
Abstract
Background
Autophagy is an essential process for breaking down macromolecules and aged/damaged cellular organelles to maintain cellular energy balance and cellular nutritional status. The idea that autophagy regulates lipid metabolism is an emerging concept with important implications for atherosclerosis. However, the potential role of autophagy and its relationship with lipid metabolism in foam cell formation remains unclear. In this study, we found that autophagy was involved in the lipopolysaccharide (LPS)-induced the formation of foam cells and was at least partially dependent on adipose differentiation-related protein (ADRP).
Method
Foam cell formation was evaluated by Oil red O staining. Autophagic activity was determined by immunofluorescence and Western blotting. ADRP gene expression of ADRP was examined by real-time PCR (RT-PCR). The protein expression of ADRP and LC3 was measured using Western blotting analysis. Intracellular cholesterol and triglyceride levels in foam cells were quantitatively measured by enzymatic colorimetric assays.
Results
LPS promoted foam cell formation by inducing lipid accumulation in macrophages. The activation of autophagy with rapamycin (Rap) decreased intracellular cholesterol and triglyceride levels, whereas the inhibition of autophagy with 3-methyladenine (3MA) enhanced the accumulation of lipid droplets. Overexpression of ADRP alone increased the formation of foam cells and consequently autophagic activity. In contrast, the inhibitory effects of ADRP activity with siRNA suppressed the activation of autophagy. Taken together, we propose a novel role for ADRP in the regulation of macrophage autophagy during LPS stimulation.
Conclusion
We defined a new molecular pathway in which LPS-induced foam cell formation is regulated through autophagy. These findings facilitate the understanding of the role of autophagy in the development of atherosclerosis.
Similar content being viewed by others


Introduction
Atherosclerosis is a chronic lipid metabolism disorder characterized by the deposition of excess lipids in large arteries. Macrophages take up excessive modified lipoproteins, leading to the accumulation of intracellular cholesterol and triglycerides in the form of lipid droplets and the formation of foam cells, which appear to be a hallmark of the atherosclerosis [1, 2]. Therefore, understanding the potential mechanism of foam cell formation is critical for elucidating the pathogenesis of atherosclerosis and treatment.
Autophagy is a highly regulated intracellular degradation process that mediates the clearance of cytoplasmic proteins, certain pathogens and organelles [3]. When autophagy is initiated, double-membraned autophagosomes form randomly in the cytoplasm and eventually fuse with a lysosome to form an autolysosome where the contents are degraded and recycled for protein synthesis [4]. During the process of autophagic membrane formation, microtubule-associated protein 1 light chain 3 (LC3) is conjugated to a lipid to generate LC3-II that is one of the best characterized structural components of the autophagosomes [5].
Recently, LPS, a potential mediator of inflammatory responses, has been found to induce the macrophage-derived foam cell formation in vitro and promote the development of atherosclerotic plaque in vivo [6, 7]. In addition, LPS could increase the expression of ADRP, a lipid droplet-associated proteins, as part of a coordinated change in macrophage physiology [8]. Studies have indicated that LPS induces autophagy in macrophages [6, 9], and autophagy has been shown to be activated in advanced (late stage) atherosclerotic plaques [10, 11], suggesting that autophagy may play an important role in LPS-induced foam cell formation.
Despite the increasing interest in the phenomenon of autophagy, the role of autophagy in atherosclerotic development and the relationship between autophagy and lipid accumulation have not been established. In this study, we describe a role for autophagy in controlling foam cell formation, which appears to be dependent on ADRP.
Materials and methods
Cell lines, antibodies and chemicals
The human monocytic cell line THP-1 was purchased from the American Type Culture Collection (ATCC). Antibodies against LC3 (Cell signaling technology), ADRP (Abcam) and β-actin (Sigma) were used. LPS, phorbol 12-myristate 13-acetate (PMA), oxidized low density lipoprotein (oxLDL), rapamycin (Rap), and 3-methyladenine (3MA) were purchased from Sigma.
