
Abstract
Lowering plasma low density lipoprotein-cholesterol (LDL-C), blood pressure, homocysteine, and preventing platelet aggregation using a combination of a statin, three blood pressure lowering drugs such as a thiazide, a β blocker, and an angiotensin converting enzyme (ACE) inhibitor each at half standard dose; folic acid; and aspirin-called as polypill- was estimated to reduce cardiovascular events by ~80%. Essential fatty acids (EFAs) and their long-chain metabolites: γ-linolenic acid (GLA), dihomo-GLA (DGLA), arachidonic acid, eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA) and other products such as prostaglandins E1 (PGE1), prostacyclin (PGI2), PGI3, lipoxins (LXs), resolvins, protectins including neuroprotectin D1 (NPD1) prevent platelet aggregation, lower blood pressure, have anti-arrhythmic action, reduce LDL-C, ameliorate the adverse actions of homocysteine, show anti-inflammatory actions, activate telomerase, and have cytoprotective properties. Thus, EFAs and their metabolites show all the classic actions expected of the "polypill". Unlike the proposed "polypill", EFAs are endogenous molecules present in almost all tissues, have no significant or few side effects, can be taken orally for long periods of time even by pregnant women, lactating mothers, and infants, children, and adults; and have been known to reduce the incidence cardiovascular diseases including stroke. In addition, various EFAs and their long-chain metabolites not only enhance nitric oxide generation but also react with nitric oxide to yield their respective nitroalkene derivatives that produce vascular relaxation, inhibit neutrophil degranulation and superoxide formation, inhibit platelet activation, and possess PPAR-γ ligand activity and release NO, thus prevent platelet aggregation, thrombus formation, atherosclerosis, and cardiovascular diseases. Based on these evidences, I propose that a rational combination of ω-3 and ω-6 fatty acids and the co-factors that are necessary for their appropriate action/metabolism is as beneficial as that of the combined use of a statin, thiazide, a β blocker, and an angiotensin converting enzyme (ACE) inhibitor, folic acid, and aspirin. Furthermore, appropriate combination of ω-3 and ω-6 fatty acids may even show additional benefits in the form of protection from depression, schizophrenia, Alzheimer's disease, and enhances cognitive function; and serve as endogenous anti-inflammatory molecules; and could be administered from childhood for life long.
Similar content being viewed by others

Introduction
Cardiovascular diseases (CVD) are responsible for significant morbidity and mortality throughout the world. Studies revealed that smoking cessation, β-blockers, anti-platelet agents, angiotensin converting enzyme (ACE) inhibitors, and lipid lowering agents such as statins, each reduce the risk of vascular events to a moderate but important degree [1–9]. In addition, observational studies suggested lower rates of fractures and dementia with statins, and lower rates of cataracts with anti-oxidant vitamins, though these observations need to be confirmed by randomised trials [9]. The results of the MRC/BHF-HPS study led to the suggestion that using a combination of aspirin, β-blockers, statins, and ACE inhibitors could prevent about two-thirds to three-quarters of future vascular events [10]. It was suggested that a combination pill (called as "polypill") consisting of atorvastatin 10 mg or simvastatin 40 mg; three blood pressure lowering drugs such as a thiazide, a β-blocker, and an ACE inhibitor, each at half standard dose; folic acid 0.8 mg; and aspirin 75 mg could reduce coronary heart disease (CHD) events by 88% (95% confidence interval 84% to 91%) and stroke by 80% (71% to 87%), and if such a combination pill is taken from age 55 years of age, at least one third of people taking it, would on an average add about 11 years of life free from an CHD event or stroke [11].
Further support to the concept of polypill for the prevention of primary and secondary cardiovascular diseases proposed by Wald and Law [11] is provided by the work of Hippisley-Cox and Coupland [12] who examined the individual and combined effects of three of the polypill ingredients-statins, aspirin, and blood pressure lowering drugs. Their analysis of 11330 patients with CHD showed that all cause mortality is lower in those taking two or three drugs compared with those taking single agents. These findings are consistent with previous studies [13, 14] that showed that a combination of two drugs-aspirin and statin-is superior to either drug alone in the secondary prevention of CHD. However, it was also noted that synergistic effects are seen with two, but not three or four, drug combinations in secondary prevention of CHD.
But concerns have been raised about the adverse effects of such a polypill. For instance, β blockers are unsuitable for subjects with bronchial asthma, and some are intolerant to aspirin and develop significant gastrointestinal side effects. It may be necessary to closely monitor to detect serious adverse effects of statins, and renal failure due to ACE inhibitors and angiotensin-II receptor antagonists. Furthermore, the efficacy of aspirin in men is established [15], but its efficacy in women is not certain [16]. A recent study showed that 2.5 mg of folic acid (the proposed polypill dosage is 0.8 mg/day) was not associated with a reduction in stroke, coronary events, and death in patients who previously had a cerebral infarction despite a moderate reduction of total homocysteine during the 2 years of follow-up [17]. Another major concern about the primary prevention strategy of the "polypill" is related to the possibility that it might lead to medicalising of the population since, it is highly cost effective to treat individuals at high risk compared to those at lower risk that is much more expensive to treat in terms of gain in quality adjusted life years. Hence, it will be worthwhile to look for alternatives to "polypill" that is less expensive, more acceptable, less likely to cause side effects and does not lead to medicalising the population. I propose that a rational combination of ω-3 and ω-6 fatty acids may, in fact, be more beneficial compared to the "polypill" in the primary and secondary prevention of cardiovascular diseases.
Metabolism of essential fatty acids
Essential fatty acids (EFAs) are essential for survival and they cannot be synthesized in the body and hence, have to be obtained in our diet and thus, are essential [18–21]. There are two types of naturally occurring EFAs in the body, the ω-6 series derived from linoleic acid (LA, 18:2) and the ω-3 series derived from α-linolenic acid (ALA, 18:3). Both ω-6 and ω-3 series are metabolized by the same set of enzymes to their respective long-chain metabolites. While some of the functions of EFAs require their conversion to eicosanoids and other products, in majority of the instances the fatty acids themselves are active.
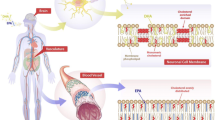
LA is converted to γ-linolenic acid (GLA, 18:3, n-6) by the action of the enzyme Δ6 desaturase (d-6-d) and GLA is elongated to form dihomo-GLA (DGLA, 20:3, n-6), the precursor of the 1 series of prostaglandins (PGs). DGLA can also be converted to arachidonic acid (AA, 20:4, n-6) by the action of the enzyme Δ5 desaturase (d-5-d). AA forms the precursor of 2 series of prostaglandins, thromboxanes and the 4 series of leukotrienes. ALA is converted to eicosapentaenoic acid (EPA, 20:5, n-3) by d-6-d and d-5-d. EPA forms the precursor of the 3 series of prostaglandins and the 5 series of leukotrienes (see Figure 1 for the metabolism of EFAs). AA and EPA give rise to their respective hydroxy acids, which, in turn, are converted to respective leukotrienes (LTs). In addition, AA, EPA, and DHA form precursor to anti-inflammatory compounds lipoxins, resolvins, and protectins (neuroprotectin D1 is one such compound derived from DHA) [22–27]. PGs, LTs, lipoxins (LXs), and resolvins are highly active, modulate inflammation, and are involved in several physiological and pathological processes [18]. In general, the term "EFAs" is used to refer to all unsaturated fatty acids: LA, GLA, DGLA, AA, ALA, EPA, and DHA; and the term polyunsaturated fatty acids (PUFAs) refer to GLA, DGLA, AA, EPA, and DHA. Although the terms EFAs and PUFAs are used interchangeably, it should be understood that all EFAs are PUFAs but all PUFAs are not EFAs. Only LA and ALA qualify to be EFAs; whereas GLA, DGLA, AA, EPA, and DHA are PUFAs. On the other hand, LA, GLA, DGLA, AA, ALA, EPA, and DHA are also called as LCPUFAs (long-chain polyunsaturated fatty acids). Many of the functions of EFAs are also brought about by PUFAs and EFA-deficiency states can be corrected to a large extent by PUFAs that suggests that PUFAs are "functional EFAs". Hence, in general, many authors use the terms EFAs and PUFAs interchangeably. This convention is followed in the present discussion.

Metabolism of essential fatty acids. Prostaglandins of 3 series are less pro-inflammatory compared to prostaglandins of 2 series. Resolvins are formed from both EPA and DHA and are known to have anti-inflammatory actions and participate in the resolution of inflammation. EPA can be converted to DHA. DHA can be retroconverted to EPA. It is estimated that about 30–40% of DHA can be retroconverted to EPA. The biochemical and/or clinical significance of this retroconversion of DHA to EPA are not known. ____ Indicates beneficial action in the form of increase in the synthesis, action, or remission of disease process. ____ Indicates decrease in the synthesis, action or enhancement of pathological process. ____ Indicates inhibition of HMG-CoA and ACE enzymes by PUFAs/EFAs.
Dietary sources of EFAs
EFAs: LA and ALA are present in human diet in abundant amounts and hence, EFA-deficiency is uncommon. In certain specific conditions such as total parenteral nutrition (TPN) and severe malabsorption occasionally EFA deficiency could be seen. The present TPN solutions contain adequate amounts of EFAs. The manifestations of EFA deficiency include: dry and scaly skin, hepatospleenomegaly, immunodeficiency, inappropriate water loss through the skin, dehydration, scalp dermatitis, alopecia, and depigmentation of hair. EFAs are widely distributed in normal human diet. The main dietary sources of EFAs are as follows.
Human breast milk is rich in all types of PUFAs [18] that explains why breast-fed children are healthier compared to bottle-fed. LA and ALA are present in significant amounts in dairy products, organ meats such as liver, and many vegetable oils such as sunflower, safflower, corn and soy. GLA is present in evening primrose oil at concentrations of 7–14% of total fatty acids; in borage seed oil it is 20–27%; and in black currant seed oil at 15–20%. GLA is also found in some fungal sources [18]. DGLA is found in liver, testes, adrenals, and kidneys. AA is present in meat, egg yolks, some seaweeds, and some shrimps. Average daily intake of AA is estimated to be ~100–200 mg/day that accounts for the total daily production of various PGs. EPA and DHA are present mainly in marine fish. Cow's milk contains very small amounts of GLA, DGLA and AA.
EFAs/PUFAs are unstable due to the presence of 2 or more double bonds in their structure. Substantial loss of EFAs/PUFAs occurs during food processing and hydrogenation of oils. Exposure to high temperatures and hydrogenation process causes denaturation of EFAs/PUFAs and their conversion to trans-fats that are harmful to the body [18]. Human diet was rich in ω-3 fatty acids in the early humans. But with the progress in industrialization and development of fast foods, the content of ω-3 fatty acids in human diet dwindled, whereas that of ω-6 fatty acids increased. The ratio of ω-3 to ω-6 fatty acids in the diet of early humans was >1, whereas this ratio is now about 10:1 to 20–25:1. It is recommended that the ratio between ω-3 to ω-6 fatty acids in the diet should be about 1 or >1 and preferably 2–3:1. This fall in the intake of ω-3 fatty acids, especially EPA and DHA in the last 50 years is believed to be responsible for the increasing incidence of atherosclerosis, CHD, hypertension, metabolic syndrome X, obesity, collagen vascular diseases and possibly, cancer. This is supported by the observation that increasing dietary α-linolenate/linoleate balance affected the ω-3/ω-6 ratio of brain phospholipid acyl chains and produced changes in general behavior as well as changes in sensitivities to drugs known to affect behavior, influenced LTs formation in polymorpho-nuclear leukocytes from AA and EPA and release of histamine from mast cells that could alter the severity of allergic and inflammatory responses. This increase in ω-3 fatty acids also resulted in an increased mean survival time of SHR-SP (spontaneously hypertensive-stroke prone) rats by lowering blood pressure and platelet aggregability, produced significant changes in Na+-K+-ATPase activity, and altered collagen-induced platelet aggregation and serotonin release in experimental animals. These results suggest that enhanced intake of ω-3 fatty acids is of significant benefit in various diseases.
Factors influencing the metabolism of EFAs
Dietary LA and ALA are metabolized by the same set of Δ6 and Δ5 desaturases and elongases to their respective metabolites (see Figure 1). These 2 fatty acids compete with one another for the same set of enzymes and Δ6 and Δ5 desaturases prefer ω-3 to ω-6. Oleic acid (OA, ω-9) that is not an EFA is also metabolized by the same desaturases. But, in view of the preference of these enzymes to LA and ALA, under normal physiological conditions, the metabolites of ω-9 are formed only in trivial amounts. Hence, presence of significant amounts of 20:3 ω-9, a metabolite of OA, in the cells and plasma indicates EFA deficiency that is utilized to detect the presence of EFA deficiency in patients, experimental animals and in vitro studies.
