
Abstract
Background
The gram-positive bacterium Streptococcus pyogenes is a common pathogen of humans that causes invasive infections, toxic-shock syndrome, rheumatic fever, necrotizing fasciitis and other diseases. Detection of antibiotic resistance in clinical isolates has renewed interest in development of new vaccine approaches for control S. pyogenes sepsis. In the study presented, a novel protein vaccine was examined. The vaccine was based on a recombinant protein fusion between streptococcal pyrogenic exotoxin B (SpeB), a cysteinyl protease expressed by all clinical isolates, and streptococcal pyrogenic exotoxin A (SpeA), a superantigen produced by a large subset of isolates.
Results
A novel protein was produced by mutating the catalytic site of SpeB and the receptor binding surface of SpeA in a fusion of the two polypeptides. Vaccination of HLA-DQ8 transgenic mice with the SpeA-SpeB fusion protein protected against a challenge with the wild-type SpeA that was lethal to naïve controls, and vaccinated mice were protected from an otherwise lethal S. pyogenes infection.
Conclusion
These results suggest that the genetically attenuated SpeA-SpeB fusion protein may be useful for controlling S. pyogenes infections. Vaccination with the SpeA-SpeB fusion protein described in this study may potentially result in protective immunity against multiple isolates of S. pyogenes due to the extensive antibody cross-reactivity previously observed among all sequence variants of SpeB and the high frequency of SpeA-producing strains.
Similar content being viewed by others


Background
Streptococcus pyogenes is a perennial human pathogen, causing mild infections and life-threatening diseases including pharyngitis, impetigo, necrotizing fasciitis, streptococcal toxic shock syndrome and rheumatic heart disease. Antibiotic-resistant strains are increasing in global distribution [1, 2], and a marked worldwide increase in the prevalence of serious invasive disease caused by S. pyogenes has occurred in the last two decades [3, 4], perhaps due to the emergence and distribution of more virulent strains. Although the incident is low, the recorded overall mortality rate is 45% among streptococcal toxic shock-like syndrome cases [5].
There are currently no licensed vaccines available for protection against diseases caused by S. pyogenes. Ideally, a vaccine should incorporate antigens from a major virulence determinant or antigens that are ubiquitously expressed by disparate bacterial strains. Streptococcal pyrogenic exotoxin A (SpeA) and other secreted superantigen toxins are potential candidates for vaccines because these proteins are associated with many outbreaks of streptococcal toxic shock syndrome and are virulence factors for invasive infections. In addition, bacteremia is commonly associated with cases of streptococcal toxic shock [6]. The secreted polypeptide of SpeA (25,700 Mr) is classified as a superantigen [7] that facilitates bacterial immune escape by targeting the primary recognition step in adaptive immunity. The cellular receptors for SpeA are human major histocompatibility complex (MHC) class II molecules, primarily HLA-DQ and HLA-DR proteins expressed on select cell lineages, and the antigen receptors of T cells (TCRs). The normal antigen-specific signal transduction of T cells is disengaged by SpeA, displacing contacts of MHC-bound antigenic peptides with antigen combining site elements of the TCR, and results in an elevated polyclonal activation of T cells. Toxic shock may ensue from pathological levels of tumor necrosis factor alpha (TNF-α) and other pro-inflammatory cytokines released in response to secreted superantigens [8, 9].
Most, if not all, S. pyogenes M protein serotypes express an extracellular cysteine protease (streptopain) historically termed streptococcal pyrogenic exotoxin B (SpeB), though not homologous in structure or function to SpeA or any other superantigen. The secreted protease SpeB is also a bacterial surface molecule with binding activity to laminin and other glycoproteins [10], making it a potential target of neutralizing antibodies. Further, SpeB is an important colonization and pathogenicity factor [11], reported to modify several host substrates. For example, the interleukin 1β precursor is cleaved by SpeB to produce active interleukin 1β [12], and the extracellular matrix proteins fibronectin and vitronectin are also cleaved [13], thus modulating entry of S. pyogenes into host cells [14]. Although multiple alleles exist, polyclonal antisera generated against SpeB from any strain react with SpeB from all S. pyogenes M1 serotypes examined [15]. Further, antibodies against SpeB are detected in patients with invasive S. pyogenes infections of either streptococcal toxic shock syndrome and/or necrotizing fasciitis [16]. The ubiquitous expression of SpeB by S. pyogenes strains and the conserved nature of the antigenic determinants recognized by antibodies are noteworthy features, thus fulfilling major criteria for a potential vaccine. Collectively, these observations prompted the presently described development of a fusion protein comprised of SpeA and SpeB that was used as a vaccine in experimental models of streptococcal toxic shock and sepsis.
