
Abstract
Background
Actinobacillus pleuropneumoniae (APP) is one of the most important swine pathogens worldwide. Identification and characterization of novel antigenic APP vaccine candidates are underway. In the present study, we use an immunoproteomic approach to identify APP protein antigens that may elicit an immune response in serotype 1 naturally infected swine and serotype 1 virulent strain S259-immunized rabbits.
Results
Proteins from total cell lysates of serotype 1 APP were separated by two-dimensional electrophoresis (2DE). Western blot analysis revealed 21 immunoreactive protein spots separated in the pH 4-7 range and 4 spots in the pH 7-11 range with the convalescent sera from swine; we found 5 immunoreactive protein spots that separated in the pH 4-7 range and 2 in the pH 7-11 range with hyperimmune sera from S259-immunized rabbits. The proteins included the known antigens ApxIIA, protective surface antigen D15, outer membrane proteins P5, subunit NqrA. The remaining antigens are being reported as immunoreactive proteins in APP for the first time, to our knowledge.
Conclusions
We identified a total of 42 immunoreactive proteins of the APP serotype 1 virulent strain S259 which represented 32 different proteins, including some novel immunoreactive factors which could be researched as vaccine candidates.
Similar content being viewed by others


Background
Actinobacillus pleuropneumoniae (APP) is one of the most important swine pathogens worldwide. Of the 15 known APP serotypes, serotype 1 is frequently associated with a range of lung diseases, including fibrinous, hemorrhagic pneumonia, and necrotizing pneumonia. High mortality has been reported in acutely infected pigs and persistent lung lesions have been observed in chronically infected pigs [1].
To control APP pathogenesis, several types of vaccines have been developed that offer various degrees of protection. The traditional chemically-[2, 3], genetically-, and irradiation-inactivated [4] whole cell vaccines used thus far have been shown to provide protection against a homologous challenge from APP, but were ineffective in staving off infection by different serotypes.
Some cross-serovar protection has been achieved with live-attenuated vaccines of mutant field isolates, such as mutant strains of APP serovar 7 [5] and serovar 1 [6, 7]. Further development of cross-serovar vaccines would benefit from a molecular understanding of APP pathogenesis, which is a complex process involving a number of different potential virulence factors. The most commonly associated virulence factors [8] i.e., ApxI, ApxII, ApxIII, and ApxIV, have been tested as subunit vaccine candidates offering potential cross-serovar protection [9, 10]. DNA vaccines encoding multiple Apx toxins offer a novel strategy for protecting against APP infection [11]. In addition to Apx toxins, several other APP proteins have been researched as vaccine candidates, such as the 48 kDa outer membrane protein [12] and the 30 kDa membrane protein of the ABC transporter family [13]. Lipopolysaccharide (LPS)-based vaccines are alternative to protein antigens [14].
The identification of novel antigens as candidate vaccines should be accelerated by modern technologies. The complete genomes of APP L20 (Serotype 5b [15]) and JL03 (serotype 3 [16]) have been sequenced, the outer membrane proteome of serotype 5b has also been characterized [17]. In this paper, we use an immunoproteomic approach to identify APP protein antigens that elicit an immune response in serotype 1 naturally infected swine and serotype 1 virulent strain S259-immunized rabbits. This approach, which we have used to study immunogenicity of other bacterial pathogens [18], combines the specificity of antibody detection with the precision of mass spectral analysis [19] for identifying antigenic bacterial proteins.
Results
Two-dimensional electrophoresis (2DE) profiles of APP bacterial proteins and western blot analysis
Proteins from total cell lysates of APP serotype 1 were separated by 2DE. Two-dimensional separation profiles are shown for separation by isoelectric point (pI) in the first dimension over a pH ranges of pH 4-7 (Figures 1A, 2A) and pH 7-11 (Figures 3A, 4A). The separation profiles were highly reproducible in 2DE experiments conducted in triplicate followed by membrane transfer and developing, yielding similar patterns of total proteins and immunoreactive proteins. Figures 1B, 2B, 3B and 4B show the western blot analysis with the convalescent sera from naturally infected APP serotype 1 swine and from hyperimmune sera from S259-immunized rabbit. No specific immunoreactive protein spots were observed when negative control sera were used.

