
Abstract
Background
Genome-wide association studies have identified numerous single nucleotide polymorphisms (SNPs) affecting high density lipoprotein (HDL) or low density lipoprotein (LDL) cholesterol levels; these SNPs may contribute to the genetic basis of vascular diseases.
Results
We assessed the impact of 34 SNPs at 23 loci on dyslipidemia, key lipid sub-phenotypes, and severe carotid artery disease (CAAD) in a case-control cohort. The effects of these SNPs on HDL and LDL were consistent with those previously reported, and we provide unbiased estimates of the percent variance in HDL (3.9%) and LDL (3.3%) explained by genetic risk scores. We assessed the effects of these SNPs on HDL subfractions, apolipoprotein A-1, LDL buoyancy, apolipoprotein B, and lipoprotein (a) and found that rs646776 predicts apolipoprotein B level while rs2075650 predicts LDL buoyancy. Finally, we tested the role of these SNPs in conferring risk for ultrasonographically documented CAAD stenosis status. We found that two loci, chromosome 1p13.3 near CELSR2 and PSRC1 which contains rs646776, and 19q13.2 near TOMM40 and APOE which contains rs2075650, harbor risk alleles for CAAD.
Conclusion
Our analysis of 34 SNPs contributing to dyslipidemia at 23 loci suggests that genetic variation in the 1p13.3 region may increase risk of CAAD by increasing LDL particle number, whereas variation in the 19q13.2 region may increase CAAD risk by promoting formation of smaller, denser LDL particles.
Similar content being viewed by others


Background
Carotid artery disease (CAAD) is an important risk factor for stroke, the third leading cause of death in the U.S. Given the high mortality, morbidity, and economic costs due to stroke, primary prevention, particularly targeted toward high risk individuals, is the most promising approach to combat stroke[1, 2]. Although medical interventions and carotid endarterectomy can potentially prevent strokes in individuals with CAAD, routine screening is not currently recommended[1]. However, it has been suggested that if high risk groups with CAAD prevalences of approximately 20% can be identified, screening may provide significant and cost effective extension to quality adjusted life years[3, 4]. Studies of siblings[5, 6], twins[7, 8], and families[9] suggest a heritable genetic contribution to carotid artery intima-media thickening and stenosis from plaque, with the heritability of ultrasonographically measured phenotypes typically ranging from 20% to 40% in population based samples[10]. Thus, identification of genetic risk factors for carotid artery stenosis, progression, and plaque instability may ultimately be useful in targeting primary prevention against stroke in patients for whom management strategies are not yet well defined.
Recently a number of large genome-wide association studies have revealed loci affecting total cholesterol, high-density lipoprotein cholesterol (HDL), low-density lipoprotein cholesterol (LDL), and triglycerides [11–15]. Because of their role in promoting dyslipidemia, these single nucleotide polymorphisms (SNPs) are strong candidates for contributing to genetic risk for atherosclerosis, and several studies have found significant impacts of these loci on coronary artery disease[11, 12, 16]. Although many clinical risk factors such as age, smoking, hypertension, and diabetes are shared between CAAD and coronary artery disease, the relative importance of these risk factors differs between these two vascular disease processes [17]. Similarly, the relative importance of risk factors varies for disease at different locations within the carotid arteries themselves[9, 10]. These discrepancies suggest that additional factors, including genetic ones, may modulate the atherosclerotic disease process differently in different anatomic locations. Thus, the impact of recently discovered dyslipidemia risk alleles on CAAD is as yet unknown.
Based on previous success in applying genetic risk scores for decreased HDL and increased LDL to the prediction of coronary artery disease[12] and the central role of these lipid fractions in evidence-based guidelines for coronary artery disease risk reduction[18], we investigated the role of SNPs affecting HDL and LDL in predicting risk for CAAD. We also sought to determine whether these SNPs alter key lipid sub-phenotypes with differential atherogenic potential. Specifically, the more efficient cholesterol efflux activity of apolipoprotein A-I (apo A-I) [19] has lead to the hypothesis that the HDL2 subfraction or apo A-I may be a better predictor of protection against atherosclerosis than HDL3 or total HDL. We also tested SNPs for their effects on apolipoprotein B (apo B), which measures LDL particle number and may be a better estimator of cardiovascular disease risk than LDL level[20, 21], LDL buoyancy which predicts the smaller, denser LDL pattern B phenotype [22] associated with increased risk of coronary artery disease[23], and lipoprotein(a) (Lp(a)) which appears to independently predict risk of coronary artery disease [24] and stroke [25]. These analyses may suggest mechanisms through which specific SNPs modulate CAAD risk beyond their effects on HDL and LDL levels.
Results
Effects of SNPs on HDL and LDL
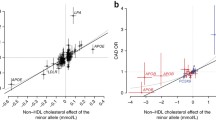
As shown in Figure 1 our data confirm the previously reported effects on HDL and LDL for the majority of SNPs tested in CLEAR study participants (see Table 1). Out of 34 SNPs tested (see Table 2), we identified 14 SNPs that showed nominally significant associations with HDL or LDL levels at a p-value of 0.05, corresponding to a FDR of 0.11 when corrected for multiple testing. These 14 SNPs correspond to those for which the 95% confidence intervals do not cross zero in Figure 1. For only three SNPs the 95% confidence intervals do not contain the previously reported effect from the literature, and for the top 25 SNPs, those indicated by closed circles, our data are more likely assuming the effect as reported in the literature (i.e.  's as given by the x's in Figure 1) than under the null ( = 0). Furthermore, for 28 out of 34 SNPs the effects on HDL or LDL in our data were in the same direction as reported previously in the literature, corresponding to significant concordance under a binomial test (p = 2.0 × 10-4).