Methods
Recombinant streptococcal proteins
Genes encoding SpeA (M19350) and SpeB (M86905) were cloned from a clinical laryngitis isolate of Streptococcus pyogenes by polymerase-chain reaction (pcr) amplification. Specific restriction enzyme motifs for cloning were introduced into the amplified DNA fragment by using the oligonucleotide primer 5' CTCG CAA GAG GTA CAT ATG CAA CAA GAC 3' to produce a unique NdeI site, and 5' GCA GTA GGT AAG CTT GCC AAA AGC 3' to produce a unique HindIII site. The amplified DNA fragment was ligated into the EcoRI site of a pcr-cloning vector (Invitrogen) and the resulting plasmid was used to transform E. coli DH5α. The HindIII/EcoRI DNA fragment containing the full-length SpeA gene minus the signal peptide was cloned into pET24 vector for expression in E. coli BL21. Although proteins were also produced with the leader peptide sequence present, deletion of the leader peptide appeared to result in a higher yield of protein.
Two different mutants of SpeA were produced by changing amino acid residue leucine 42 to either arginine or alanine by using previously described methods [17]. The first SpeA construct consists of a single mutation at residue leucine 42 [SpeA (L42R) or SpeA (L42A)], while the second construct consists of a fusion between the SpeA (L42R) and a mutant SpeB protein. The wild-type SpeB zymogen, isolated from the same strain of S. pyogenes used to clone SpeA, was truncated by PCR cloning to produce the mature protein without the prosegment domain (noncatalytic). A mutant, catalytically-inactive SpeB [SpeB (C47S)] was constructed by site-specific mutagenesis of the DNA coding sequence, altering cysteine 47 to serine. This conservative change maintains the approximate dimensions of the active-site side chain but prevents proteolytic activity. The SpeB (C47S) DNA was used as a fusion partner with SpeA (L42R) that was constructed with the following oligonucleotide primers:
1. SpeA forward primer, including NdeI site:
5' GATATACATATGCAACAAGACCCCGATCCAAGCC 3'
2. SpeA reverse primer, with SpeB overlap:
5' GAGATTTAACAACTGGTTGCTTGGTTGTTAGGTAGAC 3'
3. SpeB forward primer, with SpeA overlap:
5' GTCTACCTAACAACCAAGCAACCAGTTGTTAAATCTC 3'
4. SpeB reverse primer; adding an Amber codon:
5' GAATTCGGATCCGCTAGCCTACAACAG 3'
For cloning, the SpeA (L42R) gene was used as a PCR template and primers 1 and 2 were used to prepare a double-stranded sequence overlapping with SpeB (C47S). A separate PCR reaction with the SpeB (C47S) gene insert using primers 3 and 4 was performed to generate a double-stranded DNA fragment overlapping with SpeA (L42R). The PCR fragments were purified by agarose gel electrophoresis and mixed together for a final PCR reaction using primers 1 and 4, to create the full-length gene fusion of SpeA (L42R)-SpeB (C47S). This full-length fragment was cloned into the vector pT7Blue (Novagen) and the sequence was confirmed.
Protein production
The SpeA (L42R, L42A) and SpeA (L42R)-SpeB (C47S) fusion genes were subcloned into pET24b (+) for expression in E. coli BL21 host strains. Production of the recombinant proteins and purification methods were as previously described [17, 18]. The endotoxin levels of protein preparations were less than detection limits, as determined by a limulus amebocyte lysate assay (Cambrex, Walkersville, MD). Purified wild-type SpeA and affinity-purified rabbit antibodies specific for either SpeA or SpeB were obtained from Toxin Technology (Sarasota, FL) and used to confirm identity of the recombinant proteins by Western blots. Proteins (2 μg/lane) were electrophoresed through 12% polyacrylamide gels in the presence of SDS (1%), with dithiothreitol (2 mM). Gels were then electroblotted onto a protein-binding membrane (Amersham), and blocked (2 h, 37°C) with 0.2% casein in PBS. The membrane was then incubated (1 h, 37°C) with a 1/200 dilution of affinity-purified, rabbit anti-SpeA or SpeB (Toxin Technologies, Sarasota, FL). Unbound antibody was washed from the membrane using PBS, and bound antibody was detected with peroxidase conjugated, goat anti-rabbit antisera, using a commercial color development kit (BioRad, Richmond, CA).