Of several factors that influence the activities of desaturases and elongases [18], saturated fats, cholesterol, trans-fatty acids, alcohol, adrenaline, and glucocorticoids inhibit Δ6 and Δ5 desaturases. Pyridoxine, zinc, and magnesium are necessary co-factors for normal Δ6 desaturase activity. Insulin activates Δ6 desaturase whereas diabetics have reduced Δ6 desaturase activity. The activity of Δ6 desaturase falls with age. Oncogenic viruses and radiation inhibit Δ6 desaturase. Total fasting, protein deficiency, glucose rich diets reduce the activity of Δ6 desaturase. A fat- free diet and partial caloric restriction enhances Δ6 desaturase. Activities of Δ6 and Δ5 desaturases are decreased in diabetes mellitus, hypertension, hyperlipidemia, and metabolic syndrome X. Trans-fats, saturated fatty acids, and cholesterol interfere with EFA metabolism and promote inflammation, atherosclerosis and coronary heart disease (CHD) [18]. This implies that trans-fats, saturated fats, and cholesterol have pro-inflammatory actions whereas EFAs and PUFAs possess anti-inflammatory properties. This explains why trans-fats, saturated fats, and cholesterol are pro-atherogenic, whereas EFAs/PUFAs, especially ω-3 fatty acids are anti-atherogenic. The ability of trans-fats, saturated fats, and cholesterol to interfere with the formation of AA, EPA, and DHA from dietary LA and ALA could lead to decreased formation of LXs, resolvins, PGI2 (prostacyclin), PGI3, and other beneficial eicosanoids that prevent platelet aggregation, leukocyte chemotaxis and activation. LXs and resolvins decrease the formation of pro-inflammatory cytokines, and produce vasodilatation, events that prevent or arrest atheroslcerosis. In contrast, trans-fats, saturated fats, and cholesterol may directly activate leukocytes, induce the generation of free radicals and enhance the production and release of pro-inflammatory cytokines that facilitate atherosclerosis [18]. Trans-fats, saturated fats, and cholesterol directly activate leukocytes and macrophages to induce them to produce free radicals and pro-inflammatory cytokines:
IL-6, TNF-α, IL-1, IL-2, and MIF (macrophage migration inhibitory factor). This action of trans-fats, saturated fats, and cholesterol is in addition to their ability to suppress metabolism of EFAs to their respective long-chain metabolites. It is possible that trans-fats, saturated fats, and cholesterol may also have the ability to inhibit the formation of LXs, resolvins, PGI2, and PGI3. These studies suggest that EFAs, especially EPA and DHA are cytoprotective to endothelial cells, whereas trans-fats, saturated fats, and cholesterol produce endothelial dysfunction. AA, EPA, and DHA augment nitric oxide generation from endothelial cells [18] and thus help in the prevention of endothelial dysfunction. In contrast, trans-fats, saturated fats, and cholesterol produce endothelial dysfunction and thus, inhibit eNO production. Furthermore, NO quenches superoxide anion and thus, prevents the cytotoxic action of superoxide anion and protects endothelial cells from free radical-induced damage. This implies that for endothelial cells to be healthy they need adequate amounts of AA, EPA, and DHA so that they can generate physiological amounts of eNO not only to prevent pathological platelet aggregation and atherosclerosis but also to protect themselves from the cytotoxic actions of free radicals.
Furthermore, NO reacts with PUFAs to yield their respective nitroalkene derivatives that can be detected in plasma. These nitroalkene derivatives, termed as nitrolipids, produce vascular relaxation, inhibit neutrophil degranulation and superoxide formation, and inhibit platelet activation, and show anti-atherosclerotic properties [18]. Thus, there appears to be a close interaction between EFAs and their products and trans-fats, saturated fats, and cholesterol with regard to the ability of endothelial cells to produce PGI2, PGI3, NO, and other anti-atheroslcerotic and beneficial molecules.
Actions of PUFAs that qualify them to be the endogenous HMG-CoA reductase and ACE enzyme inhibitors, anti-arrhythmic, anti-hypertensive, anti-atherosclerotic, anti-inflammatory, cytoprotective, and cardioprotective molecules
Action on HMG-CoA reductase
PUFAs are potent inhibitors of HMG-CoA reductase enzyme and similar to statins are useful in the treatment of hyperlipidemias [28–33]. Statins enhance plasma AA levels and decrease the ratio of EPA to AA significantly [29, 30], and enhance the formation of prostacyclin (PGI2) [32]. In fact, statins and PUFAs have many overlap actions such as inhibition of IL-6 and TNF-α production and NF-κB activation, increasing the synthesis of endothelial nitric oxide (eNO); and both are anti-inflammatory in nature [18, 33–37]. In addition, a close interaction exists between NO and COX enzymes attesting to the fact that statins, PUFAs, and NO have positive and negative influences among themselves [38]. Both statins and PUFAs are useful in atherosclerosis, coronary heart disease, osteoporosis, stroke, Alzheimer's disease, and inflammatory conditions such as lupus [18, 39–52]. These evidences suggest that PUFAs mediate many, if not all, actions of statins [33] and this could be one mechanism by which they lower cholesterol levels. Recent studies revealed that statins augment concentrations of LXs in the heart [53, 54] lending support to this concept. Furthermore, when a combination of statins and PUFAs are given together a synergistic beneficial effect was seen in patients with combined hyperlipemia [55–58]. But statins cannot be given during pregnancy, whereas PUFAs have been recommended during pregnancy, lactation and infancy to improve brain growth and development, and cognitive function [18–21], [59–67], though some studies did not show improvement in cognition [68, 69]. Nevertheless, these studies reported that supplementation of PUFAs to pregnant and lactating women and infants is safe and without any side effects.
PUFAs modulate renin formation, ACE activity and endothelial nitric oxide generation
Angiotensin converting enzyme (ACE) inhibitors are useful to lower blood pressure [11]. Renin, a proteolytic enzyme, is produced and stored in the granules of the juxtaglomerular cells in the kidney. Renin acts on angiotensinogen (a circulating α2 globulin made in the liver) to form the decapeptide angiotensin-I (Ang-I). Ang-I is transformed by ACE to angiotensin-II (Ang-II). Ang-II controls blood pressure and regulates body fluid volume by modulating renin-angiotensin-aldosterone system. ACE is present in the uterus, placenta, vascular tissue, heart, brain, adrenal cortex and kidney, leukocytes, alveolar macrophages, peripheral monocytes, neuronal cells and epididymal cells [70]. Angiotensin receptors are AT1 and AT2. AT1 exists as two subtypes α and β. Actions of Ang-II are mediated by the AT1 receptor. Angiotensinases, present in several tissues, destroy Ang-II (half-life approximately 1 minute), while the half-life of renin is about 10 to 20 minutes. In addition to circulating renin-angiotensin, many tissues have a local renin-angiotensin system and thus, have the ability to produce Ang-II. Locally generated Ang-II is involved in the modulation of growth and function of many tissues including vascular smooth muscle.
Linseed oil (which contains approximately: 19% oleic acid, 24% LA, and 47% ALA) fed experimental animals showed significantly lower renal venous renin secretion rates relative to the saturated fat-fed control group. Dietary enrichment with 20 energy % PUFA lowered renin secretion by a prostaglandin-independent mechanism that might have contributed to the lower blood pressures observed compared with the saturated fat-fed control group [71]. Dietary supplementation with 3 gm/day of LA and 32 mg/day of GLA to pregnant and non-pregnant subjects showed that the diastolic pressor response to angiotensin-II was significantly less in the pregnant subjects compared to the non-pregnant subjects, suggesting that PUFAs modulate vascular tissue responses to angiotensin-II. This decreased response to angiotensin-II could be due to increased formation of PGE1 and PGI2 in fatty acid supplemented pregnant subjects [72, 73]. In a model of hypertension induced by continuous infusion of angiotensin-II in the rat, subcutaneous administration of LA and EPA and DHA were found to be equally potent in reducing, by half, the rise in systolic blood pressure induced by angiotensin-II and these anti-hypertensive effects were not accompanied by any changes in the renal synthesis of PGI2 or PGE2. Furthermore, indomethacin, a potent inhibitor of PGI2 but not of PGE2 synthesis could only partially neutralize the anti-hypertensive effects of LA and EPA/DHA, emphasizing that the anti-hypertensive effects are independent of the PG system [74]. This idea is reinforced by the observation that LA and AA inhibit renin, and thus, the overall activity of the renin-angiotensin-aldosterone cascade could be modified by alterations of plasma fatty acid concentrations [75, 76], though in two-kidney, one-dip hypertensive animals, pretreatment with indomethacin did not alter hypotensive response to LA despite the fact that LA infusion lowered blood pressure in high renin but not in low renin states and the reduction in blood pressure was not related to inhibition circulating renin or to alterations of endogenous PG biosynthesis [77]. These evidences suggest that PUFAs have modulatory influence on renin secretion and action and yet times independent of both renin secretion and PG formation. Since the anti-hypertensive actions of PUFAs seem to be independent of formation of PGs, it is likely that fatty acids themselves are able to bring about this action and/or converted to form lipoxins, resolvins and protectins that have anti-inflammatory action and also possibly, anti-hypertensive action. But this proposal needs to be verified and confirmed. Yet another possibility is that the PUFAs supplemented are incorporated into the cell membrane phospholipid fraction that is able to enhance the synthesis of eNO that is a potent vasodilator and platelet anti-aggregator.
Previously, I showed that PUFAs inhibit leukocyte ACE activity [78]. Of all the fatty acids tested, EPA was the most effective (EPA > ALA > DHA > GLA > LA > AA), whereas AA was the least effective when their ability to inhibit purified ACE activity was tested. DHA and EPA were the most effective fatty acids in inhibiting the leukocyte ACE activity (EPA > DHA > ALA = AA > LA > GLA). On the other hand, PGs (PGE1, PGE2, PGI2 and PGF2α), and free radicals: superoxide anion, hydrogen peroxide, and hydroxyl radical showed marginal (~20%) inhibitory action on ACE activity. In contrast, NO (nitric oxide) showed powerful inhibitory action on ACE activity [78], whereas PUFAs enhanced endothelial nitric oxide (eNO) generation [36, 47, 79]. The effects of PUFAs on ACE activity, and NO generation and the inability of free radicals and PGs to suppress ACE activity are interesting since there is a close interaction between platelets, leukocytes and endothelial cells that may have relevance to their involvement in CVD (cardiovascular diseases). For instance, under normal conditions, endothelial cells produce adequate amounts of PGE1 from DGLA; PGI2 from AA; LXs and resolvins from AA, EPA and DHA; and NO from L-arginine such that the pro-inflammatory and pro-atherosclerotic events such as hemodynamic forces, hyperlipidemia, hypertension, smoking are successfully abrogated. These factors induce the expression of pro-inflammatory genes that initiate and accelerate atherosclerosis at the points of shear stress, enhance infiltration of intima by leukocytes and macrophages, cause low-level activation of NF-κB and elevated expression of VCAM-1 and ICAM-1, IL-1, IL-6, MCP-1, as well as antioxidant genes glutathione peroxidase and glutathione-S- transferase 2, and pro-inflammatory eicosanoids such as TXA2, PGE2, PGF2α, LTs, and other PGs, TXs, and LTs, and increased production and release of free radicals and UCP (uncoupling proteins) expression occurs in endothelial cells, platelets, and leukocytes in atherosclerosis-susceptible regions, and endothelial cells themselves may show changes in cell shape and proliferation. These events can be prevented and atherosclerosis process and the onset of CVD can be arrested if the production of PGE1, PGI2, PGI3, LXs, resolvins, NO, and anti-inflammatory cytokines such as IL-4, IL-10, TGF-β by endothelial cells is adequate, provided there are adequate stores of respective precursors of various PUFAs and L-arginine and their respective enzymes. This suggests that under physiological conditions a delicate balance is maintained between pro- and anti-inflammatory and pro and anti-atherosclerotic factors and when this balance is tilted more towards the former atherosclerosis and CVD occurs (reviewed in [80]).
In addition, these results suggest that when tissue concentrations of PUFAs are low, the activity of ACE will be high resulting in increased formation of angiotensin-II and a simultaneous decrease in eNO. In this context, it is important to note that transgenic rats overexpressing human renin and angiotensinogen genes (dTGR) develop hypertension, inflammation, and renal failure, and showed specific renal P450-dependent AA metabolism changes that led to decreased formation epoxy-eicosatrienoic acids (5,6-, 8,9-, 11,12- and 14,15-EETs) and hydroxyeicosa-tetraenoic acids (19- and 20-HETEs). Both EETs and HETEs inhibit IL-6 and TNF-α-induced activation of NF-κB and prevent vascular inflammation [81], suggesting that AA and other PUFAs not only regulate ACE activity and Ang-II levels in the tissues but also possess anti-inflammatory properties by generating anti-inflammatory metabolites.
AA, EPA, and DHA are converted in the presence of aspirin to epi-lipoxins, lipoxins, and resolvins that possess potent anti-inflammatory actions (reviewed in [18]). Epi-lipoxins enhance the formation of eNO [18, 21, 22]. NO blocks the interaction between leukocytes and the vascular endothelium and also stimulates the formation of PGI2, a potent vasodilator and platelet anti-aggregator, from AA [82]. This suggests that the beneficial actions of aspirin could be attributed not only to its ability to enhance the formation of PGI2 and suppress the synthesis of TXA2 but also to the formation of epi-lipoxins and eNO [20, 21, 82]. Thus, PUFAs regulate renin formation and action, inhibit angiotensin-II formation by its action on ACE activity, enhance eNO formation, and form precursors to beneficial biologically active molecules such as PGE1 (from DGLA), PGI2 (from AA), PGI3 (from EPA), lipoxins (from AA, EPA, and DHA), resolvins (from AA, EPA and DHA), protectins (from DHA), and 5,6-, 8,9-, 11,12- and 14,15-EETs and hydroxyeicosa-tetraenoic acids (19- and 20-HETEs) (from AA), and thus serve as endogenous regulators of vascular tone, platelet aggregation, and blood pressure.
