MedDRA hierarchy, minor differences in ranges may occur when using this alternative classification system. Finally, we encountered several problems regarding unclear or incomplete reporting. Although it was not our aim to evaluate studies on the quality of reporting, and we did not exclude studies on these grounds, we observed that information on, for example, exclusion criteria and response rates was often lacking.
Conclusion
The current set up of ADE assessment in post-marketing studies is not adequate for countering limitations acknowledged in pre-approval trials. The assessment of non-serious ADEs is limited by the choice of methods. Many observational studies rely on methods that are inadequate for identifying all possible ADEs. Patient survey methods are underutilized, and there is a lack of studies that try to combine different methods to assess ADEs. This implies that these studies will not provide sufficient information about ADEs to clinicians and patients. Better protocols are needed on how to assess adverse drug events not only in clinical trials but also in observational studies.
Abbreviations
- ADEs:
-
adverse drug events
- CTCAE v3.0:
-
Common Terminology Criteria for Adverse Events version 3.0
- HCP:
-
health care provider
- IQR:
-
interquartile range
- OAD:
-
oral antihyperglycemic drugs
- RCTs:
-
randomized clinical trials.
References
Martin K, Bégaud B, Latry P, Miremont-Salamé G, Fourrier A, Moore N: Differences between clinical trials and postmarketing use. Br J Clin Pharmacol 2004, 57: 86–92.
Lassila R, Rothschild C, De Moerloose P, Richards M, Perez R, Gajek H: European Haemophilia Therapy Standardisation Board. Recommendations for postmarketing surveillance studies in haemophilia and other bleeding disorders. Haemophilia 2005, 11: 353–359. 10.1111/j.1365-2516.2005.01114.x
Hazell L, Shakir SAW: Under-reporting of adverse drug reactions: a systematic review. Drug Saf 2006, 29: 385–396. 10.2165/00002018-200629050-00003
Basch E: The missing voice of patients in drug-safety reporting. N Engl J Med 2010, 362: 865–869. 10.1056/NEJMp0911494
Weingart SN, Gandhi TK, Seger AC, Seger DL, Borus J, Burdick E, Leape LL, Bates DW: Patient-reported medication symptoms in primary care. Arch Intern Med 2005, 165: 234–240. 10.1001/archinte.165.2.234
Ligthelm RJ, Borzì V, Gumprecht J, Kawamori R, Wenying Y, Valensi P: Importance of observational studies in clinical practice. Clin Ther 2007, 29: 1284–1292. Spec No 10.1016/j.clinthera.2007.07.004
Mann RD: Prescription-event monitoring--recent progress and future horizons. Br J Clin Pharmacol 1998, 46: 195–201.
Bent S, Padula A, Avins AL: Brief communication: Better ways to question patients about adverse medical events: A Randomized, Controlled Trial. Ann Intern Med 2006, 144: 257–261.
Field TS, Gurwitz JH, Harrold LR, Rothschild JM, DeBellis K, Seger AC, Fish LS, Garber L, Kelleher M, Bates DW: Strategies for detecting adverse drug events among older persons in the ambulatory setting. J Am Med Inform Assoc 2004, 11: 492–498. 10.1197/jamia.M1586
European Medicines Agency Committee for medicinal products for human use (CHMP): ICH Topic E 2 E Pharmacovigilance Planning (Pvp) Note for guidance on planning pharmacovigilance activities (CPMP/ICH/5716/03) [online]. 2011. [http://www.ema.europa.eu/pdfs/human/ich/571603en.pdf]