HLA-DR/DQ binding assay
The DR1 homozygous, human B-lymphoblastoid cell line LG2 was used to detect binding of the SpeA proteins to MHC class II molecules, as previously described [17]. In brief, LG2 cells (4 × 105/50 μl) were incubated 40 min (37°C) with wild-type or mutant SpeA in Hanks balanced salt solution (HBSS) containing 0.5% bovine serum albumin. The cells were washed with HBSS and then incubated with 5 μg of affinity-purified rabbit anti-SpeA antibody (Toxin Technology) for 1 h on ice. Unbound antibody was removed, and the cells were incubated with FITC-labeled goat anti-rabbit IgG (Organon Teknika Corp., Durham, NC) on ice for 30 min. The cells were washed and analyzed by flow cytometry (FACScan; Becton Dickinson & Co., Mountain View, CA). Controls consisted of cells incubated with affinity purified anti-TSST-1 and the FITC labeled antibody without prior addition of SpeA.
T-lymphocyte responses
Lymphocyte proliferation was used to measure biological responses to the streptococcal proteins, as previously described [17]. Human peripheral blood mononuclear cells, obtained from consenting volunteers, were purified by Ficoll-hypaque (Sigma, St. Louis, MO) buoyant density gradient centrifugation. The cells were cultured in RPMI-1640 with 5% FBS for 72 h, and pulsed-labeled for 12 h with 1 μCi [3H]-thymidine (Amersham, Arlington Heights, IL). Cells were harvested onto glass fiber filters, and [3H]-thymidine incorporation into the cellular DNA was measured by a liquid scintillation counter (BetaPlate, Wallac Inc., Gaithersburg, MD).
Serum antibody
Serum levels of total IgG were determined by enzyme-linked immunosorbent assays (ELISA). Polystyrene 96-well plates (Nunc) were coated with a 1 μg/ml solution of antigen in 0.05 M sodium carbonate buffer (pH 9.6) overnight (4°C). The plates were blocked for 2 h (37°C) with 0.2% casein in PBS (138 mM NaCl, 2.7 mM KCl) and then washed three times with PBS. Serum samples were serially diluted in 0.02% casein in PBS and incubated in the antigen-coated plates for 1 h (37°C). The plates were washed three times with PBS and a 1:2000 dilution of goat anti-mouse IgG, HRP conjugated (Southern Biotechnology), was added in 0.02% casein PBS. The plates were incubated for 60 min (37°C), washed three times with PBS and then developed (30 min, 22°C) with TMB substrate (3,3',5,5'-Tetramethylbenzidine, Pierce). The reactions were stopped with the addition of 0.5 M H2SO4 and the absorbance determined at 450 nm wavelength.
Vaccinations
HLA-DQ8/human CD4+ transgenic mice were described previously [19]. Pathogen-free, 10–12-week-old BALB/c mice were obtained from Charles River (National Cancer Institute, Frederick, MD), maintained under pathogen-free conditions, and fed laboratory chow and water ad libitum. For vaccinations, mice were each injected 3 times (2 weeks between injections) intramuscularly (i.m.) with 10 μg of proteins (100 μl) combined with 100 μl MPL adjuvant (MPL™ + TDM+ CWS Emulsion, RIBI ImmunoChem Research, Inc., Hamilton, MT). This research was conducted in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adhered to the principles stated in the Guide for the Care and Use of Laboratory Animals, National Research Council, 1996.
Bacterial sepsis and toxic shock
The β-hemolytic Streptococcus pyogenes strain RIID231 (spea+, speb+), a human laryngitis isolate, was used as a source of streptococcal genes and bacteria for mouse challenges. Bacteria were propagated in Todd-Hewitt broth cultures (0.2% yeast extract) and single colonies were isolated after growth on sheep blood agar plates containing the same media to prepare bacteria for challenge studies. Streptococci were collected from broth cultures in mid-log growth phase, washed three times by gentle centrifugation in PBS and the density (A670) was adjusted by using PBS (22°C). Colony-forming units were confirmed by growth of diluted bacteria on sheep blood agar plates. For mouse challenges, bacteria diluted in PBS were injected (105 CFU in 100 μL) into tail veins using a tuberculin needle and syringe (27 g), followed 4 h later by i.p. administration of 75 μg (50 μL) of E. coli lipopolysaccharide (LPS; Difco, Detroit, MI). For challenge with SpeA, mice were injected i.p. (50 μL) with toxin diluted in PBS.