Cell culture and foam cell formation evaluation by Oil red O staining
Human THP-1 cells were seeded into six-well plates at 2 × 105 cells per well in RPMI1640 medium containing 10% fetal bovine serum (FBS), and were maintained at 37°C in a humidified incubator in a 95% air plus 5% CO2 atmosphere. The THP-1 monocytes were treated by the addition of 100 nM PMA for 24 h to facilitate monocytes differentiation into macrophages. After PMA treatment, the adherent macrophages were transformed into foam cells by incubation with 50 μg/ml oxLDL for 24 h. The cells were then treated with Rap (10 μM) or 3MA (10 μM) in the absence or presence of LPS (1 μg/ml) for 24 h. Cultured foam cells were then washed once with phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde for 30 min. After rinsing with 60% isopropanol, foam cells were stained with 0.3% Oil Red O in 60% isopropanol for 15 min and then washed again with 60% isopropanol again. Afterward, foam cells were counterstained with hematoxylin for 3 min. After washing with water, foam cells were photographed with a microscope at 400× magnification.
Measurement of intracellular cholesterol and triglyceride levels
The content of cholesterol and triglycerides in macrophage cells was quantitatively measured by enzymatic colorimetric assays with the kits from Wako (Richmond, VA) according to the manufacturer’s protocols. Briefly, 20 μl of each sample (cell lysate), standard (cholesterol 200 mg/dL) and blank (distilled water) were added into the pre-labeled tubes and added with 2 ml of color reagent (containing cholesterol oxidase, peroxidase, cholesterol ester hydrolase, ascorbate oxidase, 4-aminoantipyrine and DAOS). These reactions were mixed well and incubated at 37°C for 5 min. Finally, the measurements of the absorbance of the samples and standard against the blank were performed at 600 nm. The total cholesterol and triglycerides corresponding to the absorbance of samples were calculated from the standard calibration curve. The concentration of cellular proteins from these cells was measured with a protein assay kit from Bio-Rad (Hercules, CA).
Transfection and establishment of stable cell lines
THP-1 cells were transfected with 2 μg of the purified recombinant plasmid pEGFP-LC3 using the Lipofectamine 2000 transfection reagent (Invitrogen Life Technologies). THP-1 cells stably expressing GFP-LC3 were selected with G418 for 3-4 weeks. The pEGFP vector was used as the control for GFP-LC3.
Autophagy assays
Autophagy was evaluated in cells by fluorescence microscopy, or immunoblotting. In fluorescence microscopy experiments, autophagy was evaluated by examining the punctate forms (type II) of the autophagy marker LC3. Experiments examined either GFP-LC3 or endogenous LC3 stained with the LC3 antibody. Quantitation of autophagy was performed based on the percentage of GFP-LC3-positive autophagic vacuoles or cells with LC3 punctate dots. In all experiments, a minimum of 100 cells per sample was counted, and duplicate or triplicate samples were counted per experimental condition [12]. Statistical analysis was performed using a two-tailed Student’s t test.
Overexpression or knockdown of ADRP in THP-1 macrophages
After induced with PMA, the THP-1 cells were transiently transfected with 2 μg of pCMV5-ADRP or 100 nM siRNA against ADRP with Lipofectamine 2000 transfection reagents (Invitrogen Life Technologies) according to instructions of the manufacturer. Transfection mixtures were added to cells in Opti-MEM medium for 16 h before media was replaced with regular RPMI 1640 media supplemented with 10% FBS.
Real-time PCR
At the end of incubation, total RNA was isolated with TRIzol (TAKARA) according to the manufacturer’s instructions. Two micrograms of total RNA were reverse transcribed into cDNA. The cDNA was subsequently subjected to SYBR Green based real-time PCR using ABI 7500 real time PCR System (Applied Biosystems, Alameda, CA, USA). Primers used in real-time PCR are shown as in Table 1. The gene expression was normalized against the expression of β-actin.
Western blot analysis
Cells were lysed in RIPA buffer (25 mmol/L Tris–HCl pH 7.6, 150 mmol/L NaCl, 1% NP-40, 1% deoxycholate, 1% Triton X-100, 0.5% SDS, 2 mmol/L EDTA, 0.5 mmol/L PMSF, protease inhibitor cocktail). Protein was quantified with Bio-Rad protein assay reagents. Prestained molecular markers (Fermentas) were used to estimate the molecular weight of samples. Proteins were transferred to PVDF membranes (Millipore), and after incubation with primary and secondary antibodies, were detected by ECL regents (Amersham Biosciences). Bands were normalized to β-actin and expressed as a percent of control.
Statistical analysis
Values were shown as the mean ± SEM of measurements of at least three independently performed experiments to avoid possible variation of the cell cultures. For statistical analyses, Student’s t test was employed and P < 0.05 was considered to be statistically significant.