European Medicines Agency Committee for medicinal products for human use (CHMP): ICH Topic E1 The Extent of Population Exposure to Assess Clinical Safety for Drugs Intended for Long-Term Treatment of Non-Life-Threatening Conditions (CPMP/ICH/375/95) [online]. 2011. [http://www.ema.europa.eu/pdfs/human/ich/037595en.pdf]
US Department of Health and Human Services, Food and Drug Administration. Center for Drug Evaluation and Research (CDER): Guidance for Industry. Premarketing Risk Assessment, March 2005 [online]. 2011. [http://www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM126958.pdf]
European Medicines Agency Committee for medicinal products for human use (CHMP): ICH Topic E6 (R1) Guideline for Good Clinical Practice (CPMP/ICH/135/95) [online]. 2011. [http://www.ema.europa.eu/pdfs/human/ich/013595en.pdf]
Morimoto T, Gandhi TK, Seger AC, Hsieh TC, Bates DW: Adverse drug events and medication errors: detection and classification methods. Qual Saf Health Care 2004, 13: 306–314. 10.1136/qshc.2004.010611
National Cancer Institute. Cancer Therapy Evaluation Program: Common Terminology Criteria for Adverse Events (CTCAE) Version 3.0, August 9, 2006 [online]. 2011. [http://ctep.cancer.gov/protocolDevelopment/electronic_applications/docs/ctcaev3.pdf]
US Department of Health and Human Services, Food and Drug Administration. Center for Drug Evaluation and Research (CDER): Guidance for Industry. Diabetes Mellitus - Evaluating Cardiovascular Risk in New Antidiabetic Therapies to Treat Type 2 Diabetes December, 2008 [online]. 2011. [http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm071627.pdf]
European Medicines Agency Committee for medicinal products for human use (CHMP): Guideline on clinical investigation of medicinal products in the treatment of diabetes mellitus. Draft. CPMP/EWP/1080/00 Rev. 1. 20 Jan 2010 [online]. 2011. [http://www.ema.europa.eu/pdfs/human/ewp/108000enrev1.pdf]
Bytzer P, Talley NJ, Jones MP, Horowitz M: Oral hypoglycaemic drugs and gastrointestinal symptoms in diabetes mellitus. Aliment Pharmacol Ther 2001, 15: 137–142. 10.1046/j.1365-2036.2001.00896.x
Woodcock A, Bain S, Charlton M, Bradley C: Extent of satisfaction with tablets and food-timing in sulphonylurea-treated diabetes. Diabetes Res Clin Pract 2007, 78: 324–333. 10.1016/j.diabres.2005.07.013
Vexiau P, Mavros P, Krishnarajah G, Lyu R, Yin D: Hypoglycaemia in patients with type 2 diabetes treated with a combination of metformin and sulphonylurea therapy in France. Diabetes Obes Metab 2008,10(suppl 1):16–24.
Haugbolle LS, Sorensen EW: Drug-related problems in patients with angina pectoris, type 2 diabetes and asthma - interviewing patients at home. Pharm World Sci 2006, 28: 239–247. 10.1007/s11096-006-9023-9
Yusuff KB, Obe O, Joseph BY: Adherence to anti-diabetic drug therapy and self management practices among type-2 diabetics in Nigeria. Pharm World Sci 2008, 30: 876–883. 10.1007/s11096-008-9243-2
Klocke KR, Stauch K, Landen H: Effect of add-on acarbose to insulin therapy in routine clinical practice. Clin Drug Invest 2003, 23: 621–627. 10.2165/00044011-200323100-00001
Guagnano MT, Pace-Palitti V, Formisano S, Della Loggia F, D'Anchino M, Della Vecchia R, Merlitti D, Sensi S: Does holiday hypoglycaemia exist? Panminerva Med 2000, 42: 23–26.
Jick SS, Stender M, Myers MW: Frequency of liver disease in type 2 diabetic patients treated with oral antidiabetic agents. Diabetes Care 1999, 22: 2067–2071. 10.2337/diacare.22.12.2067
UK Hypoglycaemia Study Group: Risk of hypoglycaemia in types 1 and 2 diabetes: effects of treatment modalities and their duration. Diabetologia 2007, 50: 1140–1147.