Results
Vaccine design
The genes encoding SpeA and SpeB were cloned from a strain of S. pyogenes originating from a patient with laryngitis. The binding interface between SpeA and human MHC class II molecules consists of contacts located in the N-terminal domain that are in common with other bacterial superantigens [17]. Leucine 42 of SpeA protrudes from a reverse turn on the surface of SpeA to potentially form a major hydrophobic contact with HLA-DQ or HLA-DR receptor molecules. Mutants of SpeA were constructed to alter leucine 42 (L42) and reduce HLA-DR binding. Mutations of the SpeA amino acid residue 42 to arginine or alanine (L42R or L42A) resulted in greatly diminished interactions with cell surface HLA-class II molecules (Figure 1A), as measured by flow cytometry.

Biological activity of SpeA mutants. A. Mutations of amino acid position leucine 42 of SpeA to arginine or alanine resulted in greatly diminished interactions with cell surface MHC class II molecules, measured by laser fluorescence-activated flow cytometry and FITC-labeled rabbit anti-SpeA antibody. B. Mutations of amino acid position leucine 42 of SpeA to arginine or alanine resulted in greatly diminished activation of human lymphocytes. Human T-cell proliferation was assessed by [3H]thymidine incorporation (12 h pulse) after 60 h of culture. Each data point represents the mean of triplicate determinations; SEM ≤ 5%.
Human T-cell proliferation in response to these mutants was next assessed. Both SpeA mutations of L42 resulted in greatly diminished activation of human lymphocytes (Figure 1B). Although alanine and arginine substitutions of L42 resulted in similar levels of attenuated MHC class II binding, arginine substitution (L42R) produced the greatest reduction of T-cell responses (Figure 1B) and was therefore chosen for further study.
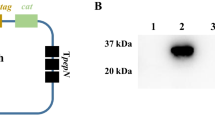
A catalytically inactive SpeB was constructed by mutating cysteine at position 47 [SpeB (C47S)] and used as a fusion partner with SpeA (L42R). The predicted 54 kDa protein was detected by polyacrylamide gel electrophoresis (Figure 2A). The SpeA (L42R)-SpeB (C47S) fusion was catalytically inactive towards peptide substrate (data not shown), using a previously reported assay [20]. In addition, rabbit antibodies specific for either SpeA or SpeB both detected SpeA (L42R)-SpeB (C47S) by Western blot analysis (Figure 2A). Although an additional recombinant protein was produced to incorporate the SpeB prosegment in the final SpeA-B fusion, this was not used further due to poor stability in solution.

Antibody recognition of SpeA (L42R)-SpeB (C47S) fusion protein. A. Antibody recognition in vitro. Coomassie Blue stain of isolated SpeA (L42R)-SpeB (C47S), lane 1; Western blot using-affinity purified, rabbit anti-SpeB (lane 2) or anti-SpeA antibody (lane 3). B. Antibody response and recognition in vivo. Mice (BALB/c) were vaccinated three times with 10 μg of each protein and adjuvant (MPL), allowing two weeks between injections. Sera from each experimental group (n = 5) were pooled for measurement of specific antibodies. Data shown are antigen-specific antibodies (ELISA units) present in a 1:100,000 dilution of pooled sera from mice vaccinated with SpeA (L42R), SpeA (L42R)-SpeB (C47S) fusion or adjuvant only.
Mouse antibody response to SpeA (L42R)-SpeB (C47S) and protection from SpeA-toxic shock
Immune recognition in vivo of the recombinant streptococcal proteins was next examined. BALB/c mice were vaccinated three times with 10 μg of SpeA (L42R) or SpeA (L42R)-SpeB (C47S), allowing two weeks between injections. Although vaccination with either SpeA (L42R) or the SpeA (L42R)-SpeB (C47S) produced high antibody titers, antibodies from SpeA (L42R) vaccination recognized only SpeA (Figure 2B), whereas, antibodies from the SpeA (L42R)-SpeB (C47S)-vaccinated mice recognized both SpeA and SpeB (Figure 2B). Seroconversion (IgG) occurred after the first vaccination with SpeA (L42R)-SpeB (C47S) compared to two injections required for the SpeA (L42R) vaccination (Figure 3). Although these data confirmed the potent immunogenicity of the SpeA constructs, the inbred mouse was an inadequate model to demonstrate protective immunity. Within reasonable physiological limits, wild-type SpeA was not lethal for several inbred mouse strains examined. Therefore, a transgenic model was used, consisting of C57BL/6 mice expressing human CD4 and HLA-DQ8 [21, 22]. Wild-type SpeA was previously shown to be lethal at relatively low concentrations for the HLA-DQ8 mice [23]. The lymphocyte response from the HLA-DQ+ mice to SpeA (data not shown) was very similar in dose effect to those obtained with human mononuclear cells. Non-vaccinated HLA-DQ8 mice succumbed to SpeA challenge, whereas, vaccination with either SpeA (L42R) or SpeA (L42R)-SpeB (C47S) fully protected HLA-DQ8 transgenic mice from challenge with the same amount of wild-type SpeA (Table 1).