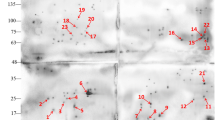
Comparison of western blot analysis with convalescent sera from swine and duplicated gels of S259 bacterial associated proteins at pH 4-7. A. Coomassie G-250-stained 2DE gel. All identified protein spots were analyzed by MALDI-TOF MS. B. Western blot analysis of proteins on 2DE gel as transferred to a PVDF membrane. The primary antibodies were convalescent sera from swine naturally infected with APP serotype 1.

Comparison of western blot analysis with rabbit hyperimmune sera and duplicated gels of S259 bacterial associated proteins at pH 4-7. B. A. Coomassie G-250-stained 2DE gel. All identified protein spots were analyzed by MALDI-TOF MS. B. Western blot analysis of proteins on 2DE gel as transferred to a PVDF membrane. The primary antibodies were hyperimmune sera from S259-immunized rabbits.

Comparison of western blot analysis with convalescent sera from swine and duplicated gel of S259 bacterial associated proteins at pH 7-11. C. A. Coomassie G-250-stained 2DE gel. All identified protein spots were analyzed by MALDI-TOF MS. B. Western blot analysis of proteins on 2DE gel as transferred to a PVDF membrane. The primary antibodies were convalescent sera from swine naturally infected with APP serotype 1.

Comparison of western blot analysis with rabbit hyperimmune sera and duplicated gels of S259 bacterial associated proteins at pH 7-11. D. A. Coomassie G-250-stained 2DE gel. All identified protein spots were analyzed by MALDI-TOF MS. B. Western blot analysis of proteins on 2DE gel as transferred to a PVDF membrane. The primary antibodies were hyperimmune sera from S259-immunized rabbits.
Alignment of total protein and immunoreactive protein images
Comparisons between 2DE gels and western blot membranes were very difficult to make because only a portion of the proteins on the transferred membrane were immunoreactive and the sensitivity of the western blot was higher than that of Coomassie brilliant blue. In this study, we overcame this challenge by using the layer function of PhotoshopCS to compare western blot and 2DE gels with the ponceaus S stain as an intermediate state. The same membranes used for western blots were also stained with Ponceau S to visualize all proteins. Digital images of the western blots and Ponceau S-stained membranes were compared with the 2DE gels by aligning the images with the layer function of Photoshop CS (Additional file 1, Figure S1, Additional file 2, Figure S2, Additional file 3, Figure S3, Additional file 4, Figure S4). Using this method to align developed membranes and 2DE gels in the pH 4-7 range (Figures 1, 2) and the pH 7-11 range (Figures 3, 4), we found 42 immunoreactive protein spots that represented 32 different proteins. The identified spots were labeled on each gel (Figures 1, 2, 3 and 4).
Identification of immunoreactive proteins
A total of 42 immunoreactive proteins were identified by immunoproteomics. Corresponding spots for immunoreactive proteins were excised from preparative 2DE gels, subjected to tryptic digestion, and analyzed by mass spectrometry and peptide mass fingerprinting (PMF). For conclusive identification of the protein corresponding with each spot, the probability score of the match (Protein scores > 83 were significant), the weight-average molecular weight (MW), pI, number of peptide matches, and percentage of the total translated open reading frame (ORF) sequence covered by the peptides were analyzed. Descriptive data for the identified spots are listed in Tables 1 and 2. Only the proteins ranking first in each matching search are shown. Western blot analysis with convalescent sera from swine naturally infected APP serotype 1, led us to identify 32 immunoreactive spots, representing 25 proteins, including 21 that separated in the pH 4-7 range (Figure 1) and 4 that separated in the pH 7-11 range (Figure 3). Western blot analysis with hyperimmune sera raised in an S259-immunized rabbit revealed 10 immunoreactive spots that represented 7 proteins, including 5 that separated in the pH 4-7 range (Figure 2) and 2 that separated in the pH 7-11 range (Figure 4).
Bioinformatics analysis
The BLASTX results are shown in Tables 1 and 2. The single top ranking proteins identified for each spot are listed in the tables. All identified proteins were predicted by PSORTb version 3.0 software http://www.psort.org/. Among the protein spots analyzed with convalescent sera from swine, 2 proteins were annotated as periplasmic proteins, 4 as outermembrane proteins, 1 as an extracellular protein, 11 as cytoplasmic proteins, 2 as cytoplasmic membrane proteins, and 5 were unknown. Meanwhile, in protein spots analyzed with hyperimmune sera from rabbit, 2 proteins were predicted as outermembrane proteins, 4 as cytoplasmic proteins, and 1 was unknown.