Results
LPS promotes foam cell formation by inducing lipid accumulation in macrophages
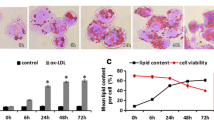
To examine the effect of LPS on macrophage lipid accumulation during foam cell formation, human THP-1 macrophage-derived foam cells were incubated with LPS (1 μg/ml) aggregates for a different periods (0, 8, 16, and 24 h), followed by visualization of lipid laden in macrophages with Oil Red O staining. As shown in Figure 1A-D, the number of lipid droplets in the cytoplasm of cells treated with LPS for 24 h were obviously induced compared to the vehicle treated cells. Then, we harvested the cells to determine cellular total cholesterol and triglyceride content. Consistent with Oil Red O staining, measurement of intracellular lipid contents indicated that the level of triglyceride and total cholesterol (Figure 1E and F) were markedly increased after LPS incubation. These data showed that LPS augmented lipid accumulation during the foam cell formation.

LPS enhances lipid accumulation during the macrophage foam cell formation. (A) Human THP-1 macrophages. (B-D) THP-1 macrophages were pre-treated with oxLDL (50 μg/ml) for 24 h and exposed to LPS (1 μg/ml) in RPMI1640 with 10% FBS for different periods (8, 16, and 24 h). Lipid droplets were visualized by Oil Red O staining. (E, F) The levels of triglcyceride (TG) and total cholesterol (TC) were evaluated. Data were presented as means ± S.E. of μM of triglyceride or cholesterol per milligram protein from 3 independent experiments. *significant difference from LPS treatment P < 0.05; **, P < 0.01.
Autophagy was activated during LPS-induced foam cell formation
To evaluate whether autophagy was significantly different after LPS treated, we analyzed distribution of LC3, typical of autophagosomes [13, 14]. Autophagy formation can be visualized with an electron microscope, which is by far the most confirmative analysis for autophagy. LC3 is recruited to autophagosome membranes during autophagy, and GFP-LC3 has been used as a marker for autophagy induction [15]. Interestingly, by transfection with GFP-LC3, we found that LPS exposure resulted in the redistribution of GFP-LC3 from a diffuse to a punctate pattern, and induced a significant increase in the percentage of cells with GFP-LC3 autophagosomes (Figure 2A). Autophagic activity in human THP-1 macrophages was either stimulated with Rap, an inhibitor of mTOR, and inhibited by using 3MA, an inhibitor of autophagy. Exposure of THP-1 macrophages to LPS increased Rap induced the LC3-GFP-labeled autophagosomes whereas 3MA decreased the number of LPS-induced autophagosomes in macrophages (Figure 2A). When autophagy occurs, cytosolic LC3 (LC3-I) is modified to its membrane-bound form (LC3-II), which is located on preautophagosomes and autophagosomes and thus commonly used as an autophagosomal marker [15]. An increased LC3II/LC3I ratio indicates enhanced autophagy activity. Consistently, Rap increased the ratio of LC3II/LC3I whereas 3MA decreased the LC3II/LC3I ratio. Exposure to LPS increased the ratio of LC3II/LC3I and the GFP-labeled autophagosomes (Figure 2B). Moreover, the time-course (0, 8, 16, and 24 h) experiments revealed that the percentage of cells expressing autophagosomes was maximal at 16 h after LPS stimulation (Figure 2C). These data indicate that LPS might activate autophagy during foam cell formation.

LPS increases the autophagic activity during the human macrophage foam cell formation. THP-1 macrophages were incubated in the absence (control) or presence of LPS (1 μg/ml), LPS plus rapamycin (Rap, an autophagy activator, 10 μM), Rap alone, LPS plus 3-methyladenine (3MA, an autophagy inhibitor, 10 μM) or 3MA alone as indicated (n = 3). (A) THP-1 human macrophages stably expressing GFP-LC3 were treated with LPS (1 μg/ml) for the indicated time points. (B) The levels of LC3II and LC3I were detected with specific antibodies by using immunoblotting and the percentage of cells with autophagosomes was calculated. (C) Representative immunofluorescence images with GFP-LC3+ in THP-1 human macrophages after exposure to 1 μg/ml LPS for the indicated time points (0, 8, 16, and 24 h) and the percentage of cells with autophagosomes were calculated. Data represent mean ± S.E. from 3 independent experiments. *significant difference from LPS treatment P < 0.05; **, P < 0.01.