's as given by the x's in Figure 1) than under the null ( = 0). Furthermore, for 28 out of 34 SNPs the effects on HDL or LDL in our data were in the same direction as reported previously in the literature, corresponding to significant concordance under a binomial test (p = 2.0 × 10-4).

Effects of SNPs on HDL and LDL. The estimated regression coefficient (circles) for the effect of each SNP on HDL or LDL (listed on the right) is shown with the associated 95% confidence interval. Previously reported effects from the literature are marked with x's. SNPs are sorted by decreasing relative likelihood under the alternative ( as given by the x) versus the null hypothesis ( = 0) with closed circles indicating SNPs for which the data are more likely under the alternative hypothesis.
Given the small marginal effects of the recently reported SNPs on HDL or LDL levels, we used a genetic risk score combining alleles additively across SNPs to better predict lipid levels. Such genetic risk scores have previously been utilized, but they are subject to upward bias when developed in the same sample used to initially detect associations. Using a risk score with SNPs weighted by the reported effect sizes from the literature (see Table 2) and after accounting for covariates, the 16 HDL SNPs explain 3.9% of the variation in HDL levels (p = 4.3 × 10-9). Using the same approach, the 18 LDL SNPs explain 3.3% of the variation in LDL levels (p = 4.4 × 10-8).
Effects of SNPs on lipid sub-phenotypes
With few exceptions we found that the same SNPs significantly associated with total HDL in our data were also associated with HDL2, HDL3, and apo A-I. At a p-value of 0.05, corresponding to an FDR of 0.11, we identified 19 significant associations between the 16 HDL SNPs and HDL2, HDL3, or apo A-I. In a principal component analysis of the t-statistics derived from tests of association between the 16 HDL SNPs and 4 phenotypes (total HDL, HDL2, HDL3, and apo A-I), we found that the first principal component was positively correlated with all 4 phenotypes and explained 94% of the variance in the t-statistics, indicating that the effects of each SNP were highly concordant across all HDL related phenotypes. Similarly, the hierarchical clustering analysis performed by Kathiresan et al[14] also shows that the current set of genetic loci affecting HDL does little to discriminate among HDL2, HDL3, and apo A-I levels.
At a p-value of 0.05, we found that rs646776, rs693, rs2228671, and rs6511720 were associated with apo B and that rs2075650 and rs2650000 were associated with LDL buoyancy. The associated FDR for these tests was quite high at 0.35, due in part to the absence of any affect of these SNPs on Lp(a). Among SNPs associated with apo B, rs646776 was by far the most statistically significant (p = 0.00035, compared with 0.017 ≤ p < 0.05 for the remaining three) with its minor allele decreasing apo B ( = -3.3). Kathiresan et al[14] found a similarly strong effect on apo B of the nearby SNP rs12740374 ( = -3.3; p = 1.2 × 10-8), which is nearly perfectly correlated with rs646776 (see Table 2). Among SNPs associated with LDL buoyancy, rs2075650 was the most statistically significant (p = 0.014), with the minor allele decreasing the relative flotation rate ( = -0.0047) leading to a more atherogenic phenotype.
Effects of SNPs on CAAD risk
After a Bonferroni correction for 34 tests, corresponding to a threshold of p = 0.0015, Figure 2 shows that two SNPs, rs646776 and rs2075650, met criteria for significant association with CAAD. For convenience SNPs have been recoded from minor allele dose to unfavorable allele dose based on their effect on HDL or LDL so that the expected effect on CAAD is >0. Due to the prior expectation that alleles that decrease HDL or increase LDL would confer increased risk for CAAD, we performed one-sided tests. The major allele of rs646776 is associated with increased LDL, increased apo B as described above, and increased risk of CAAD ( = 0.47; p = 0.0012). The minor allele of rs2075650 is associated with increased LDL, decreased LDL buoyancy as described above, and increased risk of CAAD ( = 0.56; p = 0.00091). Results were similar when we analyzed percent carotid stenosis (with controls, individuals with intermediate stenosis, and cases having phenotypes coded 0, 0.5, and 0.8 respectively) in a linear regression allowing for inclusion of 83 additional individuals with intermediate carotid stenosis with luminal narrowing of 50 to 79% (p = 0.0049 for rs646776, p = 0.0010 for rs2075650).

Effects of SNPs on CAAD risk. The estimated coefficient (circles) for each SNP in a logistic regression model of CAAD case versus control status is shown along with the associated 95% (longer vertical ticks) and 99.9% (shorter vertical ticks) confidence intervals, corresponding to one-sided p-values of 0.05 and 0.0015 respectively. Closed circles with confidence intervals in solid lines represent those SNPs that are significantly associated with CAAD after a Bonferroni correction for 34 tests.
The associations between rs646776 and rs2075650 and CAAD could not be explained by their effects on HDL and LDL levels alone. We found that including the observed HDL and LDL levels, use of lipid lowering therapy, or "pre-therapy" HDL and LDL levels (see Methods) as covariates did not dramatically change the significance or magnitude of the associations between CAAD and rs646776 or rs2075650 (data not shown). Combined with their effects on LDL particle number and buoyancy, these results raise the possibility that rs646776 and rs2075650 contribute additional information to current HDL and LDL levels alone for CAAD risk prediction.