Discussion
In this study, a total of 42 immunoreactive spots, representing 32 different proteins from APP S259 were identified by western blot analysis with convalescent sera from swine and hyperimmune sera from rabbits. Four of those proteins, ApxIIA (spot APs25,26), D15 (spot APs21,22), subunit NqrA (spot APr12), outer membrane protein P5 (spot APBs1,2,3,4,5,6,8; APBr1,2 ), have been demonstrated recently as immunogenic proteins in APP JL03 [20].
ApxIIA, which belongs to the RTX family [21], has been reported to be moderately cytotoxic and weakly hemolytic [22]. Chiang et al. designed DNA vaccines that encode ApxIA or ApxIIA. The vaccines elicited humoral immune responses and protective efficacy in mice [11]. D15 (or its precursor) is an essential component of outer membrane biogenesis and outer membrane protein assembly [23, 24]. The immunogenicity of D15 has been demonstrated in both Haemophilus ducreyi [25] and Pasteurella multocida [26]. Thus, D15 is a highly conserved antigen and may be a useful component of a universal subunit vaccine against Haemophilus infection [27]. Subunit NqrA was identified as a 48-kDa outer membrane protein with immunogenic in serotype 1 or 5A and is common to 12 APP serotypes, but is not present in related Gram-negative swine pathogen species [12]. Outer membrane protein P5 (or its precursor), which is a homolog of OmpA in Pasteurella trehalosi and Escherichia coli, may be involved in the adherence of bacteria to nasopharyn-geal mucin [28].
The ribosomal subunit proteins L7/L12 (spot APr4) and pyruvate dehydrogenase subunit E1 (Spots APr10, 11) [29] were identified as immunogenic proteins in members of the Bacillus cereus group [30], but have not previously been identified as such in APP before.
The remaining 23 proteins identified in our study are being reported here as immunoreactive proteins for the first time, to our knowledge. The majority of these immunoreactive proteins were linked to housekeeping functions, such as energy production (e.g. spot APr8, APr9), carbohydrate transport and metabolism (e.g. spot APs2, APs13), amino acid transport and metabolism (e.g. APs19), and chaperones (e.g. spot APs3, APs5 ), reflecting their importance for survival.
We ultimately concluded that the spots labeled as APs6, APs9 and APr15, APr16 were likely mixtures of several proteins; there were no corresponding sequences for these spots available for Mascot searches. Our inability to identify these spots can be attributed to several reasons, including the genome sequence of APP serotype 1 being incomplete, the spots not being evident on duplicated gels, and/or a few adjacent spots being mixed together.
Highly conserved immunogenic proteins could potentially induce protection against a wide variety of bacterial strains and are thus attractive novel vaccine candidates. Goure et al. have identified APP genes that are conserved among all 15 serotypes by comparative genomic hybridization [31]. Of these conserved genes, the genes encoding protective surface antigen D15 and outer membrane protein P5 were observed in our results. Subunit NqrA was also demonstrated to be common to 12 serotypes by Cruz et al. [12]. It is notable that outer membrane protein APBr3 was also found in different APP serotypes and showed a high level of conservation across serotypes.
The reliability of our protein identification was further confirmed by comparison of the experimental MW and pI values of the protein spots on the 2DE gels with the theoretical ones. Overall, the majority of the theoretical and experimental values matched well, though some discrepancies remained. Similar migration for several proteins has previously been described in proteomic analysis of serotype 3 APP [20] and other pathogens [32, 33]. The presence of natural isoforms, post-translational proteolytic processing and/or modification, or artifacts related to sample preparation might explain the discrepancies. In addition, 7 proteins were identified from more than one position on the gels, including outer membrane protein P5, perhaps due to the presence of horizontal isoforms.
Different western blot results were obtained with analyses from swine versus rabbit sera. Only one common protein in the two species (outer membrane protein P5) was identified. Further research is needed to illuminate the factors that may explain this divergence.
Conclusions
We optimized 2DE sample preparation for APP, and obtained clearly visualized 2DE profiles with abundant spots. This method can applied in proteome studies of other Gram-negative bacteria. Using this approach, we identified 32 proteins by western blot analysis using convalescent sera from swine and hyperimmune sera from rabbits. The newly identified immunoreactive APP serotype 1 proteins are of great interest in terms of understanding pathogen-host interaction and can be considered novel vaccine candidates.