Effects on platelets and other hemostatic indices
Aspirin is effective in the prevention and treatment of acute myocardial infarction (AMI) and in the secondary prevention of CVD [83], though the efficacy of aspirin in women is not certain [16]. One of the important constituents of the "polypill" is aspirin (75 mg). Studies revealed that low dose aspirin not only reduced risk of heart disease, but also reduced the incidence of lung, colon, and breast cancer [84]. Aspirin inhibits nuclear factor NF-κB transcription, blocks prostaglandin (PG) and thromboxane (TX) synthesis (TXA > PGI2). Aspirin does not inhibit the production of proinflammatory mediators such as leukotrienes (LTs). Although blockage of PGs and TXs accounts for many of aspirin's pharmacologic properties, recent studies revealed that aspirin evokes the formation of 15-epi-LXA4 by the acetylated PGHS-2 [prostaglandin G/H synthase (cyclooxygenase)] and 5-lipoxygenase enzymes as a result of endothelial cell-leukocyte interactions [85]. LXA4 inhibits polymorphonuclear leukocyte transmigration, modulate adhesion to endothelial cells, and inhibit chemotaxis of PMN and eosinophils. Thus, many beneficial actions of aspirin could be attributed to the formation of LXA4. In this context, it is noteworthy that AA, EPA, and DHA when used in appropriate concentrations and ratio can reproduce many of the beneficial actions of aspirin.
Both EPA and DHA, when given orally, are rapidly incorporated into platelets and compete with AA for the 2-acyl position of membrane phospholipid and as substrate for the cyclo-oxygenase (CO) and lipoxygenase (LO) enzymes. As a result, when stimulated, such platelets produce less amounts of TXA2 and more of TXA3 that is less potent in inducing platelet aggregation and thrombosis [86]. Increased intake of fish oil, a rich source of EPA and DHA, produces a lower platelet count, less platelet aggregation, a longer bleeding time, higher urinary PGI2 metabolites, and lower concentrations of thromboxane metabolites compared to those who were on Western diet [87, 88], effects that are similar to those of low-dose aspirin and qualify ω-3 EPA and DHA to be termed as an "endogenous aspirin". In general, though EPA and DHA do not have a very significant effect on blood lipids (except to lower plasma triglycerides and VLDL with no significant action on HDL-C levels), fibrinolysis and on the activity of plasminogen activity inhibitor-type-1 (PAI-1), still are effective in preventing overall mortality from CVD [18, 21, 80, 89–94].
It is believed that production of pro-inflammatory eicosanoids from AA and/or decreased synthesis of anti-inflammatory and beneficial eicosanoids from EPA/DHA could predispose an individual to cardiovascular risk and stroke. Thus, it is thought that products of AA such as TXA2, PGEs, PGFαs, and LTs contribute or initiate the process of atherosclerosis in coronary arteries and cause CHD. In contrast, it has been proposed that beneficial products or less harmful products formed from EPA and DHA are less likely to cause atherosclerosis and CHD or even regress atheroma and prevent CHD. Although this is an attractive hypothesis, hard data in support of this proposal are not forthcoming. In this context, it should be noted that not all products of AA are harmful. For instance, PGI2 formed from AA is a potent vasodilator and platelet anti-aggregator and prevents atherosclerosis and has anti-arrhythmic action [95].
Similarly, DGLA, another ω-6 fatty acid that is the precursor of AA, gives rise to PGE1, which is a vasodilator, platelet anti-aggregator and anti-arrhythmic molecule [96, 97]. These data emphasize the complexities involved in making generalizations about ascribing negative role to ω-6 fatty acids and their products in CVD. Furthermore, AA forms precursor to LXs and resolvins (See Figure 1) that have beneficial actions in resolving inflammation. In addition, Harris et al [98] noted that pooling of data from case-control or prospective cohort studies showed that none of the individual fatty acids computed across datasets were significantly different between cases and controls. EPA was 8.2% lower (p = 0.06), DHA was 8% lower in cases compared with controls, whereas DPA (docosapentaenoic acid) was virtually identical in both controls and CHD patients. In contrast and contrary to expectations, AA was 8.5% lower in cases compared to controls. The unexpected finding that AA concentrations were lower in patients with CHD suggests that it is the deficiency of AA rather than its excess that predisposes to CHD events. In addition, data from the Health Professionals' Follow-up Study [99] revealed that while there is an inverse relationship between ω-3 fatty acid intake and future risk for CHD, higher intakes of ω-6 fatty acids did not diminish the beneficial effects of ω-3 fatty acids suggesting that the absolute intakes of the ω-3 fatty acids are more important than the ratio between ω-3 fatty acids and ω-6 fatty acids. In this context, the interaction(s) between ω-3 and ω-6 fatty acids are significant.
Interaction(s) between ω-3 and ω-6 fatty acids and its relevance to CHD/CVD
In a case control study of new angina pectoris and first acute myocardial infarction, a progressive inverse relation between adipose tissue LA and the estimated relative risk of CHD was noted [100]. Wood, et al [101] observed that low concentrations of DGLA in adipose tissue showed a more significant relation to new CHD than did LA. In an extension of this study, it was noted that there is a progressive inverse relations between adipose LA and platelet-membrane EPA and the estimated relative risk of angina pectoris. These relations were statistically independent of each other and traditional CHD risk factors [100–102].
Luostarinen, et al [103] noted that the percentage of palmitic acid and LA were significantly higher and the percentage of AA and of all the other major PUFAs, both ω-3 and ω-6, was significantly lower in the total phospholipid fraction of human coronary arteries of those who had sudden cardiac death due to CHD. Felton, et al [104] reported that the concentrations of all fatty acids were increased at the edge of disrupted plaques compared with the center, but as a proportion of total fatty acids, ω-6 were lower. These results suggest that ω-6 fatty acids have a significant role in CHD and it is likely that some of the inconsistent results obtained in some studies with EPA and DHA could be attributed to inadequate provision or utilization of ω-6 fatty acids, DGLA and AA. It is possible that there is a close interaction between ω-3 and ω-6 fatty acids, which could influence one's susceptibility or resistance to CHD. In this context, it is interesting to note that EPA/DHA readily get incorporated into the atheromatous plaque, and patients treated with fish oil had more thick fibrous caps and no signs of inflammation compared with plaques in patients in the control and sunflower oil groups. Furthermore, the number of macrophages in plaques from patients receiving fish oil was lower than in the other two groups, suggesting that atherosclerotic plaques readily incorporate ω-3 PUFAs from fish-oil supplementation, inducing changes that can enhance stability of atherosclerotic plaques [105].
Studies revealed that ω-3 and ω-6 fatty acids interact with each other in such a way that one potentiates the metabolism of the other. For instance, in perfused vascular tissue, DGLA increases the conversion of EPA to PGI3, a potent vasodilator and platelet anti-aggregator [106], whereas AA augmented the conversion of EPA to PGI3 in the tissues [107–109]. EPA inhibits the activity of the enzyme Δ5 desaturase that results in an increase in the concentrations of DGLA in the tissues (especially in the endothelial cells). This increase in tissue levels of DGLA could enhance the formation of PGE1, a vasodilator and platelet anti-aggregator (see Figure 1). Thus, EPA can indirectly enhance the formation of PGE1. Furthermore, even the beneficial action of statins (HMG-CoA reductase inhibitors) and glitazones (PPARs agonists) seem to be mediated by EFAs and their metabolites such as LXs, resolvins, and neuroprotectins [28–33], [110–114], which are potent anti-inflammatory molecules [18, 21, 115–117]. Studies did suggest that ω-3 fatty acids decreased the levels of pro-inflammatory cytokines, and enhance that of IL-10, an anti-inflammatory cytokine [118–120]. This close interaction between ω-3 and ω-6 fatty acids and their ability to modify inflammatory markers, production of PGI2, PGE1, PGI3, LXs, resolvins, neuroprotectins, NO, nitrolipids, and the action of statins and glitazones on EFA metabolism and NO explains the relationship between various fatty acids and CHD and stroke (Figure 1).
PUFAs in renal function
One of the components suggested to be included in the "polypill" is a thiazide, a diuretic and an anti-hypertensive drug. If PUFAs are to be considered to function as an endogenous "polypill", then they (PUFAs) should show beneficial actions on renal function.
Healthy volunteers given EPA (3.9 gm) and DHA (2.4 gm) per day for 6 weeks showed significant increase in renal plasma flow, glomerular filtration rate, decrease in renal vascular resistance, and an increase in excretion of PGE3 with no change in blood pressure and heart rate [121]. Diet rich in evening primrose oil (a rich source of GLA and LA) and safflower oil decreased proteinuria, glomerular sclerosis, and tubular abnormalities in diabetic rats, and showed increased ratio of renal cortical production of 6-keto-PGF1α (a metabolite of PGI2) to TXB2 with no significant changes in plasma lipid composition. In contrast, fish oil feeding decreased plasma lipids and lowered 6-keto-PGF1α/TXB2 ratio without any effect on renal disease in diabetic rats [122]. Singer et al [123] observed that spontaneously hypertensive rats had significantly lower systolic blood pressure when fed fish oil (EPA and DHA), evening primrose oil (a rich source of GLA), and fish oil + evening primrose oil, suggesting that a combination of GLA, EPA, and DHA produces optimal beneficial actions with regard to renal indices and blood pressure. Vaskonen et al [124] reported that fish oil prevented rise in blood pressure induced by high-salt diet in stroke-prone spontaneously hypertensive rats. This beneficial effect on blood pressure was associated with a decrease in TXB2 formation by 75% and an increase in plasma and renal ω-3 fatty acid content.
Furthermore, EPA/DHA suppressed mesangial cell proliferation, arrested progression of IgA nephropathy, and protected against cyclosporine-induced renal damage [125–127]. These results suggest availability of optimal amounts of GLA and EPA/DHA is necessary to reduce blood pressure and preserve renal function in diabetic and hypertensive rats. Studies performed with 5/6 renal ablation rat model that developed hypertension, albuminuria, and a decline in glomerular filtration rate had significantly less glomerulosclerosis and dyslipidemia when supplemented with fish oil and flax seed oil (rich in ALA) compared with the control group at 10 and 20 wk post-surgery [128, 129]. Thus, PUFAs may show actions similar to those observed with conventional, synthetic diuretics. These beneficial actions of PUFAs can be attributed to the formation of beneficial PGA, PGE3, PGI2, PGI3, and recently identified resolvins and protectins and decrease in the production of TXA2 and LTs [130]. It is interesting that diuretic furosemide enhances endothelial synthesis and release of bradykinin and related kinins that, in turn, stimulates endothelial PGI2 formation via B2 kinin receptor activation [131] and COX-2 derived PGs interact with the renin-angiotensin system to regulate renal function [132].
PUFAs and parasympathetic nervous system
Previous studies showed that multiple-blood pressure lowering drugs have additive effects, which led Wald and Law to suggest that a β-blocker need to be added to the "polypill" composition. This is in addition to the presence of a diuretic (such as a thiazide) and an ACE-inhibitor [11]. Autonomic function is an important factor that regulates heart rate, blood pressure and cardiac rhythm. Hence, it is expected that addition of β-blocker to the composition of "polypill" will reduce sympathetic tone, blood pressure and heart rate.
Autonomic function is assessed by the measurement of heart rate variability (HRV) and the evaluation of baroreflex sensitivity (BRS). HRV reflects the physiological levels of tonic autonomic regulation, whereas BRS indicates the capacity of reflex autonomic regulation. Both low HRV and low BRS are associated with increased cardiovascular risk. Vagal stimulation by a release of acetylcholine (ACh) and adrenergic stimulation mediated by norepinephrine and epinephrine regulate the autonomic function and thus the variations in HRV and BRS. Several studies revealed that ω-3 fatty acids reduce the risk of sudden death by preventing life-threatening cardiac arrhythmias and by significantly increasing HRV [133]. Furthermore, a direct positive correlation was noted between the content of DHA in cell membranes and HRV index suggesting an anti-arrhythmic effect of ω-3 fatty acids [134]. Since increased parasympathetic tone is responsible for increase in ventricular fibrillation threshold and protects against ventricular arrhythmias, it is likely that EPA/DHA supplementation enhances parasympathetic tone. This is supported b the observation that EPA/DHA supplementation increases hippocampal ACh levels, the principal neurotransmitter of parasympathetic nerves [135]. Hence, it is likely that EPA/DHA supplementation increases the brain ACh levels leading to an increase in the parasympathetic tone and so an increase in HRV and protection from ventricular arrhythmias. Similar to EPA/DHA, AA also augments ACh release [136, 137], and thus, PUFAs enhance parasympathetic tone resulting in an increase HRV and prevention of ventricular arrhythmias.
Vagus nerve stimulation also inhibits TNF synthesis in liver and ACh significantly attenuated the release of pro-inflammatory cytokines: TNF-α, IL-16, IL-1β, and IL-18 but not anti-inflammatory cytokine IL-10 by stimulated macrophages in vitro and in vivo [138–140]. Thus, one mechanism by which PUFAs suppress inflammation could be by augmenting the release of ACh and enhancing the parasympathetic tone.
Since, normally a balance is maintained between parasympathetic and sympathetic tones, it is reasonable to suggest that whenever parasympathetic tone (vagal tone) is enhanced sympathetic tone is reduced (akin to blocking of β-receptors as it occurs in instances of use of β-blockers). Thus, indirectly PUFAs may function like β-blockers.
Folic acid and PUFAs
Hyperhomocysteinemia is a risk factor for cardiovascular diseases, and it may interact with hypertension and an unfavorable cholesterol profile to alter the risk of CVD. Hence, folic acid (0.8 mg/day) has been added as a component of the "polypill". It is important to note that folic acid increases concentrations of ω-3 PUFAs which could reduce the risk of thrombosis and CVD [141–143]. It was observed that some of the adverse effects induced by folic acid deficiency could be overcome by supplementing with ω-3 EPA and DHA [144], and in folic acid deficiency states the plasma and brain concentrations of PUFAs are decreased [145]. These results imply that some, if not all, actions of folic acid are mediated by PUFAs.
Thus, PUFAs, when given in appropriate dose and combination (containing EPA, DHA and possibly, GLA, DGLA and AA) show all the qualities of the suggested "polypill", viz., aspirin-like action, inhibit the activities of HMG-CoA and ACE enzymes, possess diuretic and anti-hypertensive actions, and indirectly show β-blocker-like action. In addition to these useful actions, PUFAs also have other beneficial actions as described below.