Genetic risk score for CAAD
Contrary to HDL and LDL, we did not find that CAAD risk prediction was improved using an additive risk score based on these SNPs. We tested both a risk score in which all SNPs were weighted equally as in Kathiresan et al[12] (p = 0.32), and one in which SNPs were weighted by the magnitude of their effects on HDL and LDL as in Aulchenko et al[11] (p = 0.63). Results were similar with or without HDL, LDL, and use of lipid lowering as covariates. To clarify this finding, we sought to determine our power to detect association between the genetic risk score and CAAD, given that the risk score based on these SNPs explains only 3.9% and 3.3% of the variance in HDL and LDL levels, respectively, and given that dyslipidemia is only one of several risk factors for CAAD. Using power simulations (see Methods), we found that the genetic risk score, acting through its effects on HDL and LDL levels alone, had an expected odds ratio for association with CAAD of 1.03 to 1.04 per unfavorable allele. The power based on these effect sizes was only 20% to 39%, suggesting that we cannot reject a model in which the overall genetic score confers risk of CAAD, but mainly through its effect on lipid levels. However, if the odds ratio per unfavorable allele averaged as little as 1.07 to 1.10 our power to detect association between the risk score and CAAD was 81% to 97%. This suggests that if a significant fraction of these 34 SNPs are predictive of CAAD risk beyond their effects on measured HDL and LDL levels, we would have had high power to detect such an association.
CAAD risk locus on chromosome 1p13.3
To further explore the CAAD association in the vicinity of rs646776 we analyzed 82 additional SNPs that were genotyped or imputed with at least 80% accuracy, and Figure 3 shows that rs646776 and neighboring SNPs in CELSR2 and PSRC1 exhibit the greatest statistical significance. To correct for multiple testing in the setting of strong linkage disequilibrium we performed permutation testing to estimate a significance threshold of p = 0.0040. Within SORT1 the intronic SNP rs4970843 shows significant association (p = 0.0030). Interestingly rs4970843 is in weak but significant long range linkage disequilibrium with rs646776 (r2 = 0.083; p = 2.5 × 10-19), and is in relatively weaker linkage disequilibrium with nearby SNPs in the highly correlated block encompassing SORT1.

Linkage disequilibrium structure and association results for the chromosome 1p13.3 region containing rs646776. The lower portion of the figure shows pairwise linkage disequilibrium in HapMap individuals with white indicating r2 = 0 and black indicating r2 = 1. Approximate boundaries of linkage disequilibrium blocks are traced. The locations of genotyped and imputed SNPs relative to the genes in the region (middle portion) are shown with black and light gray lines, respectively. Exonic regions of genes are shaded dark gray. The upper portion shows -log10 p-values for association with CAAD, with nominal significance (p = 0.05) and permutation corrected significance levels for 83 analyzed SNPs indicated by the lower and upper dashed lines, respectively. rs646776, rs599839, and rs4970843 are marked by the open triangle, square, and circle, respectively.
CAAD risk locus on chromosome 19p13.2
Figure 4 shows that rs2075650, an intronic SNP in TOMM40, displays stronger association with CAAD than rs429358, which defines the ε3/ε4 dichotomy, or rs7412, which defines the ε2/ε3 dichotomy, in the APOE ε2/ε3/ε4 polymorphism. When rs2075650 and rs429358 were analyzed conditional on one another, we found significant association with rs2075650 (p = 0.032) but not with rs429358 (p = 0.61). This did not appear to be due to the additive model being a poor fit for the effect of APOE, because when we utilized a two degree of freedom model for the ε3/ε4 effect, rs2075650 retained significance (p = 0.031) whereas neither the recessive nor dominance term for rs429358 was significant (p = 0.78 and p = 0.72, respectively). When rs2075650 and rs7412 were analyzed conditional on one another we found that both showed significant association with CAAD (p = 0.0047 for rs2075650 and p = 0.012 for rs7412). With a two degree of freedom model, rs2075650 retained significance (p = 0.0046) while the dominance term (p = 0.021) but not the recessive term (p = 0.40) was significant for rs7412. When all three SNPs were analyzed simultaneously including an interaction term for ε2/ε3 with ε3/ε4, rs2075650 was suggestively significant (p = 0.055), neither rs429358 nor its interaction with rs7412 was significant (p = 0.63 and p = 1.00, respectively), and rs7412 was significant (p = 0.036).

Linkage disequilibrium structure and association results for the chromosome 19p13.2 region containing rs2075650. See the Figure 3 caption for further description. The upper dashed line represents a Bonferroni correction for 25 analyzed SNPs. rs2075650, rs429358, and rs7412 are indicated by the open triangle, square, and circle, respectively.
Discussion
Our data confirm the effects of recently identified dyslipidemia SNPs, and we estimated that, after accounting for other covariates, genetic risk scores explain 3.9% and 3.3% of the variance in HDL and LDL, respectively. Although slightly different sets of SNPs and weighting factors were used, these values are in reasonable agreement with those of 4.8% and 3.4% as reported by Aulchenko et al[11]. Our estimates may be biased lower because of the extensive use of lipid lowering therapy in CLEAR participants with various pharmacological agents, dosing, and medication compliance which we have not attempted to fully model. In addition, while Aulchenko et al. sought to reduce upward bias by estimating weights for each SNP in independent cohorts from that in which the risk score was applied, the fact that the set of SNPs used in the risk score was apparently identified using all available cohorts suggests that a slight upward bias would still remain in their estimates. Based on the estimated effect sizes of this panel of SNPs on HDL and LDL, and the estimated effect size of dyslipidemia on CAAD, our data cannot exclude a model in which these SNPs as a group increase risk of CAAD mostly or entirely through their effects on lipids. However, our failure to identify a significant association between an overall risk score based on these SNPs and CAAD suggests that the majority of these SNPs are unlikely to contribute strongly to CAAD beyond their role in promoting dyslipidemia.