Taki H, Maki T, Iso T, Tanabe S, Kajiura T: Postmarketing study of nateglinide in Japan: treatment of medication-naive patients with type 2 diabetes. Adv Ther 2005, 22: 621–635. 10.1007/BF02849956
Taki H, Maki T, Iso T, Iwamoto K, Kajiura T: Postmarketing surveillance study of nateglinide in Japan. Adv Ther 2005, 22: 513–526. 10.1007/BF02849871
Biswas P, Wilton LV, Shakir SAW: Troglitazone and liver function abnormalities: lessons from a prescription event monitoring study and spontaneous reporting. Drug Saf 2001, 24: 149–154. 10.2165/00002018-200124020-00004
Landgraf R, Frank M, Bauer C, Dieken ML: Prandial glucose regulation with repaglinide: its clinical and lifestyle impact in a large cohort of patients with Type 2 diabetes. Int J Obes Relat Metab Disord 2000,24(suppl 3):S38–44.
Chao J, Nau DP, Aikens JE: Patient-reported perceptions of side effects of antihyperglycemic medication and adherence to medication regimens in persons with diabetes mellitus. Clin Ther 2007, 29: 177–180. 10.1016/j.clinthera.2007.01.014
Karagiannis E, Pfutzner A, Forst T, Lubben G, Roth W, Grabellus M, Flannery M, Schondorf T: The IRIS V study: pioglitazone improves systemic chronic inflammation in patients with type 2 diabetes under daily routine conditions. Diabetes Technol Ther 2008, 10: 206–212. 10.1089/dia.2008.0244
McAlister FA, Eurich DT, Majumdar SR, Johnson JA: The risk of heart failure in patients with type 2 diabetes treated with oral agent monotherapy. Eur J Heart Fail 2008, 10: 703–708. 10.1016/j.ejheart.2008.05.013
Chokrungvaranon N, Chentanez T, Arakaki RF: Clinical experience with exenatide in predominantly Asian and Pacific Islander patients with type 2 diabetes. Endocrine 2007, 32: 311–316. 10.1007/s12020-008-9042-5
Asche CV, McAdam-Marx C, Shane-McWhorter L, Sheng X, Plauschinat CA: Evaluation of adverse events of oral antihyperglycaemic monotherapy experienced by a geriatric population in a real-world setting: a retrospective cohort analysis. Drugs Aging 2008, 25: 611–622. 10.2165/00002512-200825070-00006
Hanefeld M, Pfutzner A, Forst T, Lubben G: Glycemic control and treatment failure with pioglitazone versus glibenclamide in type 2 diabetes mellitus: a 42-month, open-label, observational, primary care study. Curr Med Res Opin 2006, 22: 1211–1215. 10.1185/030079906X112598
Olansky L, Marchetti A, Lau H: Multicenter retrospective assessment of thiazolidinedione monotherapy and combination therapy in patients with type 2 diabetes: Comparative subgroup analyses of glycemic control and blood lipid levels. Clin Ther 2003,25(suppl B):B64-B80.
Blonde L, Dailey GE, Jabbour SA, Reasner CA, Mills DJ: Gastrointestinal tolerability of extended-release metformin tablets compared to immediate-release metformin tablets: results of a retrospective cohort study. Curr Med Res Opin 2004, 20: 565–572. 10.1185/030079904125003278
Hermann LS, Nilsson B, Wettre S: Vitamin B12 status of patients treated with metformin: A cross-sectional cohort study. Br J Diabetes Vasc Dis 2004, 4: 401–406. 10.1177/14746514040040060701
Feher MD, Al-Mrayat M, Brake J, Leong KS: Tolerability of prolonged-release metformin (Glucophage(registered trademark) SR) in individuals intolerant to standard metformin - Results from four UK centres. Br J Diabetes Vasc Dis 2007, 7: 225–228. 10.1177/14746514070070050501
Hussein Z, Wentworth JM, Nankervis AJ, Proietto J, Colman PG: Effectiveness and side effects of thiazolidinediones for type 2 diabetes: real-life experience from a tertiary hospital. Med J Aust 2004, 181: 536–539.
Hung YJ, Kuo SW, Wang CH, Chang HY, Hsieh SH, Landen H: Postmarketing surveillance of acarbose treatment in Taiwanese patients with type 2 diabetes mellitus. Clin Drug Invest 2006, 26: 559–565. 10.2165/00044011-200626100-00002
Twaites B, Wilton LV, Layton D, Shakir SAW: Safety of nateglinide as used in general practice in England: results of a prescription-event monitoring study. Acta Diabetol 2007, 44: 233–239. 10.1007/s00592-007-0010-y
Hershon KS, Hershon PM: Primary, secondary, tertiary, and quaternary treatment with troglitazone in type 2 diabetes mellitus in an outpatient clinical practice. Endocrine Practice 2000, 6: 20–25.