PUFAs inhibit cholesteryl ester transfer protein (CETP) activity
HDL-cholesterol (high-density lipoprotein- cholesterol, HDL-C) is an independent risk factor for CHD. Higher plasma HDL-C is associated with a decreased incidence of CHD [146] that led to the suggestion that therapeutic strategies that raise HDL-C could be of benefit in preventing CHD by increasing the movement of cholesterol from the periphery back to the liver (the so-called reverse cholesterol transport or RCT pathway).
CETP is a hydrophobic plasma glycoprotein, mainly synthesized in the liver, possessing the unique ability to facilitate the transfer of cholesteryl ester (CE). CETP circulates in the blood, bound predominantly to HDL. CETP mediates the transfer of cholesteryl esters from HDL to VLDL and LDL in exchange for triglycerides and promotes the transformation of HDL2 to HDL3, an action that could promote reverse cholesterol transport. CETP inhibition produces an increase in HDL by markedly delaying catabolism of apoA-I and A-II [147], an action that increases reverse cholesterol transport. These actions suggest that CETP inhibition could prevent atherosclerosis and prevent CHD [148–150].
In healthy, normolipidemic men, it was observed that lipid-lowering diet rich in monounsaturated fatty acid (oleic acid) decreased CETP concentrations to a significant degree [151]. In HepG2 cells, 0.5 mM of AA, EPA, and DHA reduced the levels of CETP mRNA by more than 50% of the control levels with a corresponding significant decrease in the CETP mass [152]. A significant negative correlation was found between plasma CETP activity and monounsaturated fatty acid content of plasma phospholipids or free PUFAs including ω-3 fatty acids, suggesting that PUFAs suppress CETP activity [153].
Torcetrapib, a small molecule inhibitor of CETP, is very effective at raising HDL-C and apolipoprotein A-I and decreasing levels of LDL-C and apolipoprotein-B-100 and also showed favourable effects on increasing the size of HDL-and LDL particles. In patients with familial hypercholesterolemia, torcetrapib with atorvastatin as compared with atorvastatin alone did not result in reduction of progression of atheroslcerosis as measured by carotid arterial-wall thickness despite a significant increase in HDL-C levels and decrease in levels of LDL-C and triglycerides. In fact, it was observed that administration of torcetrapib with atorvastatin was associated with progression of atherosclerosis [154], and an increase in blood pressure with no significant decrease in the progression of coronary atherosclerosis [155]. These results with torcetrapib + atorvastatin suggest that simultaneous inhibition of CETP and HMG-CoA reductase enzyme leads to an elevation of plasma HDL-C, and decrease in LDL-C and triglycerides and cholesterol but it does not arrest progression of atherosclerosis. In contrast, PUFAs, especially ω-EPA and DHA, not only inhibit CETP and HMG-CoA reductase enzyme, lower plasma triglycerides, cholesterol, and LDL-C with little or no change in HDL-C but also are effective in arresting atherosclerosis and preventing CHD [156–165]. In contrast to the results with torcetrapib + atorvastatin, Yokoyama et al [166] reported that a combination of ethyl EPA + 10 mg of pravstatin or 5 mg of simvastatin prevented major coronary events and especially non-fatal coronary events in Japanese hypercholesterolemic patients with a mean period of follow up of 4.6 years. It is interesting to note that the benefits were in addition to statin treatment, and fish oil was found to be safe and well tolerated. These results once again confirm that EPA and DHA are of benefit in the prevention and treatment of cardiovascular diseases. Thus, PUFAs appear to be superior to CETP and statins in the prevention of CVD despite the fact that they do not necessarily increase plasma HDL-C levels.
EPA, DHA, and PGI2 function as endogenous anti-arrhythmic molecules
There is reasonable evidence to suggest that EPA/DHA and PGI2 have anti-arrhythmogenic effects. Various PUFAs and PGs are present in the heart including SA (sinoatrial) node [167, 168]. I showed that PGE1, PGE2, PGI2, and TXB2 modify contractions of isolated rat cardiac muscle cells and increased the amount of 45Ca2+ exchanged by non-beating cells [169]. These results led to the suggestion that PGI2 could be an endogenous anti-arrhythmic molecule [170]. It was reported that PUFAs increased the electrical threshold for the induction of ventricular fibrillation that could reduce the risk of developing malignant cardiac arrhythmias. Mitochondrial dysfunction induced by EFA deficiency could be eliminated by the presence of normal levels of the essential fatty acids in the ω-3-enriched mitochondrial membrane phospholipids [171] that may account for the ability of EPA/DHA to decrease cardiac arrhythmias during myocardial ischemia. The recovery of mitochondrial energy metabolism and myocardial pump function during reperfusion is significantly better in ω-3 PUFA-enriched hearts, suggesting that EPA and DHA limit myocardial injury during ischemia and reperfusion [172].
EPA and DHA (at 2–10 microM) reduced the contraction rate of spontaneously beating, isolated, neonatal rat cardiac myocytes [173] without a significant change in the amplitude of the contractions. Both CO- and LO-inhibitors and antioxidants did not alter the effect of the fatty acids. The inhibitory effect of EPA and DHA on the contraction rate was similar to that produced by the class I antiarrhythmic drugs. It was also reported that lysophosphatidylcholine (LPC)- or acylcarnitine-induced arrhythmias were completely blocked by EPA, DHA, ALA, AA, and LA by inhibiting the electrical automaticity/excitability of the cardiac myocytes. Studies using whole-cell patch-clamp technique in cultured neonatal rat ventricular myocytes revealed that EPA, DHA, and to a limited extent AA can produce a concentration dependent suppression of ventricular, voltage-activated Na+ currents that may explain their anti-arrhythmic actions in vitro and in vivo [174–176].
PUFAs modulate telomere and telomerase activity
Telomere, the genetic segment that appears at the end of the chromosomes, has the special property of protecting these ends. Telomerase is the enzyme that adds telomere repeats to the ends of the chromosomes with the use of a dedicated RNA template. Inactivation of telomerase leads to telomere shortening and eventual senescence of the cells. Telomerase consists of two principal subunits: telomerase reverse transcriptase (TERT), the protein catalytic subunit, and the telomerase RNA component (TERC). Primary cells when grown in vitro, lack sufficient TERT to maintain telomeres and hence, telomeres shorten progressively with each cell division. This eventually results in shorter telomere that loses its ability to protect the ends of chromosomes and is therefore recognized by the cell's DNA repair machinery as damaged DNA. The loss of telomere results in cellular senescence since cell can no longer divide and replicate itself. In contrast, overexpression of TERT prevents telomere attrition and enables cells to proliferate indefinitely, a character of cancer cells. Thus, telomere and telomerase are central to several diseases such as cancer, aging, atherosclerosis, CHD, type 2 diabetes, hypertension, and to the biology of stem cells.
Recent reports suggested that leukocyte telomere length is a predictor of future CHD in middle-aged, high-risk men, whereas 10 mg of pravastatin, a HMG-CoA reductase inhibitor, substantially abrogated shortening of the telomere length in high-risk subjects [177–179], suggesting that patients with CHD have senescent endothelial cells. Telomere shortening has been reported in patients with type 2 diabetes mellitus, hypertension, and insulin resistance [180–184]. Diabetes mellitus, hypertension, insulin resistance, and CHD are not only low-grade systemic inflammatory conditions in which plasma levels of lipid peroxides, IL-6, and TNF-α are increased and eNO and concentrations of anti-oxidants are decreased, but also show shorter leukocyte telomere length compared to controls. NO activates telomerase [185] and delays endothelial cell senescence [186], whereas asymmetrical dimethyl arginine, an inhibitor of NO synthesis, enhances endothelial cell senescence [187, 188]. It was reported that stable expression of hTERT (human telomerase reverse transcriptase) enhances production of eNO and NO activity and renders endothelial cells to show younger phenotype [189, 190], whereas NO activates telomerase and delays endothelial cell senescence. These results imply that NO prevents whereas reactive oxygen species induce telomere shortening.
In contrast, tumor cells express increased telomerase activity. Tumor cells have relatively higher content of anti-oxidants and reduced concentrations of lipid peroxides due to PUFA deficiency. It is known that ω-3 PUFAs are of benefit in type 2 diabetes, hypertension, and hypertriglyceridemia and prevent CHD, in part, by enhancing NO generation from endothelial cells and decreasing insulin resistance [191]. Tumor cells undergo apoptosis on exposure to ω-3 PUFAs (especially in response to EPA, DHA, and GLA) due to increase in intracellular free radical generation and formation of lipid peroxides. Since, NO and lipid peroxides modify telomerase activity, it is likely that PUFAs enhance or decrease activity of TERT in endothelial cells and tumor cells respectively. This is supported by the observation that EPA and DHA inhibit hTERT activity in human colorectal adenocarcinoma cells [192, 193]. Thus, PUFAs can prevent, reverse or arrest atherosclerosis and CHD by their ability to enhance eNO synthesis that, in turn, augments hTERT activity and prevents endothelial senescence.
Conclusion
It is evident from the preceding discussion that PUFAs, especially an optimal combination of EPA, DHA and possibly, GLA, DGLA and AA show most the qualities of the suggested "polypill", viz., aspirin-like action, inhibition of HMG-CoA and ACE enzymes, and possess diuretic, anti-hypertensive, and β-blocker-like actions (see Table 1 for a summary of actions). PUFAs are naturally occurring endogenous substances, present in almost all tissues and are essential components of all mammalian cells and have been shown to be relatively safe when administered to different types of patients for long periods of time (from few months to few years). This is evident from the fact that Eskimos consume large amounts of marine fish that are rich in ω-3 fatty EPA and DHA and are not known to suffer from any significant side effects. Nevertheless, possible side effects due to long-term feeding of PUFAs need to be studied. One concern that is generally expressed about PUFAs is that their increased intake may enhance lipid peroxidation, and that these oxidized products could be harmful to tissues. But, it was reported that increased intake of EPA/DHA, in fact, reduces in vivo lipid peroxidation and oxidative stress in humans [194–196]. Furthermore, peripheral leukocytes, which are major mediators of inflammation, are capable of de novo production of catecholamines that enhance the inflammatory response [197]; whereas vagal parasympathetic signaling suppresses inflammation through cholinergic receptors on these cells [138]. This suggests that sympathetic and parasympathetic pathways and immune system cross talk with each other during inflammation. PUFAs, especially ω-3 fatty acids enhance acetylcholine levels [135–137] and increase HRV [133, 134] due to their ability to augment parasympathetic tone and thus, indirectly function as endogenous suppressors of sympathetic nervous system and thus, of β-receptor function. This is especially so since under normal conditions a balance is maintained between sympathetic and parasympathetic systems and pro- and anti-inflammatory pathways. These evidences imply that PUFAs function as endogenous enhancers of parasympathetic tone, suppress inflammatory events; and inhibit sympathetic over activity, and block β-receptor action.
In view of these beneficial actions (see Table 2 for a summary of their actions), ω-3 and ω-6 fatty acids can be given for the prevention of CVD. Since PUFAs can be given to pregnant women and lactating mothers, and children, it is suggested that a combined ω-6 and ω-3 pill could be given from childhood. Furthermore, several studies suggested that PUFAs, especially ω-3 fatty acids, are useful in the prevention and treatment of Alzheimer' disease, schizophrenia, and depression [198–209], suggesting that PUFAs have a much wider benefit compared to the "polypill". It may also be mentioned here that for their physiological/beneficial action(s) PUFAs need many co-factors such as folic acid, vitamin B12, vitamin B6, vitamin C, tetrahydrobiopterin (H4B), zinc, magnesium, calcium, L-arginine, and small amounts of selenium and vitamin E [18]. Hence, it is essential that these co-factors should also be provided in adequate amounts to bring about the beneficial action of ω-6 and ω-3 PUFAs. Since statins, glitazones, several anti-hypertensive and anti-arrhythmic drugs seem to be mediate their actions by modulating EFA/PUFA metabolism, it is possible that sub-clinical deficiency or altered metabolism of EFAs may subvert their actions/benefits. Hence, it is prudent to provide a combination of ω-6 and ω-3 PUFAs and their co-factors along with statins, glitazones, and other drugs in the treatment of CVD. Yokoyama et al [166] showed that long-term use of ethyl EPA (1800 mg/day) produced a significant reduction in non-fatal coronary events in patients with dyslipidemia compared to control. This risk reduction occurred after 2.5 years of use of ethyl EPA (mean follow up period was 4.6 years) even when PUFAs were added in addition to statin treatment lending support to the concept that ω-3 and ω-6 PUFAs can be combined with other cardiovascular drugs in the prevention and treatment of cardiovascular diseases.
Author's Contributions
I contributed to everything.