This result highlights a potential cause for caution in using a genetic risk score derived from an intermediate phenotype to predict disease. If genetic variants contributing to such a score act only through the intermediate clinical variable, then use of the clinical variable is likely to be superior. On the other hand, if genetic variants contribute to disease risk in ways not captured by the clinical variable, for example because they provide an index of lifetime exposure or because of pleiotropy with effects on the disease through multiple pathways, then a risk score may have significant clinical utility. However, when a score for an intermediate phenotype is developed using tens or hundreds of SNPs, it is likely that only a subset of the contributing SNPs will have these favorable predictive properties. Within the context of our study, we found no significant predictive power of the overall dyslipidemia risk score beyond what would be predicted by the role of dyslipidemia in CAAD alone, yet we identify two SNPs, rs646776 and rs2075650, which appear to mediate risk for CAAD beyond their effects on HDL and LDL. Based on its association with apo B, our analyses indicate that rs646776 affects LDL particle number in addition to LDL level, which may account for the additional explanatory power of this SNP. In contrast, rs2075650 appears to affect LDL buoyancy, with the minor allele contributing to the smaller, denser LDL particles that make up the more atherogenic LDL pattern B phenotype.
Based on our analyses of the 1p13.3 region containing rs646776, variation associated with CAAD is most likely located near CELSR2, a non-classical cadherin that does not interact with catenins, or PSRC1, a p53-regulated growth receptor. Although SORT1, a multi-ligand receptor present in the Golgi and on the cell surface, represents a good candidate gene because it binds and mediates degradation of lipoprotein lipase [26], increases its localization to the plasma membrane of adipocytes in response to insulin, and forms GLUT4 storage vesicles which enhance insulin sensitivity [27], our association signal was weaker in this gene. Given the strong linkage disequilibrium according to HapMap data, it is unlikely that there exists common variation in SORT1 that was not captured by our study. Although the 1p13.3 region appears to show robust association with coronary artery disease [28–30], a recent study that included 33,282 participants with a total of 503 strokes at baseline and 571 incident strokes did not identify a significant association between stroke and either rs599839 or rs4970834 in this region[31]. However, stroke is the sequela of a diverse set of underlying pathophysiologic causes which do not appear to have been distinguished in this study[32], so it is unclear whether this sample size would have sufficient power to detect an affect of the 1p13.3 region on the subset of strokes due to CAAD.
In the 19p13.2 region APOE is a stronger candidate gene than TOMM40, a channel forming subunit that is essential for protein import into the mitochondria [33]. The ε2/ε3/ε4 polymorphism is reported to be a equivocal risk factor for carotid atherosclerosis[10] although a more recent meta-analysis does support a modest association between these SNPs and carotid intima media thickness [34]. Consistent with this, our data show nominally significant protective effects of the ε2 allele and deleterious effects of the ε4 allele. However, conditioning on these SNPs did not account for the CAAD association signal in the region, and moreover the ε4 allele failed to demonstrate significant association with CAAD when rs2075650 was jointly considered. This result argues against a singular causal role for the APOE ε system in producing the CAAD association signal in this region, because one would expect the causal polymorphism to achieve greater statistical significance than, and in fact eliminate signal from, surrounding neutral variation. Instead, our data are most consistent with a causal polymorphism or collection of polymorphisms in linkage disequilibrium with both rs2075650 and the APOE ε system.
In summary, our data replicate the majority of associations reported between SNPs and HDL and LDL. Unbiased or slightly negatively biased estimates of the proportion of variance in HDL and LDL levels explained by these SNPs are 3.9% and 3.3% respectively, consistent with previous estimates[11]. The combined set of SNPs currently available does not improve CAAD risk prediction beyond what would be expected from their effects on HDL and LDL levels, but the specific SNPs rs646776 and rs2075650 are associated with CAAD risk, possibly due to their effects on LDL particle number and buoyancy, respectively.
Methods
Clear study participants
The Carotid Lesion Epidemiology And Risk (CLEAR) Study is a Seattle-based study involving the University of Washington (UW), Virginia Mason Medical Center (VM) and the Veterans Affairs Puget Sound Health Care System (VAPSHCS), focused on identifying predictors of CAAD, CAAD progression, and atherosclerotic plaque instability approved by the UW, VM, and VAPSHCS IRBs. All participants gave written informed consent. Participant characteristics are shown in Table 1. Only Caucasian males were analyzed due to under-representation of women and minorities in the cohort. Self reported ancestry was confirmed by STRUCTURE [35]. Individuals with total serum cholesterol >400 mg/dL or coagulopathy were excluded. Controls include 479 individuals with ≤15% carotid stenosis bilaterally as measured by duplex ultrasound. Individuals with vascular disease at other sites were excluded from the set of controls. Cases include 353 individuals status post carotid endarterectomy for symptomatic disease or asymptomatic individuals with ≥80% internal carotid stenosis either unilaterally or bilaterally. Individuals with intermediate stenosis have 50% to 79% luminal narrowing either unilaterally or bilaterally. Cases and controls were matched on age distribution, with censoring occurring at the time of diagnosis of vascular disease for cases or at the time of the last blood draw for controls. Hypertension was defined by treatment with antihypertensive medications. Diabetes was defined as a hemoglobin A1C≥6.5 or use of oral hypoglycemics or insulin.
Lipid phenotypes
Standard methods were used to determine total cholesterol, triglycerides, and HDL in fasting whole plasma using an Abbott Spectrum analyzer. LDL was calculated unless triglycerides were ≥400 mg/dL, in which case it was measured directly. HDL fractions 2 and 3 were determined by precipitating HDL2 from total HDL, measuring HDL3 in the supernatant, and subtracting this from total HDL to obtain HDL2. Apolipoprotein A-I, apolipoprotein B, and lipoprotein(a) were measured as described by Marcovina et al [36], Zambon et al [37], and Marcovina et al [38], respectively. LDL buoyancy was measured by the relative flotation rate Rf as described by Capell et al [22]. We utilized lipid measurements prior to initiation of lipid lowering therapy whenever possible. For 90 individuals with two to three repeated lipid measurements we used the mean of these measurements. Based on inspection of the raw phenotype and residuals distributions, we excluded 6 outlying individuals with HDL>100 mg/dL, 10 individuals with HDL2>25 mg/dL, 4 individuals with HDL3>80, and 4 individuals with apo A-I>225 mg/dL. We also excluded 4 outlying individuals with LDL>200 mg/dL and 4 individuals with LDL fraction apolipoprotein B>120 mg/dL. The positively skewed lipoprotein (a) distribution was log transformed.