Protection of transgenic HLA-DQ8 mice from Streptococcus pyogenes sepsis following vaccination with SpeA (L42R)-SpeB (C47S) fusion protein. Mice (5 per group) were vaccinated three times with 10 μg of each protein with adjuvant (MPL), allowing two weeks between injections. Three weeks after the last vaccination the mice were injected (i.v) with 10 LD50 of S. pyogenes and survival was monitored for 10 days.
Vaccination with SpeA (L42R)-SpeB (C47S) and protection from streptococcal sepsis
Inconsistent results were obtained in attempts to model S. pyogenes sepsis in several inbred mouse strains. Therefore, the HLA-DQ8 transgenic mice were also used to examine vaccine efficacy in bacterial sepsis. Mice vaccinated as above were injected (i.v.) with live bacteria followed 4 h later by i.p. administration (75 μg) of E. coli LPS. Survival was monitored for 10 d after challenge. The co-administration of LPS, as previously documented for toxic shock [24], produced a measurable fatal disease (3–7 d) in mice injected with live S. pyogenes (Figure 3). The majority (80%) of vaccinated mice were protected from lethal sepsis in contrast to the unvaccinated control mice (Figure 3). Mice vaccinated with only SpeA (L42R) were not protected from bacterial sepsis (data not shown). However, it was unclear if these results were due to a limitation of the animal model or perhaps from the necessity to also target SpeB.
Discussion
Because of the strong association between streptococcal toxic shock and invasive streptococcal infections [25], targeting SpeA is important for the development of a human vaccine for preventing or treating sepsis caused by S. pyogenes. The results presented indicated that a vaccine consisting of a fusion between the inactivated bacterial superantigen SpeA and the cysteinyl protease SpeB, formulated with an adjuvant, protected mice from lethal toxic shock syndrome induced by administration of biologically active SpeA. Further, vaccination with the SpeA (L42R)-SpeB (C47S) fusion protein protected HLA-DQ8 transgenic mice from lethal infection caused by a clinical isolate of S. pyogenes. The results from vaccinations of HLA-DQ8 transgenic mice demonstrated efficacy in a human-like, MHC class II receptor background, suggesting that SpeA (L42R)-SpeB (C47S) may be an important new vaccine for controlling streptococcal toxic shock and S. pyogenes infections. The potential advantages to this fusion protein above the isolated SpeA (L42R) are potent activation of immune responses, immune protection against a second virulence factor (SpeB), potential cost savings and simplification of vaccine production.
The rationale for selecting mutations only in the MHC-binding region of SpeA was based on previous results with staphylococcal superantigens demonstrating that the effect of mutations to the MHC binding site produced a greater attenuation of superantigen activity and lethality than mutations to the TCR-binding site [26] and that the residues involved in MHC class II binding are more conserved than those involved in binding to the TCR Vß chain [18, 27]. The mode of protection stimulated by SpeA (L42R)-SpeB (C47S) was presumed to be antibody mediated, though this was not directly ascertained in the current study. It is possible that conformational changes in the protein structures of SpeA and SpeB due to production as a single polypeptide may impact vaccine efficacy by altering recognition of the native bacterial proteins. However, antibody recognition is likely to be maintained during vaccination with the fusion product because the native biological activities of SpeB and SpeA were eliminated by site-specific mutagenesis methods that cause minimal perturbation of protein structure [28, 29].
Additional data corroborate SpeB and SpeA as rational targets for immune intervention. Both proteins were produced by M1 S. pyogenes during growth in human saliva, and growth was dependent on SpeB [30], suggesting a potential protective role for secretory antibodies. Further, it was reported that SpeB influences tissue tropism of S. pyogenes [31] and was proposed as a seromarker for infection equal in performance to standards currently in use [32]. The S. pyogenes serotype M1 that spread globally in the late 1980s and early 1990s harbored phage-borne SpeA [33] and another common superantigen SmeZ [34]. In addition, four of five recently re-emerging strains (emm49 genotype) isolated from severe invasive S. pyogenes patients in Japan were speA+ [35] and a prophage remnant encoding SpeA was noted in a macrolide-resistant strain of serotype M6 S. pyogenes [36]. It was also noted that genes encoding SpeA are commonly found in pharyngeal S. pyogenes isolates [37].