Furlong NJ, McNulty SJ, O'Brien SV, Hardy KJ: Comparison of metformin versus sulphonylurea in combination with daily NPH insulin in patients with type 2 diabetes inadequately controlled on oral hypoglycaemic agents: Median follow-up 29 months. Pract Diabetes Int 2002, 19: 245–249. 10.1002/pdi.399
Alvarez Guisasola F, Tofé Povedano S, Krishnarajah G, Lyu R, Mavros P, Yin D: Hypoglycaemic symptoms, treatment satisfaction, adherence and their associations with glycaemic goal in patients with type 2 diabetes mellitus: findings from the Real-Life Effectiveness and Care Patterns of Diabetes Management (RECAP-DM) Study. Diabetes Obes Metab 2008,10(Suppl 1):25–32.
Delea TE, Edelsberg JS, Hagiwara M, Oster G, Phillips LS: Use of thiazolidinediones and risk of heart failure in people with type 2 diabetes: a retrospective cohort study. Diabetes Care 2003, 26: 2983–2989. 10.2337/diacare.26.11.2983
Schatz H, Schoppel K, Lehwalder D, Schandry R: Efficacy, tolerability and safety of nateglinide in combination with metformin. Results from a study under general practice conditions. Exp Clin Endocrinol Diabetes 2003, 111: 262–266.
Marceille JR, Goins JA, Soni R, Biery JC, Lee TA: Chronic heart failure-related interventions after starting rosiglitazone in patients receiving insulin. Pharmacotherapy 2004, 24: 1317–1322. 10.1592/phco.24.14.1317.43155
King AB, Armstrong DU: Lipid response to pioglitazone in diabetic patients: Clinical observations from a retrospective chart review. Diabetes Technol Ther 2002, 4: 145–151. 10.1089/15209150260007354
Maru S, Koch GG, Stender M, Clark D, Gibowski L, Petri H, White AD, Simpson RJ Jr: Antidiabetic drugs and heart failure risk in patients with type 2 diabetes in the U.K. primary care setting. Diabetes Care 2005, 28: 20–26. 10.2337/diacare.28.1.20
Nichols GA, Koro CE, Gullion CM, Ephross SA, Brown JB: The incidence of congestive heart failure associated with antidiabetic therapies. Diabetes Metab Res Rev 2005, 21: 51–57. 10.1002/dmrr.480
Miller CD, Phillips LS, Ziemer DC, Gallina DL, Cook CB, El-Kebbi IM: Hypoglycemia in patients with type 2 diabetes mellitus. Arch Intern Med 2001, 161: 1653–1659. 10.1001/archinte.161.13.1653
Shaya FT, Shin JY, Mullins CD, El Khoury AC, Garber H: Risk of heart failure with the use of thiazolidinediones within a medicaid population. Pharmacy Therapeutics 2005, 30: 273–281.
Filioussi K, Bonovas S, Katsaros T: Should we screen diabetic patients using biguanides for megaloblastic anaemia? Aust Fam Physician 2003, 32: 383–384.
Grant RW, Devita NG, Singer DE, Meigs JB: Polypharmacy and medication adherence in patients with type 2 diabetes. Diabetes Care 2003, 26: 1408–1412. 10.2337/diacare.26.5.1408
Chalasani N, Teal E, Hall SD: Effect of rosiglitazone on serum liver biochemistries in diabetic patients with normal and elevated baseline liver enzymes. Am J Gastroenterol 2005, 100: 1317–1321. 10.1111/j.1572-0241.2005.41690.x
Jha AK, Kuperman GJ, Teich JM, Leape L, Shea B, Rittenberg E, Burdick E, Seger DL, Vander Vliet M, Bates DW: Identifying adverse drug events: development of a computer-based monitor and comparison with chart review and stimulated voluntary report. J Am Med Inform Assoc 1998, 5: 305–314. 10.1136/jamia.1998.0050305
Nebeker JR, Barach P, Samore MH: Clarifying adverse drug events: a clinician's guide to terminology, documentation, and reporting. Ann Intern Med 2004, 140: 795–801.