References
Pechacek TF, Asma S, Eriksen MP: Tobacco: global burden and community solutions. Evidence based cardiology. Edited by: Yusuf S, Calms A, Camm AJ, Fallen EL, Gersh BJ. 1998, 165-178. London: BMJ Books
Yusuf S, Peto R, Lewis J, Collins R, Sleight P: Beta blockade during and after myocardial infarction: an overview of the randomised trials. Prog Cardiovasc Dis. 1985, 27: 335-371. 10.1016/S0033-0620(85)80003-7
, : Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002, 324: 71-86. 10.1136/bmj.324.7336.S71
Heart Outcomes Prevention Evaluation Study Investigators: Effects of an angiotensin-converting enzyme inhibitor, ramipril, on cardiovascular events in high-risk patients. N Engl J Med. 2000, 342: 145-153. 10.1056/NEJM200001203420301
Scandinavian Simvastatin Survival Study Group: Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease: the Scandinavian Simvastatin Survival Study (4S). Lancet. 1994, 344: 1383-1399. 10.1016/S0140-6736(94)90566-5
The Long-term Intervention with Pravastatin in Ischaemic Disease (LIPID) Study Group: Prevention of cardiovascular events and death with pravastatin in patients with coronary heart disease and a broad range of initial cholesterol levels. N Engl J Med. 1998, 339: 1349-1357. 10.1056/NEJM199811053391902
Sacks FM, Pfeffer MA, Moye LA, Rouleau JL, Rutherford JD, Cole TG, Brown L, Warnica JW, Arnold JM, Wun CC, Davis BR, Braunwald E: The effect of pravastatin on coronary events after myocardial infarction in patients with average cholesterol levels. N Engl J Med. 1996, 336: 1001-1009. 10.1056/NEJM199610033351401
Plehn JF, Davis BR, Sacks FM, Rouleau JL, Pfeffer V, Cuddy TE, Moye LA, Piller LB, Rutherford J, Simpson LM, Braunwald E: Reduction of stroke incidence after myocardial infarction with pravastatin: the Cholesterol and Recurrent Events (CARE) study. The Care Investigators. Circulation. 1999, 99: 216-223.
Heart Protection Study Collaborative Group: MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20536 high-risk individuals: a randomised placebo-controlled trial. Lancet. 2002, 360: 7-22. 10.1016/S0140-6736(02)09327-3
Yusuf S: Two decades of progress in preventing vascular disease. Lancet. 2002, 360: 2-3. 10.1016/S0140-6736(02)09358-3
Wald NJ, Law MR: A strategy to reduce cardiovascular disease by more than 80%. BMJ. 2003, 326: 1419-1423. 10.1136/bmj.326.7404.1419
Hippisley-Cox J, Coupland C: Effects of combinations of drugs on all mortality in patients with ischaemic heart disease: nested case-control analysis. BMJ. 2005, 330: 1059-1063. 10.1136/bmj.330.7499.1059
Hennekens CH, Sacks FM, Tonkin A, Jukema J, Byington R, Pitt B, Berry DA, Berry SM, Ford NF, Walker AJ, Natarajan K, Sheng-Lin C, Fiedorek FT, Belder R: Additive benefits of pravastatin and aspirin to decrease risks of cardiovascular disease: randomized and observational comparisons of secondary prevention trials and their meta-analyses. Arch Intern Med. 2003, 164: 40-44. 10.1001/archinte.164.1.40. 10.1001/archinte.164.1.40
Wei L, Ebrahim S, Bartlett C, Davey PD, Sullivan FM, MacDonald TM: Statin use in the secondary prevention of coronary heart disease in primary care: cohort study and comparison of inclusion and outcome with patients in randomised trials. BMJ. 2005, 330: 821-825. 10.1136/bmj.38398.408032.8F
Eidelman R, Herbert P, Weisman S, Hennekens CH: An update on aspirin in the primary prevention of cardiovascular disease. Arch Intern Med. 2005, 163: 2006-2010. 10.1001/archinte.163.17.2006. 10.1001/archinte.163.17.2006
Ridker PM, Cook N, Lee I, Gordon D, Gaziano JM, Manson JE, Hennekens CH, Buring JE: A randomised trial of low-dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med. 2005, 352: 1293-1304. 10.1056/NEJMoa050613
Toole J, Malinow MR, Chambless LE, Spence JD, Pettigrew LC, Howard VJ, Sides EG, Wang CH, Stampfer M: Lowering homocysteine in patients with ischemic stroke to prevent recurrent stroke, myocardial infarction, and death: the Vitamin Intervention for Stroke Prevention (VISP) randomized controlled trial. JAMA. 2004, 291: 565-575. 10.1001/jama.291.5.565
Das UN: Essential fatty acids: Biochemistry, physiology, and pathology. Biotechnology J. 2006, 1: 420-439. 10.1002/biot.200600012
Lerman RH: Essential fatty acids. Altern Ther Health Med. 2006, 12: 20-29.
Horrobin DF: Omega-6 and omega-3 essential fatty acids in atherosclerosis. Semin Thromb Hemost. 1993, 19: 129-137.
Hansen HS: New biological and clinical roles for the n-6 and n-3 fatty acids. Nutr Rev. 1994, 52: 162-167.
Claria J, Serhan CN: Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc Natl Acad Sci USA. 1995, 92: 9475-9479. 10.1073/pnas.92.21.9475
Chiang N, Gronert K, Clish CB, O'Brien JA, Freeman MW, Serhan CN: Leukotriene B4 receptor transgenic mice reveal novel protective roles for lipoxins and aspirin-triggered lipoxins in reperfusion. J Clin Invest. 1999, 104: 309-316. 10.1172/JCI7016
Levy BD, Clish CB, Schmidt B, Gronert K, Serhan CN: Lipid mediator class switching during acute inflammation signals in resolution. Nat Immunol. 2001, 2: 612-619. 10.1038/89759
Bandeira-Melo C, Serra MF, Diaz BI, Cordeiro RSB, Silva PMR, Lenzi HL, Bakhle YS, Serhan CN, Martins MA: Cyclooxygenase -2-derived prostaglandin E2 and lipoxin A4 accelerate resolution of allergic edema in Angiostronglus costaricensis-infected rats: relationship with concurrent eosinophilia. J Immunol. 2000, 164: 1029-1036.
Ariel A, Serhan CN: Resolvins and protectins in the termination program of acute inflammation. Trends Immunol. 2007, 28: 176-183. 10.1016/j.it.2007.02.007
Levy BD, Kohli P, Gotlinger K, Haworth O, Hong S, Kazani S, Israel E, Haley KJ, Serhan CN: Protectin D1 is generated in asthma and dampens airway inflammation and hyperresponsiveness. J Immunol. 2007, 178: 496-502.
El-Sohemy A, Archer MC: Regulation of mevalonate synthesis in low density lipoprotein receptor knockout mice fed n-3 or n-6 polyunsaturated fatty acids. Lipids. 1999, 34: 1037-1043. 10.1007/s11745-999-0455-8
Nakamura N, Hamazaki T, Jokaji H, Minami S, Kobayashi M: Effect of HMG-CoA reductase inhibitors on plasma polyunsaturated fatty acid concentration in patients with hyperlipidemia. Int J Clin Lab Res. 1998, 28: 192-195. 10.1007/s005990050043
Duncan RE, El-Sohemy A, Archer MC: Regulation of HMG-CoA reductase in MCF-7 cells by genistein, EPA, and DHA, alone and in combination with mevastatin. Cancer Lett. 2005, 224: 221-228. 10.1016/j.canlet.2004.11.031
El-El-Sohemy A, Archer MC: Regulation of mevalonate synthesis in rat mammary glands by dietary n-3 and n-6 polyunsaturated fatty acids. Cancer Res. 1997, 57: 3685-3687.
Levine L: Statins stimulate arachidonic acid release and prostaglandin I2 production in rat liver cells. Lipids Health Dis. 2003, 2: 1- 10.1186/1476-511X-2-1
Das UN: Essential fatty acids as possible mediators of the actions of statins. Prostaglandins Leukot Essen Fatty Acids. 2001, 65: 37-40. 10.1054/plef.2001.0285
Dobrucki LW, Kalinowski L, Dobrucki IT, Malinski T: Statin-stimulated nitric oxide release from endothelium. Med Sci Monit. 2001, 7: 622-627.
McGown CC, Brookes ZL: Beneficial effects of statins on the microcirculation during sepsis: the role of nitric oxide. Br J Anaesth. 2007, 98: 163-175. 10.1093/bja/ael358
Okuda Y, Kawashima K, Sawada T, Tsurumaru K, Asano M, Suzuki S, Soma M, Nakajima T, Yamashita K: Eicosapentaenoic acid enhances nitric oxide production by cultured human endothelial cells. Biochem Biophys Res Commun. 1997, 232: 487-491. 10.1006/bbrc.1997.6328
Yasuda H, Yuen PS, Hu X, Zhou H, Star RA: Simvastatin improves sepsis-induced mortality and acute kidney injury via renal vascular effects. Kidney Int. 2006, 69: 1535-1542. 10.1038/sj.ki.5000300
Sierra S, Lara-Villoslada F, Comalada M, Olivares M, Xaus J: Dietary fish oil n-3 fatty acids increase regulatory cytokine production and exert anti-inflammatory effects in two murine models of inflammation. Lipids. 2006, 41: 1115-1125. 10.1007/s11745-006-5061-2
Salvemini D, Misko TP, Masferrer JL, Seibert K, Currie MG, Needleman P: Nitric oxide activates cyclooxygenase enzymes. Proc Natl Acad Sci USA. 1993, 90: 7240-7244. 10.1073/pnas.90.15.7240
Rosenson RS: Statins in atherosclerosis: lipid-lowering agents with antioxidant capabilities. Atherosclerosis. 2004, 173: 1-12. 10.1016/S0021-9150(03)00239-9
Colquhoun D, Keech A, Hunt D, Marschner I, Simes J, Glaziou P, White H, Barter P, Tonkin A, : Effects of pravastatin on coronary events in 2073 patients with low levels of both low-density lipoprotein cholesterol and high-density lipoprotein cholesterol: results from the LIPID study. Eur Heart J. 2004, 25: 771-777. 10.1016/j.ehj.2004.03.013
Majima T, Komatsu Y, Fukao A, Ninomiya K, Matsumura T, Nakao K: Short-term Effects of Atorvastatin on Bone Turnover in Male Patients with Hypercholesterolemia. Endocr J. 2007, 54: 145-151. 10.1507/endocrj.K06-127
Briel M, Studer M, Glass TR, Bucher HC: Effects of statins on stroke prevention in patients with and without coronary heart disease: a meta-analysis of randomized controlled trials. Am J Med. 2004, 117: 596-606. 10.1016/j.amjmed.2004.04.022
Si ML, Long C, Yang DI, Chen MF, Lee TJ: Statins prevent beta-amyloid inhibition of sympathetic alpha7-nAChR-mediated nitrergic neurogenic dilation in porcine basilar arteries. J Cereb Blood Flow Metab. 2005, 25: 1573-1585. 10.1038/sj.jcbfm.9600232
Aprahamian T, Bonegio R, Rizzo J, Perlman H, Lefer DJ, Rifkin IR, Walsh K: Simvastatin treatment ameliorates autoimmune disease associated with accelerated atherosclerosis in a murine lupus model. J Immunol. 2006, 177: 3028-3034.
Ma J, Folsom AR, Lewis L, Eckfeldt JH: Relation of plasma phospholipid and cholesterol ester fatty acid composition to carotid artery intima-media thickness: the Atherosclerosis Risk in Communities (ARIC) Study. Am J Clin Nutr. 1997, 65: 551-559.
Psota TL, Gebauer SK, Kris-Etherton P: Dietary omega-3 fatty acid intake and cardiovascular risk. Am J Cardiol. 2006, 98 (suppl): 3i-18i. 10.1016/j.amjcard.2005.12.022. 10.1016/j.amjcard.2005.12.022
Harris WS: Omega-3 fatty acids and cardiovascular disease: a case for omega-3 index as a new risk factor. Pharmacol Res. 2007, 55: 217-223. 10.1016/j.phrs.2007.01.013
Bouzan C, Cohen JT, Connor WE, Kris-Etherton PM, Gray GM, Konig A, Lawrence RS, Savitz DA, Teusch SM: A quantitative analysis of fish consumption and stroke risk. Am J Prev Med. 2005, 29: 347-352. 10.1016/j.amepre.2005.07.002
Green KN, Martinez-Coria H, Khashwji H, Hall EB, Yurko-Mauro KA, Ellis L, LaFerla FM: Dietary docosahexaenoic acid and docosapentaenoic acid ameliorate amyloid-beta and tau pathology via a mechanism involving presenilin 1 levels. J Neurosci. 2007, 27: 4385-4395. 10.1523/JNEUROSCI.0055-07.2007
Kotani S, Sakaguchi E, Warashina S, Matsukawa N, Ishikura Y, Kiso Y, Sakakibara M, Yoshimoto T, Guo J, Yamashima T: Dietary supplementation of arachidonic and docosahexaenoic acids improves cognitive dysfunction. Neurosci Res. 2006, 56: 159-164. 10.1016/j.neures.2006.06.010
Reifen R, Blank M, Afek A, Kopilowiz Y, Sklan D, Geshwin ME, German B, Yoshida S, Shoenfeld Y: Dietary polyunsaturated fatty acids decrease anti-dsDNA and anti-cardiolipin antibodies production in idiotype induced mouse model of systemic lupus erythematosus. Lupus. 1998, 7: 192-197. 10.1191/096120398678919985
Levy BD: Myocardial 15-epi-lipoxin A4 generation provides a new mechanism for the immunomodulatory effects of statins and thiazolidinediones. Circulation. 2006, 114: 873-875. 10.1161/CIRCULATIONAHA.106.647925
Birnbaum Y, Ye Y, Lin Y, SY Freeberg, Nishi SP, Martinez JD, Huang MH, Uretsky BF, Perez-Polo JR: Augmentation of myocardial production of 15-epi-lipoxin A4 by pioglitazone and atorvastatin in the rat. Circulation. 2006, 114: 929-935. 10.1161/CIRCULATIONAHA.106.629907
Nordoy A, Bonaa KH, Sandset PM, Hansen JB, Nilsen H: Effect of omega-3 fatty acids and simvastatin on hemostatic risk factors and postprandial hyperlipemia in patients with combined hyperlipemia. Arterioscler Thromb Vasc Biol. 2000, 20: 259-265.
Nordoy A, Hansen JB, Brox J, Svensson B: Effects of atorvastatin and omega-3 fatty acids on LDL subfractions and postprandial hyperlipemia in patients with combined hyperlipemia. Nutr Metab Cardiovasc Dis. 2001, 11: 7-16.