Methods
Strain and culture conditions
APP virulent strain S259 was purchased from the Control Institute of Veterinary Bioproducts and Pharmaceuticals of China. The cells were cultured in Trypticase Soy Broth (TSB, Merck, Germany); nicotinamide adenine dinucleotide (NAD, Sinopharm, Shanghai, China) was added to the medium at a final concentration of 0.01%. One-hundred-milliliter cultures were shaken overnight at 37°C on a rotary incubation shaker running at 180 rpm until they reached the late stage of exponential phase. Cells were harvested by centrifugation at 10,000 ×g for 10 min at 4°C and washed three times with phosphate buffered saline (PBS).
Sera
Convalescent serum from swine naturally infected with APP serotype 1 were screened by two steps. Step 1, seeking convalescent sera against APP. A total of 769 swine serum samples were collected from 29 herds in different provinces of China in 2009. Then ApxIV-ELISA was performed to screen convalescent serum from naturally infections as reported previously [34]. The ApxIV-ELISA has the advantage over other serological tests that it does not show cross-reactions with other bacterial species and that it is highly sensitive and allows also the serological detection of pigs infected by the different serotypes of APP or carrying the agent without apparent clinical signs or symptoms of infection. Furthermore the test allows clear differentiation between pigs infected with APP and healthy pigs that were vaccinated against APP. In addition, we have traced the background of corresponding swine. These swine were never immunized any vaccines against APP infection and were apparently healthy. Step 2, seeking convalescent sera against APP serotype 1 with S259 whole cell ELISA as described elsewhere [18]. As negative controls, we tested sera from newly born piglets using the same methods.
To prepare hyperimmune serum, rabbits were immunized with formaldehyde-inactivated APP S259 vaccine, with Montanide ISA 206 VG (SEPPIC, France) used as an adjuvant. Two doses of 1.0 × 109 cells/rabbit were administered by intramuscular injections with a 3-week interval. Sera from negative control and immunized rabbits were collected before the first and after the second immunization. Serum titers were evaluated with S259 whole cell ELISA.
Our experimental research has been performed with the approval of Institute of Veterinary Medicine, Jiangsu Academy of Agricultural Sciences(SYXK 2010-0005)
Protein sample extraction
Washed APP S259 cell pellets were re-suspended in 5-mL sample preparation solutions (7 M urea, 2 M thiourea, 4% w/v CHAPS, and 40 mM DTT) and sonicated in an ice bath for 50 cycles (5 s on, 10 s off) at a power setting of 200 W. The cell lysate was incubated for 30 min at 25°C to solubilize proteins (vortexing every 10 min) and centrifuged at 10,000 ×g for 20 min at 25°C to pellet the insoluble components. To precipitate proteins, the cleared supernatants were treated with pre-chilled 100% trichloroacetic acid (TCA) to a final concentration of 10% and incubated in ice water for 30 min. Precipitated protein was collected by centrifugation at 10,000 ×g for 10 min at 25°C and washed twice with pre-chilled acetone. The final pellet was air-dried.
Isoelectric focusing (IEF)
The dried pellet was dissolved in sample preparation solution, then incubated for 30 min at 25°C (vortexing every 10 min) and centrifuged at 10,000 ×g for 20 min at 25°C. Before rehydration, the supernatant was treated with a 2-D Clean-up Kit (GE Healthcare, Shanghai, China) to remove contaminants that interfere with IEF. Then IEF was performed using the Ettan IPGphor 3 IEF system (GE Healthcare, Shanghai, China) with 13-cm (Immobiline DryStripk, pH 4-7; GE Healthcare, Shanghai, China) as well as 7-cm (Immobiline DryStrip, pH 7-11; GE Healthcare, Shanghai, China) immobilized pH gradient (IPG) gel strips. IPG strips were rehydrated overnight at room temperature with rehydration solution [7 M urea, 2 M thiourea, 2% w/vCHAPS, 0.2% w/vDTT, 0.5% v/vIPG buffer (same range as the IPG strip), and 0.002% w/v bromophenol blue]. Each of the 13-cm and 7-cm strips was loaded with 200 μg and 100 μg of protein, respectively. IEF was carried out at 20°C for 11.5 h (maximum voltage of 8,000 V, maximum current of 50 μA/IPG strip, total 28,000 Vh) with 13-cm IPG strips or for 5.5 h (maximum voltage of 5,000 V, maximum current of 50 μA/IPG strip, total 22,000 Vh) with 7-cm IPG strips, after 12 h of active rehydration.