Pakhomov SV, Jacobsen SJ, Chute CG, Roger VL: Agreement between patient-reported symptoms and their documentation in the medical record. Am J Manag Care 2008, 14: 530–539.
Honigman B, Light P, Pulling RM, Bates DW: A computerized method for identifying incidents associated with adverse drug events in outpatients. Int J Med Inform 2001, 61: 21–32. 10.1016/S1386-5056(00)00131-3
Golomb BA, McGraw JJ, Evans MA, Dimsdale JE: Physician response to patient reports of adverse drug effects: implications for patient-targeted adverse effect surveillance. Drug Saf 2007, 30: 669–675. 10.2165/00002018-200730080-00003
Fromme EK, Eilers KM, Mori M, Hsieh YC, Beer TM: How accurate is clinician reporting of chemotherapy adverse effects? A comparison with patient-reported symptoms from the Quality-of-Life Questionnaire C30. J Clin Oncol 2004, 22: 3485–3490. 10.1200/JCO.2004.03.025
Van Grootheest K, van Puijenbroek EP, de Jong-van den Berg LT: Do pharmacists' reports of adverse drug reactions reflect patients' concerns? Pharm World Sci 2004, 26: 155–159.
Van Grootheest K, de Jong-van den Berg LT: Patients' role in reporting adverse drug reactions. Expert Opin Drug Saf 2004, 3: 363–368. 10.1517/14740338.3.4.363
Jarernsiripornkul N, Kakaew W, Loalukkana W, Krska J: Adverse drug reaction monitoring: comparing doctor and patient reporting for new drugs. Pharmacoepidemiol Drug Saf 2009, 18: 240–245. 10.1002/pds.1708
Mitchell AS, Henry DA, Sanson-Fisher R, O'Connell DL: Patients as a direct source of information on adverse drug reactions. BMJ 1988, 297: 891–893. 10.1136/bmj.297.6653.891
Ferrari A, Spaccapelo L, Gallesi D, Sternieri E: Focus on headache as an adverse reaction to drugs. J Headache Pain 2009, 10: 235–239. 10.1007/s10194-009-0127-1
Aagaard L, Nielsen LH, Hansen EH: Consumer reporting of adverse drug reactions: a retrospective analysis of the Danish adverse drug reaction database from 2004 to 2006. Drug Saf 2009, 32: 1067–1074. 10.2165/11316680-000000000-00000
Jarernsiripornkul N, Krska J, Capps PA, Richards RM, Lee A: Patient reporting of potential adverse drug reactions: a methodological study. Br J Clin Pharmacol 2002, 53: 318–325. 10.1046/j.0306-5251.2001.01547.x
Foster JM, van der Molen T, Caeser M, Hannaford P: The use of questionnaires for measuring patient-reported side effects of drugs: its importance and methodological challenges. Pharmacoepidemiol Drug Saf 2008, 17: 278–296. 10.1002/pds.1533
Ten Berg MJ, Huisman A, van den Bemt PM, Schobben AF, Egberts AC, van Solinge WW: Linking laboratory and medication data: new opportunities for pharmacoepidemiological research. Clin Chem Lab Med 2007, 45: 13–19. 10.1515/CCLM.2007.009
Honigman B, Lee J, Rothschild J, Light P, Pulling RM, Yu T, Bates DW: Using computerized data to identify adverse drug events in outpatients. J Am Med Inform Assoc 2001, 8: 254–266. 10.1136/jamia.2001.0080254
Eichler HG, Pignatti F, Flamion B, Leufkens H, Breckenridge A: Balancing early market access to new drugs with the need for benefit/risk data: a mounting dilemma. Nat Rev Drug Discov 2008, 7: 818–826. 10.1038/nrd2664
Egberts TC, Smulders M, de Koning FH, Meyboom RH, Leufkens HG: Can adverse drug reactions be detected earlier? A comparison of reports by patients and professionals. BMJ 1996, 313: 530–531.