Nordoy A, Svensson B, Hansen JB: Atorvastatin and omega-3 fatty acids protect against activation of the coagulation system in patients with combined hyperlipemia. J Thromb Haemost. 2003, 1: 690-697. 10.1046/j.1538-7836.2003.00140.x
Nordoy A: Statins and omega-3 fatty acids in the treatment of dyslipidemia and coronary heart disease. Minerva Med. 2002, 93: 357-363.
Xiang M, Harbige LS, Zetterstrom R: Long-chain polyunsaturated fatty acids in Chinese and Swedish mothers: diet, breast milk and infant growth. Acta Paediatr. 2005, 94: 1543-1549. 10.1080/08035250500251601
Amusquivar E, Ruperez FJ, Barbas C, Herrera E: Low arachidonic acid rather than alpha-tocopherol is responsible for the delayed postnatal development in offspring of rats fed fish oil instead of olive oil during pregnancy and lactation. J Nutr. 2000, 130: 2855-2865.
Auestad N, Scott DT, Janowsky JS, Jacobsen C, Carroll RE, Montalto MB, Halter R, Qiu W, Jacobs JR, Connor WE, Connor SL, Taylor JA, Neuringer M, Fitzgerald KM, Hall RT: Visual, cognitive, and language assessments at 39 months: a follow-up study of children fed formulas containing long-chain polyunsaturated fatty acids to 1 year of age. Pediatrics. 2003, 112 (3 Pt 1): e177-183. 10.1542/peds.112.3.e177
Judge MP, Harel O, Lammi-Keefe CJ: A Docosahexaenoic acid-functional food during pregnancy benefits infant visual acuity at four but not six months of age. Lipids. 2007, 42: 117-122. 10.1007/s11745-006-3007-3
Helland IB, Smith L, Saarem K, Saugstad OD, Drevon CA: Maternal supplementation with very-long-chain n-3 fatty acids during pregnancy and lactation augments children's IQ at 4 years of age. Pediatrics. 2003, 111: e39-e44. 10.1542/peds.111.1.e39
Helland IB, Saugstad OD, Smith L, Saarem K, Solvoll K, Ganes T, Drevon CA: Similar effects on infants of n-3 and n-6 fatty acids supplementation to pregnant and lactating women. Pediatrics. 2001, 108: E82- 10.1542/peds.108.5.e82
Dunstan JA, Simmer K, Dixon G, Prescott SL: Cognitive assessment at 21/2 years following fish oil supplementation in pregnancy: a randomized controlled trial. Arch Dis Child Fetal Neonatal Ed. 2006, 93: F45-F50. 10.1136/adc.2006.099085
Lauritzen L, Jorgensen MH, Olsen SF, Straarup EM, Michaelsen KF: Maternal fish oil supplementation in lactation: effect on developmental outcome in breast-fed infants. Reprod Nutr Dev. 2005, 45: 535-547. 10.1051/rnd:2005044
Auestad N, Halter R, Hall RT, Blatter M, Bogle ML, Burks W, Erickson JR, Fitzgerald KM, Dobson V, Innis SM, Singer LT, Montalto MB, Jacobs JR, Qiu W, Bernstein MH: Growth and development in term infants fed long-chain polyunsaturated fatty acids: a double-masked, randomized, parallel, prospective, multivariate study. Pediatrics. 2001, 108: 372-381. 10.1542/peds.108.2.372
Tofail F, Kabir I, Hamadani JD, Chowdhury F, Yesmin S, Mehreen F, Huda SN: Supplementation of fish-oil and soy-oil during pregnancy and psychomotor development of infants. J Health Popul Nutr. 2006, 24: 48-56.
de Groot RH, Adam J, Jolles J, Hornstra : Alpha-linolenic acid supplementation during human pregnancy does not effect cognitive functioning. Prostaglandins Leukot Essent Fatty Acids. 2004, 70: 41-47. 10.1016/j.plefa.2003.08.004
Dezso B, Jacobson J, Poulsen K: Evidence for the presence of angiotensins in normal, unstimulated alveolar macrophages and monocytes. J Hypertens. 1989, 7 (1): 5-11.
Codde JP, Croft KD, Barden A, Mathews E, Vandongen R, Beilin LJ: An inhibitory effect of dietary polyunsaturated fatty acids on renin secretion in the isolated perfused rat kidney. J Hypertens. 1984, 2: 265-270.
O'Brien PM, Morrison R, Broughton Pipkin F: The effect of dietary supplementation with linoleic and gammalinolenic acids on the pressor response to angiotensin II – a possible role in pregnancy-induced hypertension?. Br J Clin Pharmacol. 1985, 19: 335-342.
Scholkens BA, Gehring D, Schlotte V, Weithmann U: Evening primrose oil, a dietary prostaglandin precursor, diminishes vascular reactivity to renin and angiotensin II in rats. Prostaglandins Leukot Med. 1982, 8: 273-285. 10.1016/0262-1746(82)90050-6
Hui R, St-Louis J, Falardeau P: Antihypertensive properties of linoleic acid and fish oil omega-3 fatty acids independent of the prostaglandin system. Am J Hypertens. 1989, 2: 610-617.
Talwalkar RT, Kotchen TA: Inhibition of renin by plasma linoleic acid. Am J Med Sci. 1988, 296 (3): 192-197. 10.1097/00000441-198809000-00008
Kotchen TA, Welch WJ, Talwalkar RT: In vitro and in vivo inhibition of renin by fatty acids. Am J Physiol. 1978, 234: E593-E599.
Reddy SR, Talwalkar R, Downs J, Kotchen TA: Effect of linoleic acid infusion on blood pressure in normotensive and hypertensive rats. Am J Physiol. 1989, 257 (2 Pt 2): H611-H617.
Das UN: Effect of cis-unsaturated fatty acids, prostaglandins, and free radicals on angiotensin-converting enzyme activity in vitro. Proc Soc Exp Biol Med. 1997, 214: 374-379.
Li Q, Zhang Q, Wang M, Zhao S, Ma J, Luo N, Li N, Li Y, Xu G, Li J: Eicosapentaenoic acid modifies lipid composition in caveolae and induces translocation of endothelial nitric oxide synthase. Biochimie. 2007, 89: 169-177. 10.1016/j.biochi.2006.10.009
Das UN: A defect in the activity of Δ6 and Δ5 desaturases may be a factor in the initiation and progression of atherosclerosis. Prostaglandins Leukot Essen Fatty Acids. 2007, 76: 251-268. 10.1016/j.plefa.2007.03.001. 10.1016/j.plefa.2007.03.001
Kaergel E, Muller DN, Honeck H, Theuer J, Shagdarsuren E, Mullally A, Luft FC, Schunck W-H: P450-dependent arachidonic acid metabolism and angiotensin-II-induced renal damage. Hypertension. 2002, 40: 273-279. 10.1161/01.HYP.0000029240.44253.5E
Wang W, Diamond SL: Does elevated nitric oxide production enhance the release of prostacyclin from shear stressed aortic endothelial cells?. Biochem Biophys Res Commun. 1997, 233: 748-751. 10.1006/bbrc.1997.6548
Antithrombotic Trialists' Collaboration: Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002, 324: 71-86. 10.1136/bmj.324.7336.S71
Schreinemachers DM, Everson RB: Aspirin use and lung, colon, and breast cancer incidence in a prospective study. Epidemiology. 1994, 5: 138-146. 10.1097/00001648-199403000-00003
Claria J, Serhan CN: Aspirin triggers previously undescribed bioactive eicosanoids by human endothelial cell-leukocyte interactions. Proc Natl Acad Sci USA. 1995, 92: 9475-9479. 10.1073/pnas.92.21.9475
Leaf A, Weber PC: Cardiovascular effects of n-3 fatty acids. N Engl J Med. 1988, 322: 697-698.
Kristensen SD, Schmidt EB, Dyerberg J: Dietary supplementation with n-3 polyunsaturated fatty acids and human platelet function: a review with particular emphasis on implications for cardiovascular disease. J Intern Med Suppl. 1989, 731: 141-150.
Scheurlen M, Kirchner M, Clemens MR, Jaschonek K: Fish oil preparations rich in docosahexaenoic acid modify platelet responsiveness to prostaglandin-endoperoxide/thromboxane A2 receptor agonists. Biochem Pharmacol. 1993, 46: 245-249. 10.1016/0006-2952(93)90410-X
Psota TL, Gebauer SK, Kris-Etherton P: Dietary omega-3 fatty acid intake and cardiovascular risk. Am J Cardiol. 2006, 98 (suppl): 3i-18i. 10.1016/j.amjcard.2005.12.022. 10.1016/j.amjcard.2005.12.022
Davidson MH: Mechanisms for the hypotriglyceridemic effect of marine omega-3 fatty acids. Am J Cardiol. 2006, 98 (suppl): 27i-33i. 10.1016/j.amjcard.2005.12.024
Robinson JG, Stone NJ: Antiatherosclerotic and antithrombotic effects of omega-3 fatty acids. Am J Cardiol. 2006, 98 (suppl): 39i-49i. 10.1016/j.amjcard.2005.12.026
Reiffel JA, McDonald A: Antiarrhythmic effects of omega-3 fatty acids. Am J Cardiol. 2006, 98 (suppl): 50i-60i. 10.1016/j.amjcard.2005.12.027. 10.1016/j.amjcard.2005.12.027
Jacobson TA: Secondary prevention of coronary heart disease with omega-3 fatty acids. Am J Cardiol. 2006, 98 (suppl): 61i-70i. 10.1016/j.amjcard.2005.12.028
Das UN: Beneficial effect(s) of n-3 fatty acids in cardiovascular diseases: but, why and how?. Prostaglandins Leukot Essen Fatty Acids. 2000, 63: 351-362. 10.1054/plef.2000.0226. 10.1054/plef.2000.0226
Wang L: The role of prostaglandins in the antiarrhythmic effect of ischemic preconditioning. J Biomed Sci. 2001, 8: 406-410. 10.1007/BF02255949
Das UN: Prostacyclin as an endogenous anti-arrhythmic agent. Basic Res Cardiol. 1983, 78: 716-718. 10.1007/BF01907219
Somberg JC, Yelamanchi V, Molnar J, Aschermann M, Bernik PJ, Caspi A, Marmor A, Rabinowitz B, Reisin L, Ruzyllo W: Prostaglandin E1: electrophysiological safety in patients with congestive heart failure and peripheral arterial occlusive disease. The Alprostadil Investigators. Am J Ther. 1997, 4: 401-414.
Harris WS, Assaad B, Poston WC: Tissue omega-6/omega-3 fatty acid ratio and risk for coronary artery disease. Am J Cardiol. 2006, 98 (suppl): 19i-26i. 10.1016/j.amjcard.2005.12.023. 10.1016/j.amjcard.2005.12.023
Mozaffarian D, Ascherio A, Hu FB, Stampfer MJ, Willett WC, Siscovick DS, Rimm EB: Interplay between different polyunsaturated fatty acids and risk of coronary heart disease in men. Circulation. 2005, 111: 166-173 10.1161/01.CIR.0000152099.87287.83
Wood DA, Riemersma RA, Butler S, Thomson M, Macintyre C, Elton RA, Oliver MF: Linoleic and eicosapentaenoic acids in adipose tissue and platelets and risk of coronary heart disease. Lancet. 1987, i: 177-183. 10.1016/S0140-6736(87)90001-8
Wood DA, Butler S, Riemersma RA, Thomson M, Oliver MF, Fulton M, Birtwhistle A, Elton R: Adipose tissue and platelet fatty acids and coronary heart disease in Scottish men. Lancet. 1984, ii: 117-121. 10.1016/S0140-6736(84)91044-4
Roberts TL, Wood DA, Riemersma RA, Gallagher PJ, Lampe FC: Linoleic acid and risk of sudden cardiac death. Br Heart J. 1993, 70: 524-529. 10.1136/hrt.70.6.524
Luostarinen R, Boberg M, Saldeen T: Fatty acid composition in total phospholipids of human coronary arteries in sudden cardiac death. Atherosclerosis. 1993, 99: 187-193. 10.1016/0021-9150(93)90021-L
Felton CV, Crook D, Davies MJ, Oliver MF: Relation of plaque lipid composition and morphology to the stability of human aortic plaques. Arterioscler Thromb Vasc Biol. 1997, 17: 1337-1345.
Thies F, Garry JM, Yaqoob P, Rerkasem K, Williams J, Shearman CP, Gallagher PJ, Calder PC, Grimble RF: Association of n-3 polyunsaturated fatty acids with stability of atherosclerotic plaques: a randomised controlled trial. Lancet. 2003, 361: 477-485. 10.1016/S0140-6736(03)12468-3
Juan H, Sametz W: Dihomo-gamma-linolenic acid increases the metabolism of eicosapentaenoic acid in perfused vascular tissue. Prostaglandins Leukotrienes Med. 1985, 19: 79-86. 10.1016/0262-1746(85)90162-3
Dunn MJ, Grone HJ: The relevance of prostaglandins in human hypertension. Adv Prostaglandin Thromboxane Leukot Res. 1985, 13: 179-87.
Bordet JC, Guichardant M, Lagarde M: Hydroperoxides produced by n-6 lipoxygenation of arachidonic and linoleic acids potentiate synthesis of prostacyclin related compounds. Biochim Biophys Acta. 1988, 958: 460-468.