Sodium dodecyl sulfate polyacrylamide gel electrophoresis(SDS-PAGE)
Before running SDS-PAGE, each IPG strip was equilibrated for 15 min with 10 mg/ml DTT and 40 mg/ml iodoacetamide in equilibration buffer (6 M urea, 75 mM Tris-HCl pH 8.8, 29.3% v/v glycerol, 2% w/v SDS, 0.002% w/v bromophenol blue). Each IPG strip plus an SDS-PAGE molecular weight standard (Invitrogen, Shanghai, China) was loaded on a 12% polyacrylamide gel and sealed with 1% agarose. Electrophoresis was performed at 15°C at an initial voltage of 110 V for 30 min, followed by 220 V until the tracking dye reached the gel bottom. Pairs of gels were run simultaneously, one for Coomassie G-250 stain and the other for western blot analysis. Each IEF/SDS-PAGE experiment was repeated three times.
Western blot analysis
Protein samples from SDS-PAGE gels were transferred onto PVDF membranes (GE Healthcare, Shanghai, China) using a semi-dry blotting apparatus (TE77, GE Healthcare, Shanghai, China) for 2 h at 0.65 mA/cm2. Membrane-bound proteins were detected by staining with Ponceau S. Briefly, the PVDF membranes were submerged in Ponceau S stain solution (0.1% w/v Ponceau S, 5% v/v acetic acid) with gentle agitation for 5 min. The membranes were washed several times with distilled water (dH2O) until the protein spots were visible; the Ponceau S-stained membranes were digitally scanned with a Umax scanner (TE77, GE Healthcare, Shanghai, China). Ponceau S stain was removed by rinsing the membranes in dH2O with gentle agitation. After removing Ponceau S, the membranes were blocked with 5% w/v skim milk in 50 mM Tris-HCl buffer (pH 7.4) containing 0.05% Tween 20 (TBST) for 2 h at room temperature. The blocked-membrane was then incubated with convalescent sera from swine or hyperimmune sera from rabbit at room temperature for 2 h (1:1000 dilution with blocking buffer) and then washed three times with TBST for 15 min per wash. The membranes were incubated with Staphylococcal protein A labeled with horseradish peroxidase (Boster, Wuhan, China) which has been widespread applied as one kind of broad-spectrum secondary antibodies of most mammalian species [35–38] at room temperature for 1 h (1:10000 dilution with blocking buffer), washed three times with TBST, and developed by adding 3,3'-Diaminobenzidine (Tiangen, Beijing, China) until the optimum color was obtained. For each sample, the western blot was repeated three times.
Alignment of total and immunoreactive protein images
Digital images of PVDF membranes stained with Ponceau S and immunoblotting analysis with sera from swine and rabbit were aligned using the layer function of Photoshop CS (Adobe). The corresponding protein spots and blots on each image were aligned precisely. The Ponceau S-stained image was also compared with the duplicated gel.
Matrix Assisted Laser Desorption Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF MS) and database searches
Immunoreactive proteins in the western blots were identified and excised from a duplicate SDS-PAGE gels and sent to Nanjing Ji'ao BioTechnologies Co., Ltd for in-gel trypsin digestion and MALDI-TOF MS. PMF data were analyzed using the MASCOT server http://www.matrixscience.com. In MASCOT searches, protein scores > 83 were significant (p < 0.05). The protein scores as well as the original PMF data such as extent of sequence coverage, number of peptides matched were used to accept protein identifications. The remaining proteins with scores < 83 were either verified manually or rejected.
Bioinformatics analysis
Sequences of the identified proteins were searched in the BLASTX server http://blast.ncbi.nlm.nih.gov/ to find homologous sequences and searched in the PSORT server http://www.psort.org/ to predict protein subcellular localization.
References
Sebunya TN, Saunders JR: Haemophilus pleuropneumoniae infection in swine: a review. J Am Vet Med Assoc 1983, 182: 1331–1337.
Haesebrouck F, Pasmans F, Chiers K, Maes D, Ducatelle R, Decostere A: Efficacy of vaccines against bacterial diseases in swine: what can we expect? Vet Microbiol 2004, 100: 255–268. 10.1016/j.vetmic.2004.03.002
Beskow P, Robertsson JA, Soderlind O: Testing of remedial measures in fattening pig herds affected with subclinical infections of Actinobacillus pleuropneumoniae serotype 2. Zentralbl Veterinarmed B 1993, 40: 549–558.