Golder S, McIntosh HM, Duffy S, Glanville J, Centre for Reviews and Dissemination and UK Cochrane Centre Search Filters Design Group: Developing efficient search strategies to identify reports of adverse effects in MEDLINE and EMBASE. Health Info Libr J 2006, 23: 3–12.
McIntosh HM, Woolacott NF, Bagnall AM: Assessing harmful effects in systematic reviews. BMC Med Res Methodol 2004, 4: 19. 10.1186/1471-2288-4-19
Donekal S, Shomali ME: Effectiveness of the novel anti-diabetes medication exenatide in everyday practice: Comparison with clinical trials. Diabetes Res Clin Pract 2008, 80: e4-e6.
Redondo-Capafons S, Garriga Biosca MR, Pla Poblador R: Follow-up of the use of metformin among the high risk population. Farm Hosp 2005, 29: 364–366. 10.1016/S1130-6343(05)73697-7
Burk M, Morreale AP, Cunningham F: Conversion from troglitazone to rosiglitazone or pioglitazone in the VA: A multicenter DUE. Formulary 2004, 39: 310–317.
Manley HJ, Allcock NM: Thiazolidinedione safety and efficacy in ambulatory patients receiving hemodialysis. Pharmacotherapy 2003, 23: 861–865. 10.1592/phco.23.7.861.32727
Swislocki AL, Khuu Q, Liao E, Wu E, Beza F, Lopez J, Kwan G, Noth RH: Safety and efficacy of metformin in a restricted formulary. Am J Manag Care 1999, 5: 62–68.
Tang WH, Francis GS, Hoogwerf BJ, Young JB: Fluid retention after initiation of thiazolidinedione therapy in diabetic patients with established chronic heart failure. Am Coll Cardiol 2003, 41: 1394–1398. 10.1016/S0735-1097(03)00159-1
Fehmann HC: The alpha-glucosidase inhibitor miglitol for the treatment of type 2 diabetes mellitus in the doctor's office. MMW Fortschr Med 2001,119(Suppl 2):55–61. [in German]
Kane MP, Busch RS, Bakst G, Hamilton RA: Substitution of pioglitazone for troglitazone in patients with type 2 diabetes. Endocr Pract 2004, 10: 18–23.
Kawamori R, Kadowaki T, Onji M, Seino Y, Akanuma Y, on behalf of the PRACTICAL Study Group: Hepatic safety profile and glycemic control of pioglitazone in more than 20,000 patients with type 2 diabetes mellitus: postmarketing surveillance study in Japan. Diabetes Res Clin Pract 2007, 76: 229–235. 10.1016/j.diabres.2006.08.017
Mertes G: Safety and efficacy of acarbose in the treatment of Type 2 diabetes: data from a 5-year surveillance study. Diabetes Res Clin Pract 2001, 52: 193–204. 10.1016/S0168-8227(01)00221-2
Pan CY, Landen H: Post-marketing surveillance of acarbose treatment in patients with type 2 diabetes mellitus and subjects with impaired glucose tolerance in China. Clin Drug Invest 2007, 27: 397–405. 10.2165/00044011-200727060-00003
Rosak C, Petzoldt R, Wolf R, Reblin T, Dehmel B, Seidel D: Rosiglitazone plus metformin is effective and well tolerated in clinical practice: results from large observational studies in people with type 2 diabetes. Int J Clin Pract 2005, 59: 1131–1136. 10.1111/j.1368-5031.2005.00652.x
Scholz GH, Schneider K, Knirsch W, Becker G: Efficacy and tolerability of glimepiride in daily practice: A non-interventional observational cohort study. Clin Drug Invest 2001, 21: 597–604. 10.2165/00044011-200121090-00001
Slama G, Eschwège E, Bernard MH, Grimaldi A, Oppert JM, Pouchain D, Bégaud B, pour le groupe des investigateurs de l'étude Avantage: One-year follow-up in real clinical practice conditions of type 2 diabetic patients treated with rosiglitazone: the Avantage study. Ann Endocrinol 2008, 69: 36–46. [in French] 10.1016/j.ando.2007.10.024
Spengler M, Schmitz H, Landen H: Evaluation of the efficacy and tolerability of acarbose in patients with diabetes mellitus a postmarketing surveillance study. Clin Drug Invest 2005, 25: 651–659. 10.2165/00044011-200525100-00004
Kasliwal R, Wilton LV, Shakir SAW: Monitoring the safety of pioglitazone: Results of a prescription-event monitoring study of 12 772 patients in England. Drug Saf 2008, 31: 839–850. 10.2165/00002018-200831100-00003
Kubota K, Kawabe E, Hinotsu S, Hamada C, Ohashi Y, Kurokawa K: Pilot study of prescription-event monitoring in Japan comparing troglitazone with alternative oral hypoglycemics. Eur J Clin Pharmacol 2000, 56: 831–838.