Previous reports have suggested other surface proteins of S. pyogenes as potential candidates for vaccines [38, 39], but strain variation is generally a complication. For example, though the M protein is universally expressed by S. pyogenes, antibody against one strain may not be protective against other strains due to varying susceptibility to opsonophagocytosis [40] resulting from differences in M protein structure. Vaccination with a fusion product of multiple M epitopes was reported as an alternative means to induce antibodies specific for dominant serotypes [39]. In contrast, mice actively immunized with SpeB resulted in non-type-specific immunity to challenge with heterologous S. pyogenes[41]. It is anticipated that vaccination with the SpeA-SpeB fusion protein described in the present study may result in protective immunity against multiple isolates of S. pyogenes due to the extensive antibody cross-reactivity previously observed among all sequence variants of SpeB [15] and the high frequency of SpeA-producing strains.
References
Reinert RR, Lutticken R, Al-Lahham A: High-level fluoroquinolone resistance in a clinical Streptoccoccus pyogenes isolate in Germany. Clin Microbiol Infect. 2004, 10: 659-662. 10.1111/j.1469-0691.2004.00890.x.
Buxbaum A, Forsthuber S, Sauermann R, Gattringer R, Graninger W, Georgopoulos A: Development of macrolide-resistance and comparative activity of telithromycin in streptococci in Austria, 1996–2002. Int J Antimicrob Agents. 2004, 24: 397-400. 10.1016/j.ijantimicag.2004.04.010.
Stevens DL: Streptococcal toxic shock syndrome associated with necrotizing fasciitis. Annu Rev Med. 2000, 51: 271-288. 10.1146/annurev.med.51.1.271.
Musser JM, Krause RM: The revival of group A streptococcal diseases with a commentary on staphylococcal toxic shock syndrome. Emerging infections. Edited by: Krause RM, Fauci A. 1998, New York, Academic Press, 185-218.
Hasegawa T, Hashikawa SN, Nakamura T, Torii K, Ohta M: Factors determining prognosis in streptococcal toxic shock-like syndrome: results of a nationwide investigation in Japan. Microbes Infect. 2004, 6: 1073-1077. 10.1016/j.micinf.2004.06.001.
Stevens DL: Invasive group A streptococcus infections. Clin Infect Dis. 1992, 14: 2-13.
Ulrich RG, Bavari S, Olson M: Bacterial superantigens in human diseases: Structure, Function and Diversity. Trends in Microbiol. 1995, 3: 463-468. 10.1016/S0966-842X(00)89011-3.
Norrby-Teglund A, Pauksens K, Norgren M, Holm SE: Correlation between serum TNF alpha and IL-6 levels and severity of group A streptococcal infections. Scand J Infect Dis. 1995, 27: 125-130. 10.3109/00365549509018991.
Stevens DL, Bryant AE, Hackett SP, Chang A, Peer G, Kosanke S, Emerson T, Hinshaw L: Group A streptococcal bacteremia: the role of tumor necrosis factor in shock and organ failure. J Infect Dis. 1996, 173: 619-626.
Hytonen J, Haataja S, Gerlach D, Podbielski A, Finne J: The SpeB virulence factor of Streptococcus pyogenes, a multifunctional secreted and cell surface molecule with strepadhesin, laminin-binding and cysteine protease activity. Mol Microbiol. 2001, 39: 512-519. 10.1046/j.1365-2958.2001.02269.x.
Kuo CF, Wu JJ, Lin KY, Lei HY, Lin YS: Role of Streptococcal Pyrogenic Exotoxin B in the Mouse Model of Group A Streptococcal Infection. Infect Immun. 1998, 66: 3931-3935.
Kapur V, Majesky MW, Li LL, Black RA, Musser JM: Cleavage of interleukin 1 beta (IL-1 beta) precursor to produce active IL-1 beta by a conserved extracellular cysteine proteae from Streptococcus pyogenes. Proc Natl Acad Sci USA. 1993, 90: 7676-7680. 10.1073/pnas.90.16.7676.
Kapur V, Topouzis S, Majesky MW, Li LL, Hamrick MR, Hamill RJ, Patti JM, Musser JM: A conserved Streptococcal pyogenes extracellular cysteine protease cleaves human fibronectin and degrades vitronectin. Microb Path. 1993, 15: 327-346. 10.1006/mpat.1993.1083.