Genotyping and SNPs
Genotypes were measured using the Illumina HumanCVD Genotyping BeadChip using an Illumina BeadStation Laboratory System platform[39, 40]. Duplicate genotyping for 34 individuals showed 99.7% consistency in calls. The APOE ε2/ε3/ε4 polymorphism was genotyped as previously described [41]. Additional SNPs in the chromosome 1p13.3 and 19p13.2 regions were genotyped using TaqMan Assays by Design on an Applied Biosystems 7900HT System [42]. Using unphased reference genotypes from release 27 of the HapMap project [43]. we performed imputation for untyped SNPs using BIMBAM [44, 45]. Although inaccurate genotype imputation is expected to cause false negatives rather than false positives, to avoid spurious conclusions we sought to determine which HapMap SNPs could be accurately imputed relying on the SNPs genotyped in the CLEAR study. We selected a random set of 10 individuals from the HapMap CEU sample and set to missing those SNPs not genotyped in the CLEAR study. We then imputed these missing SNPs using the remaining SNPs that had been genotyped in the CLEAR study. For each imputed SNP we computed the correlation between the imputed mean genotypes and the true genotypes for those HapMap individuals in whom genotypes had been masked. We repeated this procedure 20 times, selecting a different set of 10 individuals for genotype masking each time, and we report the imputation accuracy as the mean correlation over these 20 iterations. Only SNPs with >90% imputation accuracy were included in downstream analyses.
Statistical analyses
All analyses were performed in R[46]. Unless otherwise specified, tests for genetic association were performed assuming an additive model with the homozygous genotypes coded as 0 or 2 and the heterozygous genotype coded as 1. Analyses of lipid phenotypes were performed in cases, individuals with intermediate stenosis, and controls using linear regression with censored age, body mass index (BMI), hypertension, diabetes, and use of lipid lowering therapy as covariates. Current cigarette usage was included as a covariate for analyses of HDL. Unless otherwise specified analyses of CAAD were performed using logistic regression with case status (≥80% stenosis) coded as 1 and control status (≤15% stenosis) coded as 0 and censored age, current cigarette usage, pack-years smoked, BMI, hypertension, and diabetes as covariates. To account for multiple testing we estimated false discovery rates (FDR) using the Benjamini-Hochberg procedure[47].
Analysis of CAAD risk in the setting of lipid lowering therapy
Consistent with guidelines[18], cases with CAAD in the CLEAR study are generally treated with lipid lowering therapy to a target of LDL<100 mg/dL, whereas controls, who are without CAAD, coronary artery disease, or risk equivalents, are managed with a target LDL of <130 mg/dL or <160 mg/dL. Thus, including HDL, LDL, or lipid lowering therapy as covariates is problematic because it leads to a model in which the dependent variable, CAAD case control status, is causal for these independent variables. However, in order to study the effects of SNPs on CAAD in the context of lipid risk factors, we attempted to estimate "pre-therapy" HDL and LDL values for those individuals on lipid lowering therapy. We based on these values on the lipid altering effects of statins, the drug class for 92% of all lipid draws when therapy was in use. For HDL, we estimated that the post-therapy values were 2.1% to 9.6% higher than pre-therapy[48], and for LDL 30% to 63% lower than pre-therapy[48, 49]. We also estimated the percent change in HDL (13% increase) and LDL (28% decrease) from 41 individuals in the CLEAR study who had measurements both prior to and following initiation of lipid lowering therapy.
Power to detect association between genetic risk scores and CAAD
We performed simulations to determine our power to detect association between the genetic risk score and CAAD, given the percent variation in HDL and LDL levels explained by the genetic risk score and given the effect size of dyslipidemia as a risk factor for CAAD. Using the estimated "pre-therapy" HDL and LDL values described above, we first permuted the observed genotypes so that an additive score for the 16 HDL SNPs explained on average 3.9% of the variance in HDL levels and so that an additive score for the 18 LDL SNPs explained on average 3.3% of the variance in LDL levels. We then combined the HDL and LDL scores to form the overall genetic risk score. Next we simulated CAAD status by sampling binomial random variables with underlying probabilities given by the fitted effects of a logistic regression model that included "pre-therapy" HDL and LDL levels as well as all other covariates. For the three different "pre-therapy" HDL and LDL estimates described above, the odds ratios for association with CAAD in this model ranged from 0.95 to 0.96 per mg/dL change in HDL and 1.010 to 1.011 per mg/dL change in LDL. Finally, we tested for association between the simulated genetic risk score and simulated CAAD status in the setting of the usual covariates without HDL or LDL levels in the model. To estimate the expected odds ratio for the genetic risk score and the power, we performed 1000 such simulations for each of the three different estimates of "pre-therapy" HDL and LDL levels.