Katinger A, Lubitz W, Szostak MP, Stadler M, Klein R, Indra A, Huter V, Hensel A: Pigs aerogenously immunized with genetically inactivated (ghosts) or irradiated Actinobacillus pleuropneumoniae are protected against a homologous aerosol challenge despite differing in pulmonary cellular and antibody responses. J Biotechnol 1999, 73: 251–260. 10.1016/S0168-1656(99)00143-1
Prideaux CT, Pierce L, Krywult J, Hodgson AL: Protection of mice against challenge with homologous and heterologous serovars of Actinobacillus pleuropneumoniae after live vaccination. Curr Microbiol 1998, 37: 324–332. 10.1007/s002849900386
Liu J, Chen X, Lin L, Tan C, Chen Y, Guo Y, Jin M, Guo A, Bei W, Chen H: Potential use an Actinobacillus pleuropneumoniae double mutant strain DeltaapxIICDeltaapxIVA as live vaccine that allows serological differentiation between vaccinated and infected animals. Vaccine 2007, 25: 7696–7705. 10.1016/j.vaccine.2007.07.053
Lin L, Bei W, Sha Y, Liu J, Guo Y, Liu W, Tu S, He Q, Chen H: Construction and immunogencity of a DeltaapxIC/DeltaapxIIC double mutant of Actinobacillus pleuropneumoniae serovar 1. FEMS Microbiol Lett 2007, 274: 55–62. 10.1111/j.1574-6968.2007.00813.x
Xu F, Chen X, Shi A, Yang B, Wang J, Li Y, Guo X, Blackall PJ, Yang H: Characterization and immunogenicity of an apxIA mutant of Actinobacillus pleuropneumoniae. Vet Microbiol 2006, 118: 230–239. 10.1016/j.vetmic.2006.07.013
Bosse JT, Janson H, Sheehan BJ, Beddek AJ, Rycroft AN, Kroll JS, Langford PR: Actinobacillus pleuropneumoniae: pathobiology and pathogenesis of infection. Microbes Infect 2002, 4: 225–235. 10.1016/S1286-4579(01)01534-9
Seah JN, Frey J, Kwang J: The N-terminal domain of RTX toxin ApxI of Actinobacillus pleuropneumoniae elicits protective immunity in mice. Infect Immun 2002, 70: 6464–6467. 10.1128/IAI.70.11.6464-6467.2002
Chiang CH, Huang WF, Huang LP, Lin SF, Yang WJ: Immunogenicity and protective efficacy of ApxIA and ApxIIA DNA vaccine against Actinobacillus pleuropneumoniae lethal challenge in murine model. Vaccine 2009, 27: 4565–4570. 10.1016/j.vaccine.2009.05.058
Cruz WT, Nedialkov YA, Thacker BJ, Mulks MH: Molecular characterization of a common 48-kilodalton outer membrane protein of Actinobacillus pleuropneumoniae. Infect Immun 1996, 64: 83–90.
Martin PR, Mulks MH: Cloning and characterization of a gene encoding an antigenic membrane protein from Actinobacillus pleuropneumoniae with homology to ABC transporters. FEMS Immunol Med Microbiol 1999, 25: 245–254. 10.1111/j.1574-695X.1999.tb01349.x
Rioux S, Dubreuil D, Begin C, Laferriere C, Martin D, Jacques M: Evaluation of protective efficacy of an Actinobacillus pleuropneumoniae serotype 1 lipopolysaccharide-protein conjugate in mice. Comp Immunol Microbiol Infect Dis 1997, 20: 63–74. 10.1016/S0147-9571(96)00022-7
Foote SJ, Bosse JT, Bouevitch AB, Langford PR, Young NM, Nash JH: The complete genome sequence of Actinobacillus pleuropneumoniae L20 (serotype 5b). J Bacteriol 2008, 190: 1495–1496. 10.1128/JB.01845-07
Xu Z, Zhou Y, Li L, Zhou R, Xiao S, Wan Y, Zhang S, Wang K, Li W, Jin H, Kang M, Dalai B, Li T, Liu L, Cheng Y, Zhang L, Xu T, Zheng H, Pu S, Wang B, Gu W, Zhang XL, Zhu GF, Wang S, Zhao GP, Chen H: Genome biology of Actinobacillus pleuropneumoniae JL03, an isolate of serotype 3 prevalent in China. PLoS One 2008, 3: e1450. 10.1371/journal.pone.0001450
Chung JW, Ng-Thow-Hing C, Budman LI, Gibbs BF, Nash JH, Jacques M, Coulton JW: Outer membrane proteome of Actinobacillus pleuropneumoniae: LC-MS/MS analyses validate in silico predictions. Proteomics 2007, 7: 1854–1865. 10.1002/pmic.200600979
Zhang W, Lu CP: Immunoproteomic assay of membrane-associated proteins of Streptococcus suis type 2 China vaccine strain HA9801. Zoonoses Public Health 2007, 54: 253–259. 10.1111/j.1863-2378.2007.01056.x
Hess JL, Blazer L, Romer T, Faber L, Buller RM, Boyle MD: Immunoproteomics. J Chromatogr B Analyt Technol Biomed Life Sci 2005, 815: 65–75. 10.1016/j.jchromb.2004.07.047
Liao Y, Deng J, Zhang A, Zhou M, Hu Y, Chen H, Jin M: Immunoproteomic analysis of outer membrane proteins and extracellular proteins of Actinobacillus pleuropneumoniae JL03 serotype 3. BMC Microbiol 2009, 9: 172. 10.1186/1471-2180-9-172
Welch RA: Pore-forming cytolysins of gram-negative bacteria. Mol Microbiol 1991, 5: 521–528. 10.1111/j.1365-2958.1991.tb00723.x
Kamp EM, Popma JK, Anakotta J, Smits MA: Identification of hemolytic and cytotoxic proteins of Actinobacillus pleuropneumoniae by use of monoclonal antibodies. Infect Immun 1991, 59: 3079–3085.