Nyberg P, Rasmussen M, Von Pawel-Rammingen U, Bjorck L: SpeB modulates fibronectin-dependent internalization of Streptococcus pyogenes by efficient proteolysis of cell-wall-anchored protein F1. Microbiology. 2004, 150: 1559-1569. 10.1099/mic.0.27076-0.
Gubba S, Low DE, Musser JM: Expression and characterization of group A Streptococcus extracellular cysteine protease recombinant mutant proteins and documentation of seroconversion during human invasive disease episodes. Infect Immun. 1998, 66: 765-770.
Akesson P, Rasmussen M, Mascini E, von Pawel-Rammingen U, Janulczyk R, Collin M, Olsen A, Mattsson E, Olsson ML, Bjorck L, Christensson B: Low antibody levels against cell wall-attached proteins of Streptococcus pyogenes predispose for severe invasive disease. J Infect Dis. 2004, 189: 797-804. 10.1086/381982.
Ulrich RG, Bavari S, Olson MA: Staphylococcal enterotoxins A and B share a common structural motif for binding class II major histocompatibility complex molecules. Nat Struct Biol. 1995, 2: 554-560. 10.1038/nsb0795-554.
Coffman JD, Zhu J, Roach JM, Bavari S, Ulrich RG, Giardina SL: Production and purification of a recombinant staphylococcal enterotoxin B vaccine candidate expressed in Escherichia coli. Prot Express Purif. 2002, 24: 302-312. 10.1006/prep.2001.1556.
Nabozny GH, Baisch JM, Cheng S, Cosgrove D, Griffiths MM, Luthra HS, David CS: HLA-DQ8 transgenic mice are highly susceptible to collagen-induced arthritis: a novel model for human polyarthritis. J Exp Med. 1996, 183: 27-37. 10.1084/jem.183.1.27.
Nomizu M, Pietrzynski G, Kato T, Lachance P, Menard R, Ziomek E: Substrate Specificity of the Streptococcal Cysteine Protease. J Biol Chem. 2001, 276: 44551-44556. 10.1074/jbc.M106306200.
Taneja V, David CS: HLA class II transgenic mice as models of human diseases. Immunol Rev. 1999, 169: 67-79. 10.1111/j.1600-065X.1999.tb01307.x.
Nabozny GH, Baisch JM, Cheng S, Cosgrove D, Griffiths MM, Luthra HS, David CS: HLA-DQ8 transgenic mice are highly susceptible to collagen-induced arthritis: a novel model for human polyarthritis. J Exp Med. 1996, 183: 27-37. 10.1084/jem.183.1.27.
Welcher BC, Carra JH, DaSilva L, Hanson J, David CS, Aman MJ, Bavari S: Lethal shock induced by streptococcal pyrogenic exotoxin A in mice transgenic for human leukocyte antigen-DQ8 and human CD4 receptors: implications for development of vaccines and therapeutics. J Infect Dis. 2002, 186: 501-510. 10.1086/341828.
Stiles BG, Bavari S, Krakauer T, Ulrich RG: Toxicity of staphylococcal enterotoxins potentiated by lipopolysaccharide: major histocompatibility complex class II molecule dependency and cytokine release. Infect Immun. 1993, 61: 5333-5338.
Stevens DL: Streptococcal toxic shock syndrome. Clin Microbiol Infect. 2002, 8: 133-136. 10.1046/j.1469-0691.2002.00387.x.
Bavari S, Dyas B, Ulrich RG: Superantigen vaccines: A comparative study of genetically attenuated receptor-binding mutants of staphylococcal enterotoxin A. J Infect Dis. 1996, 174: 338-345.
Ulrich RG: Evolving superantigens of Staphylococcus aureus. FEMS Microbiol. 2000, 27: 1-7. 10.1111/j.1574-695X.2000.tb01404.x.
Krupka HI, Segelke B, Ulrich RG, Ringhofer S, Knapp M, Rupp B: Structural basis for abrogated binding between staphylococcal enterotoxin A superantigen vaccine and MHC-II. J Prot Science. 2002, 11: 642-651. 10.1110/ps.39702.
Ulrich RG, Olson M, Bavari S: Bacterial superantigen vaccines. Vaccines 96, Molecular Approaches to the Control of Infectious Diseases. Edited by: Brown F, Norrby E, Burton D, Mekalanos J. 1996, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, 135-139.
Shelburne SA, Granville C, Tokuyama M, Sitkiewicz I, Patel P, Musser JM: Growth characteristics of and virulence factor production by group A Streptococcus during cultivation in human saliva. Infect Immun. 2005, 73: 4723-4731. 10.1128/IAI.73.8.4723-4731.2005.