References
Goldstein LB, Adams R, Alberts MJ, Appel LJ, Brass LM, Bushnell CD, Culebras A, Degraba TJ, Gorelick PB, Guyton JR, Hart RG, Howard G, Kelly-Hayes M, Nixon JV, Sacco RL, American Heart Association/American Stroke Association Stroke Council; Atherosclerotic Peripheral Vascular Disease Interdisciplinary Working Group; Cardiovascular Nursing Council; Clinical Cardiology Council; Nutrition, Physical Activity, and Metabolism Council; Quality of Care and Outcomes Research Interdisciplinary Working Group; American Academy of Neurology: Primary prevention of ischemic stroke: a guideline from the American Heart Association/American Stroke Association Stroke Council: cosponsored by the Atherosclerotic Peripheral Vascular Disease Interdisciplinary Working Group; Cardiovascular Nursing Council; Clinical Cardiology Council; Nutrition, Physical Activity, and Metabolism Council; and the Quality of Care and Outcomes Research Interdisciplinary Working Group: the American Academy of Neurology affirms the value of this guideline. Stroke. 2006, 37: 1583-1633.
Goldstein LB, Adams R, Becker K, Furberg CD, Gorelick PB, Hademenos G, Hill M, Howard G, Howard VJ, Jacobs B, Levine SR, Mosca L, Sacco RL, Sherman DG, Wolf PA, del Zoppo GJ: Primary prevention of ischemic stroke: A statement for healthcare professionals from the Stroke Council of the American Heart Association. Stroke. 2001, 32: 280-299.
Derdeyn CP, Powers WJ: Cost-effectiveness of screening for asymptomatic carotid atherosclerotic disease. Stroke. 1996, 27: 1944-1950.
Whitty CJ, Sudlow CL, Warlow CP: Investigating individual subjects and screening populations for asymptomatic carotid stenosis can be harmful. J Neurol Neurosurg Psychiatry. 1998, 64: 619-623. 10.1136/jnnp.64.5.619
North KE, MacCluer JW, Devereux RB, Howard BV, Welty TK, Best LG, Lee ET, Fabsitz RR, Roman MJ: Heritability of carotid artery structure and function: the Strong Heart Family Study. Arterioscler Thromb Vasc Biol. 2002, 22: 1698-1703. 10.1161/01.ATV.0000032656.91352.5E
Zannad F, Visvikis S, Gueguen R, Sass C, Chapet O, Herbeth B, Siest G: Genetics strongly determines the wall thickness of the left and right carotid arteries. Hum Genet. 1998, 103: 183-188. 10.1007/s004390050804
Jartti L, Ronnemaa T, Kaprio J, Jarvisalo MJ, Toikka JO, Marniemi J, Hammar N, Alfredsson L, Saraste M, Hartiala J, Koskenvuo M, Raitakari OT: Population-based twin study of the effects of migration from Finland to Sweden on endothelial function and intima-media thickness. Arterioscler Thromb Vasc Biol. 2002, 22: 832-837. 10.1161/01.ATV.0000013313.70875.A7
Lange LA, Bowden DW, Langefeld CD, Wagenknecht LE, Carr JJ, Rich SS, Riley WA, Freedman BI: Heritability of carotid artery intima-medial thickness in type 2 diabetes. Stroke. 2002, 33: 1876-1881. 10.1161/01.STR.0000019909.71547.AA
Fox CS, Polak JF, Chazaro I, Cupples A, Wolf PA, D'Agostino RA, O'Donnell CJ: Genetic and environmental contributions to atherosclerosis phenotypes in men and women: heritability of carotid intima-media thickness in the Framingham Heart Study. Stroke. 2003, 34: 397-401. 10.1161/01.STR.0000048214.56981.6F
Manolio TA, Boerwinkle E, O'Donnell CJ, Wilson AF: Genetics of ultrasonographic carotid atherosclerosis. Arterioscler Thromb Vasc Biol. 2004, 24: 1567-1577. 10.1161/01.ATV.0000138789.11433.c1
Aulchenko YS, Ripatti S, Lindqvist I, Boomsma D, Heid IM, Pramstaller PP, Penninx BW, Janssens AC, Wilson JF, Spector T: Loci influencing lipid levels and coronary heart disease risk in 16 European population cohorts. Nat Genet. 2009, 41: 47-55. 10.1038/ng.269
Kathiresan S, Melander O, Anevski D, Guiducci C, Burtt NP, Roos C, Hirschhorn JN, Berglund G, Hedblad B, Groop L, Altshuler DM, Newton-Cheh C, Orho-Melander M: Polymorphisms associated with cholesterol and risk of cardiovascular events. N Engl J Med. 2008, 358: 1240-1249. 10.1056/NEJMoa0706728
Kathiresan S, Melander O, Guiducci C, Surti A, Burtt NP, Rieder MJ, Cooper GM, Roos C, Voight BF, Havulinna AS, Wahlstrand B, Hedner T, Corella D, Tai ES, Ordovas JM, Berglund G, Vartiainen E, Jousilahti P, Hedblad B, Taskinen MR, Newton-Cheh C, Salomaa V, Peltonen L, Groop L, Altshuler DM, Orho-Melander M: Six new loci associated with blood low-density lipoprotein cholesterol, high-density lipoprotein cholesterol or triglycerides in humans. Nat Genet. 2008, 40: 189-197. 10.1038/ng.75
Kathiresan S, Willer CJ, Peloso GM, Demissie S, Musunuru K, Schadt EE, Kaplan L, Bennett D, Li Y, Tanaka T: Common variants at 30 loci contribute to polygenic dyslipidemia. Nat Genet. 2009, 41: 56-65. 10.1038/ng.291
Willer CJ, Sanna S, Jackson AU, Scuteri A, Bonnycastle LL, Clarke R, Heath SC, Timpson NJ, Najjar SS, Stringham HM: Newly identified loci that influence lipid concentrations and risk of coronary artery disease. Nat Genet. 2008, 40: 161-169. 10.1038/ng.76
Samani NJ, Deloukas P, Erdmann J, Hengstenberg C, Kuulasmaa K, McGinnis R, Schunkert H, Soranzo N, Thompson J, Tiret L, Ziegler A: Large scale association analysis of novel genetic loci for coronary artery disease. Arterioscler Thromb Vasc Biol. 2009, 29: 774-780. 10.1161/ATVBAHA.108.181388
Sharrett AR, Sorlie PD, Chambless LE, Folsom AR, Hutchinson RG, Heiss G, Szklo M: Relative importance of various risk factors for asymptomatic carotid atherosclerosis versus coronary heart disease incidence: the Atherosclerosis Risk in Communities Study. Am J Epidemiol. 1999, 149: 843-852.