Voulhoux R, Bos MP, Geurtsen J, Mols M, Tommassen J: Role of a highly conserved bacterial protein in outer membrane protein assembly. Science 2003, 299: 262–265. 10.1126/science.1078973
Gentle I, Gabriel K, Beech P, Waller R, Lithgow T: The Omp85 family of proteins is essential for outer membrane biogenesis in mitochondria and bacteria. J Cell Biol 2004, 164: 19–24. 10.1083/jcb.200310092
Thomas KL, Leduc I, Olsen B, Thomas CE, Cameron DW, Elkins C: Cloning, overexpression, purification, and immunobiology of an 85-kilodalton outer membrane protein from Haemophilus ducreyi. Infect Immun 2001, 69: 4438–4446. 10.1128/IAI.69.7.4438-4446.2001
Chaudhuri P, Goswami PP: Cloning of 87 kDa outer membrane protein gene of Pasteurella multocida P52. Res Vet Sci 2001, 70: 255–256. 10.1053/rvsc.2001.0470
Loosmore SM, Yang YP, Coleman DC, Shortreed JM, England DM, Klein MH: Outer membrane protein D15 is conserved among Haemophilus influenzae species and may represent a universal protective antigen against invasive disease. Infect Immun 1997, 65: 3701–3707.
Zhou M, Zhang A, Guo Y, Liao Y, Chen H, Jin M: A comprehensive proteome map of the Haemophilus parasuis serovar 5. Proteomics 2009, 9: 2722–2739. 10.1002/pmic.200800717
Dallo SF, Kannan TR, Blaylock MW, Baseman JB: Elongation factor Tu and E1 beta subunit of pyruvate dehydrogenase complex act as fibronectin binding proteins in Mycoplasma pneumoniae. Mol Microbiol 2002, 46: 1041–1051. 10.1046/j.1365-2958.2002.03207.x
Delvecchio VG, Connolly JP, Alefantis TG, Walz A, Quan MA, Patra G, Ashton JM, Whittington JT, Chafin RD, Liang X, et al.: Proteomic profiling and identification of immunodominant spore antigens of Bacillus anthracis, Bacillus cereus, and Bacillus thuringiensis. Appl Environ Microbiol 2006, 72: 6355–6363. 10.1128/AEM.00455-06
Goure J, Findlay WA, Deslandes V, Bouevitch A, Foote SJ, MacInnes JI, Coulton JW, Nash JH, Jacques M: Microarray-based comparative genomic profiling of reference strains and selected Canadian field isolates of Actinobacillus pleuropneumoniae. BMC Genomics 2009, 10: 88. 10.1186/1471-2164-10-88
Molloy MP, Herbert BR, Slade MB, Rabilloud T, Nouwens AS, Williams KL, Gooley AA: Proteomic analysis of the Escherichia coli outer membrane. Eur J Biochem 2000, 267: 2871–2881. 10.1046/j.1432-1327.2000.01296.x
Walz A, Mujer CV, Connolly JP, Alefantis T, Chafin R, Dake C, Whittington J, Kumar SP, Khan AS, DelVecchio VG: Bacillus anthracis secretome time course under host-simulated conditions and identification of immunogenic proteins. Proteome Sci 2007, 5: 11. 10.1186/1477-5956-5-11
Dreyfus A, Schaller A, Nivollet S, Segers RP, Kobisch M, Mieli L, Soerensen V, Hussy D, Miserez R, Zimmermann W, et al.: Use of recombinant ApxIV in serodiagnosis of Actinobacillus pleuropneumoniae infections, development and prevalidation of the ApxIV ELISA. Vet Microbiol 2004, 99: 227–238. 10.1016/j.vetmic.2004.01.004
Biberfield P, Ghetie V, Sjoquist J: Demonstration and assaying of IgG antibodies in tissues and on cells by labeled staphylococcal protein A. J Immunol Methods 1975, 6: 249–259. 10.1016/0022-1759(75)90068-X
Richman DD, Cleveland PH, Oxman MN, Johnson KM: The binding of staphylococcal protein A by the sera of different animal species. J Immunol 1982, 128: 2300–2305.