Svensson MD, Scaramuzzino DA, Sjobring U, Olsen A, Frank C, Bessen DE: Role for a secreted cysteine proteinase in the establishment of host tissue tropism by group A streptococci. Mol Microbiol. 2000, 38: 242-253. 10.1046/j.1365-2958.2000.02144.x.
Batsford S, Brundiers M, Schweier O, Horbach E, Mönting JS: Antibody to streptococcal cysteine proteinase as a seromarker of group A Streptococcal (Streptococcus pyogenes) infections. Scand J Infect Dis. 2002, 34: 407-412. 10.1080/00365540110080674.
Cleary PP, LaPenta D, Vessela R, Lam H, Cue D: A globally disseminated M1 subclone of group A streptococci differs from other subclones by 70 kilobases of prophage DNA and capacity for high-frequency intracellular invasion. Infect Immun. 1998, 66: 5592-5597.
Vlaminckx BJ, Mascini EM, Schellekens J, Schouls LM, Paauw A, Fluit AC, Novak R, Verhoef J, Schmitz FJ: Site-specific manifestations of invasive group a streptococcal disease: type distribution and corresponding patterns of virulence determinants. J Clin Microbiol. 2003, 41: 4941-4949. 10.1128/JCM.41.11.4941-4949.2003.
Ikebe T, Endo M, Ueda Y, Okada K, Suzuki R, Minami T, Tanaka H, Nakanishi N, Tomita M, Nishie H, Ishii N, Sasaki E, Miura Y, Yamamura T, Watanabe H: The Genetic Properties of Streptococcus pyogenes emm49 Genotype Strains Recently Emerged among Severe Invasive Infections in Japan. Jpn J Infect Dis. 2004, 57: 187-188.
Banks DJ, Porcella SF, Barbian KD, Beres SB, Philips LE, Voyich JM, DeLeo FR, Martin JM, Somerville GA, Musser JM: Progress toward characterization of the group A Streptococcus metagenome: complete genome sequence of a macrolide-resistant serotype M6 strain. J Infect Dis. 2004, 190: 727-738. 10.1086/422697.
Haukness HA, Tanz RR, Thomson RB, Pierry DK, Kaplan EL, Beall B, Johnson D, Hoe NP, Musser JM, Shulman ST: The heterogeneity of endemic community pediatric group a streptococcal pharyngeal isolates and their relationship to invasive isolates. J Infect Dis. 2002, 185: 915-920. 10.1086/339407.
Olive C, Hsien K, Horvath A, Clair T, Yarwood P, Toth I, Good MF: Protection against group A streptococcal infection by vaccination with self-adjuvanting lipid core M protein peptides. Vaccine. 2005, 23: 2298-2303. 10.1016/j.vaccine.2005.01.041.
Hall MA, Stroop SD, Hu MC, Walls MA, Reddish MA, Burt DS, Lowell GH, Dale JB: Intranasal immunization with multivalent group A streptococcal vaccines protects mice against intranasal challenge infections. Infect Immun. 2004, 72: 2507-2512. 10.1128/IAI.72.5.2507-2512.2004.
Villasenor SA, McShan WM, Salmi D, Kaplan EL, Johnson DR, Stevens DL: Variable susceptibility to opsonophagocytosis of group A streptococcus M-1 strains by human immune sera. J Infect Dis. 1999, 180: 1921-1928. 10.1086/315120.
Kapur V, Maffei JT, Greer RS, Li LL, Adams GJ, Musser JM: Vaccination with streptococcal extracellular cysteine protease (interleukin-1 beta convertase) protects mice against challenge with heterologous group A streptococci. Microb Pathol. 1994, 16: 443-450. 10.1006/mpat.1994.1044.
Acknowledgements
The author thanks M. Afroz Sultana for technical assistance, Dwayne Lunsford (Southern Research Institute) for performing sepsis studies with inbred mice, and Sina Bavari for providing transgenic mice. Opinions, interpretations, conclusions, and recommendations are those of the author and are not necessarily endorsed by the U.S. Government.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author declares that he has no competing interests.
Authors' contributions
RGU conceived and performed the study. RGU wrote and approved the final manuscript draft.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ulrich, R.G. Vaccine based on a ubiquitous cysteinyl protease and streptococcal pyrogenic exotoxin A protects against Streptococcus pyogenessepsis and toxic shock. J Immune Based Ther Vaccines 6, 8 (2008). https://doi.org/10.1186/1476-8518-6-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-8518-6-8