National Cholesterol Education Program (NCEP) Expert Panel on Detection Evaluation and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III): Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002, 106: 3143-3421.
Huang Y, von Eckardstein A, Wu S, Assmann G: Cholesterol efflux, cholesterol esterification, and cholesteryl ester transfer by LpA-I and LpA-I/A-II in native plasma. Arterioscler Thromb Vasc Biol. 1995, 15: 1412-1418.
Barter PJ, Ballantyne CM, Carmena R, Castro Cabezas M, Chapman MJ, Couture P, de Graaf J, Durrington PN, Faergeman O, Frohlich J, Furberg CD, Gagne C, Haffner SM, Humphries SE, Jungner I, Krauss RM, Kwiterovich P, Marcovina S, Packard CJ, Pearson TA, Reddy KS, Rosenson R, Sarrafzadegan N, Sniderman AD, Stalenhoef AF, Stein E, Talmud PJ, Tonkin AM, Walldius G, Williams KM: Apo B versus cholesterol in estimating cardiovascular risk and in guiding therapy: report of the thirty-person/ten-country panel. J Intern Med. 2006, 259: 247-258. 10.1111/j.1365-2796.2006.01616.x
Benn M, Nordestgaard BG, Jensen GB, Tybjaerg-Hansen A: Improving prediction of ischemic cardiovascular disease in the general population using apolipoprotein B: the Copenhagen City Heart Study. Arterioscler Thromb Vasc Biol. 2007, 27: 661-670. 10.1161/01.ATV.0000255580.73689.8e
Capell WH, Zambon A, Austin MA, Brunzell JD, Hokanson JE: Compositional differences of LDL particles in normal subjects with LDL subclass phenotype A and LDL subclass phenotype B. Arterioscler Thromb Vasc Biol. 1996, 16: 1040-1046.
St-Pierre AC, Ruel IL, Cantin B, Dagenais GR, Bernard PM, Despres JP, Lamarche B: Comparison of various electrophoretic characteristics of LDL particles and their relationship to the risk of ischemic heart disease. Circulation. 2001, 104: 2295-2299. 10.1161/hc4401.098490
Danesh J, Collins R, Peto R: Lipoprotein(a) and coronary heart disease. Meta-analysis of prospective studies. Circulation. 2000, 102: 1082-1085.
Smolders B, Lemmens R, Thijs V: Lipoprotein (a) and stroke: a meta-analysis of observational studies. Stroke. 2007, 38: 1959-1966. 10.1161/STROKEAHA.106.480657
Nielsen MS, Jacobsen C, Olivecrona G, Gliemann J, Petersen CM: Sortilin/neurotensin receptor-3 binds and mediates degradation of lipoprotein lipase. J Biol Chem. 1999, 274: 8832-8836. 10.1074/jbc.274.13.8832
Shi J, Kandror KV: Sortilin is essential and sufficient for the formation of Glut4 storage vesicles in 3T3-L1 adipocytes. Dev Cell. 2005, 9: 99-108. 10.1016/j.devcel.2005.04.004
Muendlein A, Geller-Rhomberg S, Saely CH, Winder T, Sonderegger G, Rein P, Beer S, Vonbank A, Drexel H: Significant impact of chromosomal locus 1p13.3 on serum LDL cholesterol and on angiographically characterized coronary atherosclerosis. Atherosclerosis. 2009, 206 (2): 494-9. 10.1016/j.atherosclerosis.2009.02.040
Samani NJ, Braund PS, Erdmann J, Gotz A, Tomaszewski M, Linsel-Nitschke P, Hajat C, Mangino M, Hengstenberg C, Stark K, Ziegler A, Caulfield M, Burton PR, Schunkert H, Tobin MD: The novel genetic variant predisposing to coronary artery disease in the region of the PSRC1 and CELSR2 genes on chromosome 1 associates with serum cholesterol. J Mol Med. 2008, 86: 1233-1241. 10.1007/s00109-008-0387-2
Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, Dixon RJ, Meitinger T, Braund P, Wichmann HE, Barrett JH, König IR, Stevens SE, Szymczak S, Tregouet DA, Iles MM, Pahlke F, Pollard H, Lieb W, Cambien F, Fischer M, Ouwehand W, Blankenberg S, Balmforth AJ, Baessler A, Ball SG, Strom TM, Braenne I, Gieger C, Deloukas P, Tobin MD, Ziegler A, Thompson JR, Schunkert H, WTCCC and the Cardiogenics Consortium : Genomewide association analysis of coronary artery disease. N Engl J Med. 2007, 357: 443-453. 10.1056/NEJMoa072366
Karvanen J, Silander K, Kee F, Tiret L, Salomaa V, Kuulasmaa K, Wiklund PG, Virtamo J, Saarela O, Perret C, Perola M, Peltonen L, Cambien F, Erdmann J, Samani NJ, Schunkert H, Evans A, MORGAM Project: The impact of newly identified loci on coronary heart disease, stroke and total mortality in the MORGAM prospective cohorts. Genet Epidemiol. 2009, 33: 237-246. 10.1002/gepi.20374
Asplund K, Karvanen J, Giampaoli S, Jousilahti P, Niemela M, Broda G, Cesana G, Dallongeville J, Ducimetriere P, Evans A, Ferrières J, Haas B, Jorgensen T, Tamosiunas A, Vanuzzo D, Wiklund PG, Yarnell J, Kuulasmaa K, Kulathinal S, MORGAM Project: Relative risks for stroke by age, sex, and population based on follow-up of 18 European populations in the MORGAM Project. Stroke. 2009, 40: 2319-2326. 10.1161/STROKEAHA.109.547869
Humphries AD, Streimann IC, Stojanovski D, Johnston AJ, Yano M, Hoogenraad NJ, Ryan MT: Dissection of the mitochondrial import and assembly pathway for human Tom40. J Biol Chem. 2005, 280: 11535-11543. 10.1074/jbc.M413816200
Paternoster L, Martinez Gonzalez NA, Lewis S, Sudlow C: Association between apolipoprotein E genotype and carotid intima-media thickness may suggest a specific effect on large artery atherothrombotic stroke. Stroke. 2008, 39: 48-54. 10.1161/STROKEAHA.107.488866
Pritchard JK, Stephens M, Donnelly P: Inference of population structure using multilocus genotype data. Genetics. 2000, 155: 945-959.