Marshall V, Wilton L, Shakir SAW: Safety profile of repaglinide as used in general practice in England: results of a prescription-event monitoring study. Acta Diabetol 2006, 43: 6–13. 10.1007/s00592-006-0203-9
Casscells SW, Granger E, Swedorske J, Goldhammer R, Shaheen M, Dorris J, Hong A, Wiktor M: A comparison of select cardiovascular outcomes by antidiabetic prescription drug classes used to treat type 2 diabetes among Military Health System beneficiaries, fiscal year 2003–2006. Am J Ther 2008, 15: 198–205. 10.1097/MJT.0b013e31817534d5
Rajagopalan R, Iyer S, Perez A: Comparison of pioglitazone with other antidiabetic drugs for associated incidence of liver failure: no evidence of increased risk of liver failure with pioglitazone. Diabetes Obes Metab 2005, 7: 161–169. 10.1111/j.1463-1326.2004.00382.x
Monster TB, de Jong PE, de Jong-van den Berg LT: Drug-induced renal function impairment: a population-based survey. Pharmacoepidemiol Drug Saf 2003, 12: 135–143. 10.1002/pds.811
Abbasi AA, Kasmikha R, Sotingeanu DG: Metformin-induced lacticacidemia in patients with type 2 diabetes mellitus. Endocr Pract 2000, 6: 442–446.
Gavin LA, Barth J, Arnold D, Shaw R: Troglitazone add-on therapy to a combination of sulfonylureas plus metformin achieved and sustained effective diabetes control. Endocr Pract 2000, 6: 305–310.
Taki H, Maki T, Iso T, Iwamoto K, Kajiura T: Study of nateglinide in Japan: long-term treatment of patients with type 2 diabetes. Adv Ther 2006, 23: 307–324. 10.1007/BF02850136
Acknowledgements
This study was performed as a part of PhD project, funded by Dutch Top Institute Pharma (TIPharma). TIPharma did not participate in the literature search, data analysis or interpretation of the results. There are no conflicts of interest. The authors thank Truus van Ittersum for her assistance with the literature search.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
LH conducted the literature search, participated in the data extraction and analysis, and drafted the manuscript. FHR conceived of the study, and participated in its design and in the data extraction and analysis. DdZ participated in the conception and design of the study. DD participated in the data extraction and analysis. PD participated in the conception and design of the study, in the data extraction and analysis, and edited the final manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12955_2011_858_MOESM1_ESM.PDF
Additional file 1: Search strategy used for eligible studies. Provides the domains, terms and boolean operators used in the systematic search of Medline and Embase for observational studies reporting on ADEs in patients with diabetes. (PDF 69 KB)
12955_2011_858_MOESM2_ESM.DOC
Additional file 2: Description of the studies included in the review. Provides the following data for each included study: data collection method employed for ADE assessment, publication year, country, study design, type of ADEs included, sample size, follow up period, patients selection. (DOC 206 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Hakobyan, L., Haaijer-Ruskamp, F.M., de Zeeuw, D. et al. A review of methods used in assessing non-serious adverse drug events in observational studies among type 2 diabetes mellitus patients. Health Qual Life Outcomes 9, 83 (2011). https://doi.org/10.1186/1477-7525-9-83
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1477-7525-9-83