Lindmark R, Thoren-Tolling K, Sjoquist J: Binding of immunoglobulins to protein A and immunoglobulin levels in mammalian sera. J Immunol Methods 1983, 62: 1–13. 10.1016/0022-1759(83)90104-7
Zhou J, Liao YX, Chen Z, Li YC, Gao LL, Chen YX, Cai LG, Chen Q, Yu SY: [Development of a SPA-ELISA method for detecting anti-coronavirus IgG antibodies in serum samples from fulvous fruit bats]. Nan Fang Yi Ke Da Xue Xue Bao 2008, 28: 736–738.
Acknowledgements
This work was supported by a grant from the Special Fund for Public Welfare Industry of Chinese Ministry of Agriculture (No. 200803020) and the Priority Academic Program Development of Jiangsu Higher Education Institutions
Author information
Authors and Affiliations
Corresponding authors
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
WZ conceived the idea of immunoproteomic study, participated in its design, performed major portion of the data analysis, drafted manuscript; JS carried out protein preparation, 2DE, western blot analysis and contributed in the preparation of the manuscript. GL assisted in protein sample extraction and western blot analysis; FT prepared of hyperimmune serum; YL contributed to the bioinformatics analysis; ZZ carried out ELISA; YW and ZW participated in the design, implementation, and coordination of the study; CL and HY reviewed the design plan, participated in revision of the final version of the manuscript. All authors read and approved the final manuscript.
Wei Zhang, Jing Shao contributed equally to this work.
Electronic supplementary material
12953_2010_256_MOESM1_ESM.PPT
Additional file 1: Figure S1. Comparison of western blot analysis with swine convalescent sera and ponceaus S stain, as an intermediate state, at pH 4-7. In Photoshop, the immunoblot was used as the background layer and ponceaus S stain as the surface layer. The 0%, 20%, 40%, 60%, 80% and 100% transparency data are shown in A, B, C, D, E and F, respectively. (PPT 9 MB)
12953_2010_256_MOESM2_ESM.PPT
Additional file 2: Figure S2, Comparison of western blot analysis with rabbit hyperimmune sera and ponceaus S stain, as an intermediate state, at pH 4-7. In Photoshop, the immunoblot was used as the background layer and ponceaus S stain as the surface layer. The 0%, 20%, 40%, 60%, 80% and 100% transparency data are shown in A, B, C, D, E and F, respectively. (PPT 595 KB)
12953_2010_256_MOESM3_ESM.PPT
Additional file 3: Figure S3. Comparison of western blot analysis with swine convalescent sera and ponceaus S stain, as an intermediate state, at pH 7-11. In Photoshop, the immunoblot was used as the background layer and ponceaus S stain as the surface layer. The 0%, 20%, 40%, 60%, 80% and 100% transparency data are shown in A, B, C, D, E and F, respectively. (PPT 3 MB)
12953_2010_256_MOESM4_ESM.PPT
Additional file 4: Figure S4. Comparison of western blot analysis with rabbit hyperimmune sera and ponceaus S stain, as an intermediate state, at pH 7-11. In Photoshop, the immunoblot was used as the background layer and ponceaus S stain as the surface layer. The 0%, 20%, 40%, 60%, 80% and 100% transparency data are shown in A, B, C, D, E and F, respectively. (PPT 84 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Zhang, W., Shao, J., Liu, G. et al. Immunoproteomic analysis of bacterial proteins of Actinobacillus pleuropneumoniae serotype 1. Proteome Sci 9, 32 (2011). https://doi.org/10.1186/1477-5956-9-32
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1477-5956-9-32