Marcovina SM, Albers JJ, Henderson LO, Hannon WH: International Federation of Clinical Chemistry standardization project for measurements of apolipoproteins A-I and B. III. Comparability of apolipoprotein A-I values by use of international reference material. Clin Chem. 1993, 39: 773-781.
Zambon A, Austin MA, Brown BG, Hokanson JE, Brunzell JD: Effect of hepatic lipase on LDL in normal men and those with coronary artery disease. Arterioscler Thromb. 1993, 13: 147-153.
Marcovina SM, Albers JJ, Gabel B, Koschinsky ML, Gaur VP: Effect of the number of apolipoprotein(a) kringle 4 domains on immunochemical measurements of lipoprotein(a). Clin Chem. 1995, 41: 246-255.
Fan JB, Oliphant A, Shen R, Kermani BG, Garcia F, Gunderson KL, Hansen M, Steemers F, Butler SL, Deloukas P, Galver L, Hunt S, McBride C, Bibikova M, Rubano T, Chen J, Wickham E, Doucet D, Chang W, Campbell D, Zhang B, Kruglyak S, Bentley D, Haas J, Rigault P, Zhou L, Stuelpnagel J, Chee MS: Highly parallel SNP genotyping. Cold Spring Harb Symp Quant Biol. 2003, 68: 69-78. 10.1101/sqb.2003.68.69
Keating BJ, Tischfield S, Murray SS, Bhangale T, Price TS, Glessner JT, Galver L, Barrett JC, Grant SF, Farlow DN: Concept, design and implementation of a cardiovascular gene-centric 50 k SNP array for large-scale genomic association studies. PLoS One. 2008, 3: e3583- 10.1371/journal.pone.0003583
Yu CE, Devlin B, Galloway N, Loomis E, Schellenberg GD: ADLAPH: A molecular haplotyping method based on allele-discriminating long-range PCR. Genomics. 2004, 84: 600-612. 10.1016/j.ygeno.2004.06.003
De la Vega FM, Lazaruk KD, Rhodes MD, Wenz MH: Assessment of two flexible and compatible SNP genotyping platforms: TaqMan SNP Genotyping Assays and the SNPlex Genotyping System. Mutat Res. 2005, 573: 111-135.
The International HapMap Consortium: The International HapMap Project. Nature. 2003, 426: 789-796. 10.1038/nature02168
Guan Y, Stephens M: Practical issues in imputation-based association mapping. PLoS Genet. 2008, 4: e1000279- 10.1371/journal.pgen.1000279
Scheet P, Stephens M: A fast and flexible statistical model for large-scale population genotype data: applications to inferring missing genotypes and haplotypic phase. Am J Hum Genet. 2006, 78: 629-644. 10.1086/502802
R: A language and environment for statistical computing. http://www.R-project.org
Benjamini Y, Hochberg Y: Controlling the false discovery rate: a practical and powerful approach to multiple testing. J Roy Statist Soc Ser B. 1995, 57: 289-300.
Jones PH, Davidson MH, Stein EA, Bays HE, McKenney JM, Miller E, Cain VA, Blasetto JW: Comparison of the efficacy and safety of rosuvastatin versus atorvastatin, simvastatin, and pravastatin across doses (STELLAR* Trial). Am J Cardiol. 2003, 92: 152-160. 10.1016/S0002-9149(03)00530-7
Jones P, Kafonek S, Laurora I, Hunninghake D: Comparative dose efficacy study of atorvastatin versus simvastatin, pravastatin, lovastatin, and fluvastatin in patients with hypercholesterolemia (the CURVES study). Am J Cardiol. 1998, 81: 582-587. 10.1016/S0002-9149(97)00965-X
Acknowledgements
Most importantly we would like to thank the CLEAR study participants, without whom this work would be impossible. We would also like to thank the CLEAR study staff. JR would like to thank the American Heart Association for postdoctoral award 09POST2250623 (declined). Major support for this study came from NIH R01 HL67406 and P01 HL30086. This research was also supported in part by resources from the VA Puget Sound Health Care System, Seattle, Washington.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
JR and GPJ designed the study and wrote the manuscript. JR performed all statistical analyses with guidance from RR, PJH, and GPJ. JER performed the experimental work. JKM and TSH recruited and phenotyped study participants. All authors contributed to the interpretation of the data and approved the final version of this manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Ronald, J., Rajagopalan, R., Ranchalis, J.E. et al. Analysis of recently identified dyslipidemia alleles reveals two loci that contribute to risk for carotid artery disease. Lipids Health Dis 8, 52 (2009). https://doi.org/10.1186/1476-511X-8-52
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-511X-8-52