Bordet JC, Guichardant M, Lagarde M: Arachidonic acid strongly stimulates prostaglandin I3 (PGI3) production from eicosapentaenoic acid in human endothelial cells. Biochem Biophys Res Commun. 1986, 135: 403-410. 10.1016/0006-291X(86)90009-4
Das UN: Oestrogen, statins and essential fatty acids: Similarity in their actions and benefits-is there a common link?. Nutrition. 2002, 18: 178-188. 10.1016/S0899-9007(01)00719-5
Jula A, Marniemi J, Ronnemaa T, Virtanen A, Huupponen R: Effects of diet and simvastatin on fatty acid composition in hypercholesterolemic men: a randomized controlled trial. Arterioscler Thromb Vasc Biol. 2005, 25: 1952-1959. 10.1161/01.ATV.0000177812.84927.fa
Harris JI, Hibbeln JR, Mackey RH, Muldoon MF: Statin treatment alters serum n-3 and n-6 fatty acids in hypercholesterolemic patients. Prostaglandins Leukot Essent Fatty Acids. 2004, 71: 263-269. 10.1016/j.plefa.2004.06.001
Bellini MJ, Polo MP, de Alaniz MJ, de Bravo MG: Effect of simvastatin on the uptake and metabolic conversion of palmitic, dihomo-gamma-linoleic and alpha-linolenic acids in A549 cells. Prostaglandins Leukot Essent Fatty Acids. 2003, 69: 351-367. 10.1016/S0952-3278(03)00149-2
Rise P, Pazzucconi F, Sirtori CR, Galli C: Statins enhance arachidonic acid synthesis in hypercholesterolemic patients. Nutr Metab Cardiovasc Dis. 2001, 11: 88-94.
Morris T, Stables M, Gilroy DW: New perspectives on aspirin and the endogenous control of acute inflammatory resolution. ScientificWorldJournal. 2006, 6: 1048-1065. 10.1100/tsw.2006.192
Schwab JM, Serhan CN: Lipoxins and new lipid mediators in the resolution of inflammation. Curr Opin Pharmacol. 2006, 6: 414-420. 10.1016/j.coph.2006.02.006
Serhan CN: Novel omega-3-derived local mediators in anti-inflammation and resolution. Pharmacol Ther. 2005, 105: 7-21. 10.1016/j.pharmthera.2004.09.002
Lopez-Garcia E, Schultze MB, Manson JE, Meigs JB, Albert CM, Rifai N, Willett WC, Hu FB: Consumption of (n-3) fatty acids is related to plasma biomarkers of inflammation and endothelial activation in women. J Nutr. 2004, 134: 1806-1811.
Sierra S, Lara-Villoslada F, Olivares M, Junenez J, Boza J, Xaus J: IL-10 expression is involved in the regulation of the immune response by omega 3 fatty acids. Nutr Hosp. 2004, 19: 376-382.
Kumar GS, Das UN: Effect of prostaglandins and their precursors on the proliferation of human lymphocytes and their secretion of tumor necrosis factor and various interleukins. Prostaglandins Leukot Essent Fatty Acids. 1994, 50: 331-334. 10.1016/0952-3278(94)90242-9
Dusing R, Struck A, Gobel BO, Weisser B, Vetter H: Effects of n-3 fatty acids on renal function and renal prostaglandin E metabolism. Kidney Int. 1990, 38: 315-319. 10.1038/ki.1990.202
Barcelli UO, Weiss M, Beach D, Motz A, Thompson B: High linoleic acid diets ameliorate diabetic nephropathy in rats. Am J Kidney Dis. 1990, 16: 244-251.
Singer P, Berger I, Moritz V, Forster D, Taube C: N-6 and N-3 PUFA in liver lipids, thromboxane formation and blood pressure from SHR during diets supplemented with evening primrose, sunflower seed or fish oil. Prostaglandins Leukot Essen Fatty Acids. 1990, 39: 207-211. 10.1016/0952-3278(90)90073-T
Vaskonen T, Laakso J, Mervaala E, Sievi E, Karppanen H: Interrelationships between salt and fish oil in stroke-prone spontaneously hypertensive rat. Blood Press. 1996, 5: 178-189. 10.3109/08037059609062127
Grande JP, Walker HJ, Holub BJ, Warner GM, Keller DM, Haugen JD, Donadio JV, Dousa TP: Suppressive effects of fish oil on mesangial cell proliferation in vitro and in vivo. Kidney Int. 2000, 57: 1027-1040. 10.1046/j.1523-1755.2000.00930.x
Donadio JV, Grande JP: The role of fish oil/omega-3 fatty acids in the treatment of IgA nephropathy. Semin Nephrol. 2004, 24: 225-243. 10.1016/j.semnephrol.2004.01.004
Heide Homan van der JJ, Bilo HJ, Tegzess AM, Donker AJ: The effects of dietary supplementation with fish oil on renal function in cyclosporine-treated renal transplant recipients. Transplantation. 1990, 49: 523-527. 10.1097/00007890-199003000-00010
Clark WF, Parbtani A, Philbrick DJ, Holub BJ, Huff MW: Chronic effects of omega-3 fatty acids (fish oil) in a rat 5/6 renal ablation model. J Am Soc Nephrol. 1991, 1: 1343-1353.
Ingram AJ, Parbtani A, Clark WF, Spanner E, Huff MW, Philbrick DJ, Holub BJ: Effects of flaxseed and flax oil diets in a rat-5/6 renal ablation model. Am J Kidney Dis. 1995, 25: 320-329. 10.1016/0272-6386(95)90015-2
Duffield JS, Hong S, Vaidya VS, Lu Y, Fredman G, Serhan CN, Bonventre JV: Resolvin D series and protectin D1 mitigate acute kidney injury. J Immunol. 2006, 177: 5902-5911.
Wiemer G, Fink E, Linz W, Hropot M, Scholkens BE, Wohlfart P: Furosemide enhances the release of endothelial kinins, nitric oxide and prostacyclin. J Pharmacol Exp Ther. 1994, 271: 1611-1615.
Harris RC, Zhang MZ, Cheng HF: Cyclooxygenase-2 nd the renal renin-angiotensin system. Acta Physiol Scand. 2004, 181: 543-547. 10.1111/j.1365-201X.2004.01329.x
Christensen JH, Gustenhoff P, Korup E, Aaroe J, Toft E, Moller JM, Rasmussen K, Dyerberg J, Schmidt EB: n-3 polyunsaturated fatty acids, heart rate variability and ventricular arrhythmias in post-AMI-patients. A clinical controlled trial. Ugeskr Laeger. 1997, 159: 5525-5529.
Christensen JH, Christensen MS, Dyerberg J, Schmidt EB: Heart rate variability and fatty acid content of blood cell membranes: a dose-response study with n-3 fatty acids. Am J Clin Nutr. 1999, 70: 331-337.
Minami M, Kimura S, Endo T, Hamaue N, Hirafuji M, Togashi H, Yoshioka M, Saito H, Watanabe S, Kobayashi T, Okuyama H: Dietary docosahexaenoic acid increases cerebral acetylcholine levels and improves passive avoidance performance in stroke-prone spontaneously hypertensive rats. Pharmacol Biochem Behav. 1997, 58: 1123-1129. 10.1016/S0091-3057(97)00300-6
DeGeorge JJ, Morell P, McCarthy KD, Lapetina EG: Cholinergic stimulation of arachidonic acid and phosphatidic acid metabolism in C62B glioma cells. J Biol Chem. 1986, 261: 3428-3433.
Almeida T, Cunha RA, Ribeiro JA: Facilitation by arachidonic acid of acetylcholine release from the rat hippocampus. Brain Res. 1999, 826: 104-111. 10.1016/S0006-8993(99)01267-6
Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ: Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000, 405: 458-462. 10.1038/35013070
Bernik TR, Friedman SG, Ochani M, DiRaimo R, Ulloa L, Yang H, Sudan S, Czura CJ, Ivanova SM, Tracey CJ: Pharmacological stimulation of the cholinergic antiinflammatory pathway. J Exp Med. 2002, 195: 781-788. 10.1084/jem.20011714
Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susaria S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ: Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003, 421: 384-388. 10.1038/nature01339
Pita ML, Delgado MJ: Folic acid administration increases N-3 polyunsaturated fatty acids in rat plasma and tissue lipids. Thromb Haemost. 2000, 84: 420-423.
Durand P, Prost M, Blache D: Pro-thrombotic effects of a folic acid deficient diet in rat platelets and macrophages related to elevated homocysteine and decreased n-3 polyunsaturated fatty acids. Atherosclerosis. 1996, 121: 231-243. 10.1016/0021-9150(95)06724-8
Das UN: Folic acid says NO to vascular diseases. Nutrition. 2003, 19: 686-692. 10.1016/S0899-9007(02)01044-4
Joshi S, Rao S, Girigosavi S, Daware M, Kale A, Hegde M: Differential effects of fish oil and folic acid supplementation during pregnancy in rats on cognitive performance and serum glucose in their offspring. Nutrition. 2004, 20: 465-472. 10.1016/j.nut.2004.01.012
Rao S, Joshi S, Kale A, Hegde M, Mahadik S: Maternal folic acid supplementation to dams on marginal protein level alters brain fatty acid levels of their adult offspring. Metabolism. 2006, 55: 628-634. 10.1016/j.metabol.2005.12.008
Rhoads GG, Gulbrandsen CL, Kagan A: Serum lipoproteins and coronary heart disease in a population study of Hawaii Japanese men. N Engl J Med. 1976, 294: 293-298.
Ikewaki K, Rader DJ, Sakamoto T, Nishiwaki M, Wakimoto N, Schaefer JR, Ishikawa T, Fairwell T, Zech LA, Nakamura H, Nagano M, Brewer HB: Delayed catabolism of high density lipoprotein apolipoprotein A-I and A-II in human cholesteryl ester transfer protein deficiency. J Clin Invest. 1993, 92: 1650-1658. 10.1172/JCI116750
Barter PJ, Brewer HB, Chapman MJ, Hennekens CH, Rader DJ, Tall AR: Cholesteryl ester transfer protein: a novel target for raising HDL and inhibiting atherosclerosis. Arterioscler Thromb Vasc Biol. 2003, 23: 160-167. 10.1161/01.ATV.0000054658.91146.64
Inazu I, Koizumi J, Mabuchi H: Cholesteryl ester transfer protein and atherosclerosis. Curr Opin Lipidol. 2000, 11: 389-396. 10.1097/00041433-200008000-00008
Watts GF: The yin and yang of cholesteryl ester transfer protein and atherosclerosis. Clin Sci. 2002, 103: 595-597.
Jansen S, Lopez-Miranda J, Castro P, Lopez-Segura F, Marin C, Ordovas JM, Paz E, Jimenez-Pereperez J, Fuentes F, Perez-Jimenez F: Low-fat and high-monounsaturated fatty acid diets decrease plasma cholesterol ester transfer protein concentrations in young, healthy, normolipemic men. Am J Clin Nutr. 2000, 72: 36-41.
Hirano R, Igarashi O, Kondo K, Itakura H, Matsumoto A: Regulation by long-chain fatty acids of the expression of cholesteryl ester transfer protein in HepG2 cells. Lipids. 2001, 36: 401-406. 10.1007/s11745-001-0735-3
Smaoui M, Hammami S, Attia N, Chaaba R, Abid N, Kilani N, Kchaou H, Mahjoub S, Abid M, Hammami M: Modulation of plasma cholesteryl ester transfer protein activity by unsaturated fatty acids in Tunisian type 2 diabetic women. Nutr Metab Cardiovasc Dis. 2006, 16: 44-53. 10.1016/j.numecd.2005.05.011
Kastelein JJP, van Leuven AI, Burgess L, Evans GW, Kuivenhoven JA, Barter PJ, Revkin JH, Grobbee DE, Riley WA, Shear CL, Duggan WT, Bots ML, : Effect of torcetrapib on carotid atherosclerosis in familial hypercholesterolemia. N Engl J Med. 2007, 356: 1620-1630. 10.1056/NEJMoa071359
Nissen SE, Tardif JC, Nicholls SJ, Revkin JH, Shear CL, Duggan WT, Ruzyllo W, Bachinsky WB, Lasala GP, Tuzcu EM, : Effect of ACAT Inhibition on the Progression of Coronary Atherosclerosis. N Engl J Med. 2007, 356: 1304-1316. 10.1056/NEJMoa070635
Prichard BNC, Smith CCT, Ling KLE, Betteridge DJ: Fish oils and cardiovascular disease. BMJ. 1995, 310: 819-820.
Kagawa Y, Nishizawa M, Suzuki M: Eicosapolyenoic acids of serum lipids of Japanese islanders with low incidence of cardiovascular diseases. J Nutr Sci Vitaminol (Tokyo). 1982, 28: 441-453.
Kromhout D, Bosschieter EB, de Lezenne Coulander C: The inverse relation between fish consumption and 20-year mortality from coronary heart disease. N Engl J Med. 1985, 312: 1205-1209.
Erkkila AT, Lehto S, Pyorala K, Uusitupa MI: n-3 fatty acids and 5-y risks of death and cardiovascular disease events in patients with coronary artery disease. Am J Clin Nutr. 2003, 78: 65-71.
He K, Song Y, Daviglus ML, Liu K, Van Horn L, Dyer AR, Greenland P: Accumulated evidence on fish consumption and coronary heart disease mortality: a meta-analysis of cohort studies. Circulation. 2004, 109: 2705-2711. 10.1161/01.CIR.0000132503.19410.6B
Dyerberg J, Eskesen DC, Andersen PW, Astrup A, Buermann B, Christensen JH, Clausen P, Rasmussen BF, Schmidt EB, Tholstrup T: Effects of trans- and n-3 unsaturated fatty acids on cardiovascular risk markers in healthy males: an 8 weeks dietary intervention study. Eur J Clin Nutr. 2004, 58: 1062-1070. 10.1038/sj.ejcn.1601934
Hu FB, Bronner LL, Willett WC, Stampfer MJ, Rexrode KM, Albert CM, Hunter D, Manson JE: Fish and omega-3 fatty acid intake and risk of coronary heart disease in women. JAMA. 2002, 287: 1815-1821. 10.1001/jama.287.14.1815
Daviglus ML, Stamler J, Orencia AJ, Dyer AR, Liu K, Greenland P, Walsh MK, Morris D, Shekelle RB: Fish consumption and the 30-year risk of fatal myocardial infarction. N Engl J Med. 1997, 336: 1046-1053. 10.1056/NEJM199704103361502
Bucher HC, Hengstler P, Schindler C, Meier G: N-3 polyunsaturated fatty acids in coronary heart disease: a meta-analysis of randomized controlled trials. Am J Med. 2002, 112: 298-304. 10.1016/S0002-9343(01)01114-7
GISSI Prevenzione Investigators: Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: results of the GISSI-Prevenzione trial. Lancet. 1999, 354: 447-455. 10.1016/S0140-6736(99)07072-5
Yokoyama M, Origasa H, Matsuzaki M: Effects of eicosapentaenoic acid (EPA) on major coronary events in hypercholesterolemic patients (JELIS): a randomized open-label blinded endpoint analysis. Lancet. 2007, 369: 1090-1098. 10.1016/S0140-6736(07)60527-3
Courtney KR, Colwell WT, Jensen RA: Prostaglandins and pacemaker activity in isolated guinea pig SA node. Prostaglandins. 1978, 16: 451-459. 10.1016/0090-6980(78)90224-1
Das UN: Cardiac pacemaking, cardiac glycosides, anti-arrhythmic drugs and prostaglandins. Speculations Sci Tech. 1979, 2: 381-384.
Das UN, Lee AM, Barritt GJ: Prostanoids can modify response to electrical stimulus and 45Ca2+ exchange in isolated myocardial muscle cells. Prostaglandins Leukot Med. 1983, 12: 305-314. 10.1016/0262-1746(83)90009-4
Das UN: Prostacyclin as an endogenous anti-arrhythmic agent. Basic Res Cardiol. 1983, 78: 716-718. 10.1007/BF01907219
McMillin JB, Bick RJ, Benedict CR: Influence of dietary fish oil on mitochondrial function and response to ischemia. Am J Physiol. 1992, 263 (5 Pt 2): H1479-485.
Demaison L, Sergiel JP, Moreau D, Grynberg A: Influence of the phospholipid n-6/n-3 polyunsaturated fatty acid ratio on the mitochondrial oxidative metabolism before and after myocardial ischemia. Biochim Biophys Acta. 1994, 1227: 53-59.
Kang JX, Leaf A: Effects of long-chain polyunsaturated fatty acids on the contraction of neonatal rat cardiac myocytes. Proc Natl Acad Sci USA. 1994, 91: 9886-9890. 10.1073/pnas.91.21.9886
Kang JX, Leaf A: Protective effects of free polyunsaturated fatty acids on arrhythmias induced by lysophosphatidylcholine or palmitoylcarnitine in neonatal rat cardiac myocytes. Eur J Pharmacol. 1996, 297: 97-106. 10.1016/0014-2999(95)00701-6
Xiao YF, Kang JX, Morgan JP, Leaf A: Blocking effects of polyunsaturated fatty acids on Na+ channels of neonatal rat ventricular myocytes. Proc Natl Acad Sci USA. 1995, 92: 11000-11004. 10.1073/pnas.92.24.11000
Xiao YF, Ke Q, Chen Y, Morgan JP, Leaf A: Inhibitory effect of n-3 fish oil fatty acids on cardiac Na+/Ca2+ exchange currents in HEK293t cells. Biochem Biophys Res Commun. 2004, 321: 116-123. 10.1016/j.bbrc.2004.06.114
Brouilette SW, Moore JS, McMahon AD, Thompson JR, Ford I, Shepherd J, Packard CJ, Samani NJ, : Telomere length, risk of coronary heart disease, and statin treatment in the West of Scotland Primary Prevention Study: a nested case-control study. Lancet. 2007, 369: 107-114. 10.1016/S0140-6736(07)60071-3
Obana N, Takagi S, Kinouchi Y, Takita Y, Sekikawa A, Takahashi S, Hiwatashi N, Oikawa S, Shimosegawa T: Telomere shortening of peripheral blood mononuclear cells in coronary disease patients with metabolic disorders. Intern Med. 2003, 42: 150-153. 10.2169/internalmedicine.42.150
Nakashima H, Ozono R, Suyama C, Sueda T, Kambe M, Oshima T: Telomere attrition in white blood cell correlating with cardiovascular damage. Hypertens Res. 2004, 27: 319-325. 10.1291/hypres.27.319
Jeanclos E, Krolewski A, Skurnick J, Kimura M, Aviv H, Warram JH, Aviv A: Shortened telomere length in white blood cells of patients with IDDM. Diabetes. 1998, 47: 482-486. 10.2337/diabetes.47.3.482
Sampson MJ, Winterborne MS, Hughes JC, Dozio N, Hughes DA: Monocyte telomere shortening and oxidative DNA damage in type 2 diabetes. Diabetes Care. 2006, 29: 283-289. 10.2337/diacare.29.02.06.dc05-1715
Benetos A, Gardner JP, Zureik M, Labat C, Xiaobin L, Adampoulos C, Temmar M, Bean KE, Thomas F, Aviv A: Short telomeres are associated with increased carotid atherosclerosis in hypertensive subjects. Hypertension. 2004, 43: 182-185. 10.1161/01.HYP.0000113081.42868.f4
Demissie S, Levy D, Benjamin EJ, Cupples LA, Gardner JP, Herbert A, Kimura M, Larson MG, Meigs JB, Keaney JF, Aviv A: Insulin resistance, oxidative stress, hypertension, and leukocyte telomere length in men from the Framingham Heart Study. Aging Cell. 2006, 5: 325-330. 10.1111/j.1474-9726.2006.00224.x
Gardner JP, Li S, Srinivasan SR, Chen W, Kimura M, Lu X, Berenson GS, Aviv A: Rise in insulin resistance is associated with escalated telomere attrition. Circulation. 2006, 111: 2171-2177. 10.1161/01.CIR.0000163550.70487.0B
Vasa M, Breitschopf K, Zeiher AM, Dimmeler S: Nitric oxide activates telomerase and delays endothelial cell senescence. Circ Res. 2000, 87: 540-542.
Hayashi T, Matsui-Hirai H, Miyazaki-Akita A, Fukatsu A, Funami J, Ding QF, Kamalanathan S, Hattori Y, Ignarro LJ, Iguchi A: Endothelial cellular senescence is inhibited by nitric oxide: implications in atherosclerosis associated with menopause and diabetes. Proc Natl Acad Sci USA. 2006, 103: 17018-17023. 10.1073/pnas.0607873103
Scalera F, Borlak J, Beckmann B, Martens-Lobenhoffer J, Thum T, Tager M, Bode-Boger SM: Endogenous nitric oxide synthesis inhibitor asymmetric dimethyl L-arginine accelerates endothelial cell senescence. Arterioscler Thromb Vasc Biol. 2004, 24: 1816-1822. 10.1161/01.ATV.0000141843.77133.fc
Bode-Boger SM, Scalera F, Martens-Lobenhoffer J: Asymmetric dimethylarginine (ADMA) accelerates cell senescence. Vasc Med. 2005, 10 (Suppl 1): S65-S71. 10.1191/1358863x05vm606oa
Minamino T, Miyauchi H, Yoshida T, Ishida Y, Yoshida H, Komuro I: Endothelial cell senescence in human atherosclerosis: role of telomere in endothelial dysfunction. Circulation. 2002, 105: 1541-1544. 10.1161/01.CIR.0000013836.85741.17
Matsushita H, Chang E, Glassford AJ, Cooke JP, Chiu CP, Sao PS: eNOS activity is reduced in senescent human endothelial cells: Preservation by hTERT immortalization. Circ Res. 2001, 89: 793-798. 10.1161/hh2101.098443
Das UN: Beneficial actions of polyunsaturated fatty acids in cardiovascular diseases: But, how and why?. Current Nutr Food Sci. 2008, 4: 2-31. 10.2174/157340108783497418. 10.2174/157340108783497418
Eitsuka T, Nakagawa K, Suzuki T, Miyazawa T: Polyunsaturated fatty acids inhibit telomerase activity in DLD-1 human colorectal adenocarcinoma cells: a dual mechanism approach. Biochim Biophys Acta. 2005, 1737: 1-10.
Eitsuka T, Nakagawa K, Miyazawa T: Dual mechanisms for telomerase inhibition in DLD-1 human colorectal adenocarcinoma cells by polyunsaturated fatty acids. Biofactors. 2004, 21: 19-21.
Mori TA, Woodman RJ, Burke V, Puddey IB, Croft KD, Beilin LJ: Effect of eicosapentaenoic acid and docosahexaenoic acid on oxidative stress and inflammatory markers in treated-hypertensive type 2 diabetic subjects. Free Radic Biol Med. 2003, 35: 772-781. 10.1016/S0891-5849(03)00407-6
Mori TA, Puddey IB, Burke V, Croft KD, Dunstan DW, Rivera JH, Beilin LJ: Effect of omega 3 fatty acids on oxidative stress in humans: GC-MS measurement of urinary F2-isoprostane excretion. Redox Rep. 2000, 5: 45-46.
Barden A, Mori TA, Dunstan JA, Taylor AL, Thornton CA, Croft KD, Beilin LJ, Prescott SL: Fish oil supplementation in pregnancy lowers F2-isoprostanes in neonates at high risk of atopy. Free Radic Res. 2004, 38: 233-239. 10.1080/10715760310001656722
Flier MA, Rittirsch D, Nadeau BA, Chen AJ, Sarma JV, Zetoune FS, McGuire SR, List RP, Day DE, Hoesel LM, Gao H, Van Rooijen N, Huber-Lang MS, Neubig RR, Ward PA: Phagocyte-derived catecholamines enhance acute inflammatory injury. Nature. 2007, 449: 721-726. 10.1038/nature06185
Appleton KM, Hayward RC, Gunnell G, Peters TJ, Rogers PJ, Kessler D, Ness AR: Effects of n-3 long-chain polyunsaturated fatty acids on depressed mood: systematic review of published trials. Am J Clin Nutr. 2006, 84: 1308-1316.
Su KP, Huang SY, Chiu CC, Shen WW: Omega-3 fatty acids in major depressive disorder: A preliminary double-blind, placebo-controlled trial. Eur Neuropscychopharmacol. 2003, 13: 267-271. 10.1016/S0924-977X(03)00032-4
Puri BK, Leavitt BR, Hayden MR, Ross CA, Rosenblatt A, Greenamyre JT, Hersch S, Vaddadi KS, Sword A, Horrobin DF, Manku M, Murck H: Ethyl-EPA in Huntington disease: a double-blind, randomized, placebo-controlled trial. Neurology. 2005, 65: 286-292. 10.1212/01.wnl.0000169025.09670.6d
Arvindakshan M, Ghate M, Ranjekar PK, Evans DR, Mahadik SP: Supplementation with a combination of omega-3 fatty acids and antioxidants (vitamins E and C) improves the outcome of schizophrenia. Schizophr Res. 2003, 62: 195-204. 10.1016/S0920-9964(02)00284-0
Green KN, Martinez-Coria H, Khashwji H, Hall EB, Yurko-Mauro KA, Ellis L, LaFerla FM: Dietary docosahexaenoic acid and docosapentaenoic acid ameliorate amyloid-beta and tau pathology via a mechanism involving presenilin 1 levels. J Neurosci. 2007, 27: 4385-4395. 10.1523/JNEUROSCI.0055-07.2007
Das UN: Can perinatal supplementation of long-chain polyunsaturated fatty acids prevent atopy, bronchial asthma and other inflammatory conditions?. Med Sci Monit. 2006, 12: RA99-RA111.
Das UN: Can perinatal supplementation of long-chain polyunsaturated fatty acids prevents schizophrenia in adult life?. Med Sci Monit. 2004, 10: HY33-HY37.
Das UN: Perinatal supplementation of long-chain polyunsaturated fatty acids, immune response, and adult diseases. Med Sci Monit. 2004, 10: HY19-HY25.
Das UN: Can endogenous lipid molecules serve as predictors and prognostic markers of coronary heart disease?. Lipids Health Dis. 2008, 7: 19- 10.1186/1476-511X-7-19
Das UN: Can essential fatty acids reduce the burden of disease(s)?. Lipids Health Dis. 2008, 7: 9- 10.1186/1476-511X-7-9
Das UN: A defect in the activity of Δ6 and Δ5 desaturases may be a factor predisposing to the development of insulin resistance syndrome. Prostaglandins Leukotrienes Essen Fatty Acids. 2005, 72: 343-350. 10.1016/j.plefa.2005.01.002
Das UN: Folic acid and polyunsaturated fatty acids improve cognitive function and prevent depression, dementia, and Alzheimer's disease – but how and why?. Prostaglandins Leukot Essent Fatty Acids. 2008, 78: 11-19. 10.1016/j.plefa.2007.10.006
Acknowledgements
UND was in receipt of Ramalingaswami Fellowship of Department of Biotechnology, New Delhi, India during the tenure of this study.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
UND owns and runs the biotech company UND Life Sciences that specialises in developing lipid-based drugs for cancer, diabetes melltus and hypertension.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Das, U.N. Essential fatty acids and their metabolites could function as endogenous HMG-CoA reductase and ACE enzyme inhibitors, anti-arrhythmic, anti-hypertensive, anti-atherosclerotic, anti-inflammatory, cytoprotective, and cardioprotective molecules. Lipids Health Dis 7, 37 (2008). https://doi.org/10.1186/1476-511X-7-37
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-511X-7-37