Modulation of autophagic activity can alter the accumulation of lipid droplets during the foam cell formation
Autophagy has recently been implicated in the control of lipid accumulation [16–20]. To examine the direct effect of autophagy during lipid accumulation in macrophages, autophagy activity in human THP-1 macrophages was either stimulated with Rap or inhibited by using 3MA. As shown in Figure 3A, lipid accumulation was increased by the exposure to LPS, and the increase was enhanced by Rap. In contrast, inhibition of autophagy with 3MA prevented both the basal and the LPS-induced lipid accumulation (Figure 3A), whereas Rap alone did not alter the level of total cholesterol or triglyceride level. In contrast, inhibition of autophagy with 3MA decreased triglyceride in macrophages that were either treated with LPS or not (Figure 3B). Together, these results demonstrate that autophagic activity on lipid droplets plays an important role in the accumulation of lipids during foam cell formation.

Modulation of autophagic activity alters the accumulation of lipid droplets induced by the exposure to LPS in THP-1 macrophages. (A) THP-1 macrophages were pre-treated with oxLDL (50 μg/ml) for 24 h, then incubated in the absence (control) or presence of 1 μg/ml LPS, LPS plus Rap (10 μM), Rap alone, LPS plus 3-methyladenine (3MA, an autophagy inhibitor, 10 μM) or 3MA alone for 24 h as indicated (n = 3). Cells were fixed and stained with Oil Red O to visualize the lipid droplets. (B,C) The levels of total cholesterol and triglcyceride were evaluated by enzymatic colorimetric assays. Results were presented as mean ± SEM of 3 independent experiments, each in triplicate. *significant difference from LPS treatment P < 0.05; **, P < 0.01.
The level of ADRP is correlated with the autophagic activity in LPS-treated macrophages
ADRP is a lipid droplet-associated proteins that plays a critical role in the homeostasis of cytosolic lipid droplets in various types of cells [7]. ADRP is also coated on lipid droplets (LDs) in the macrophage and foam cells, and its levels is directly correlated with cellular neutral lipid content in the foam cells of atherosclerosis [21, 22]. To examine whether ADRP is involved in LPS-induced autophagy, macrophages were incubated with LPS aggregates for a different time (0, 8, 16, and 24 h) or for 24 h with different amounts of LPS (10–1000 ng/ml), the total RNA was then extracted, and gene expression was measured by real-time PCR analysis. As shown in Figure 4A and B, exposure to LPS stimulated ADRP in a time-and dose-dependent manner. Furthermore, treatment with Rap upregulated the levels of ADRP. In contrast, 3MA inhibited ADRP protein levels as shown by western blotting analysis (Figure 4C and D). These results demonstrate that the level of ADRP in macrophages is positively correlated with the autophagic activity after treatment with LPS.

Autophagy altered the expression of ADRP in LPS-stimulated THP-1 macorphages. (A, B) Macrophages were incubated with LPS for a different time (0, 8, 16, and 24 h) or with different amounts of LPS (10-1000 ng/ml) for 24 h. Real-time PCR analysis ADRP gene expression. (C) THP-1 cells were treated with 1 μg/ml LPS in the absence or presence of Rap (10 μM), or 3MA (10 μM) as noted for 24 h. The level of ADRP was then quantified by real-time PCR. (D) The levels of ADRP and β-actin proteins were detected by using immunoblotting with specific antibodies. Results were presented as mean ± S.E. of 3 independent experiments. *significant difference from LPS treatment P < 0.05; **, P < 0.01.
ADRP enhances autophagy in the LPS-induced macrophage foam cell
To determine the role of ADRP in the LPS-induced autophagy during the foam cell formation, ADRP was overexpressed in THP-1 macrophages by transfection with pCMV5-ADRP plasmid. We confirmed that the LC3-II/LC3-I ratio increased when ADRP was overexpressed in macrophages, indicating that overexpressed ADRP could enhance LPS-induced autophagy (Figure 5A). Furthermore, small interfering RNA (siRNA) against ADRP was used to knockdown ADRP in THP-1 cells stably expressing GFP-LC3. Subjected to LPS treatment and evaluated for autophagy, we found that knockdown of ADRP with specific siRNA inhibited LPS-induced LC3-II levels in comparison with a scrambled nontargeted siRNA (Figure 5B). In summary, ADRP could enhance the LPS-induced autophagic activity on lipid droplets in THP-1 macrophages.

ADRP overexpression increases the LPS-induced autophagic activity, while ADRP knockdown downregulates the autophagic activity in THP-1 macrophages. (A) ADRP overexpression by adenoviral gene transfer augmented LPS-induced LC3 protein levels as determined by western blotting and the percentage of cells with autophagosomes were calculated. (B) Specific knockdown of ADRP with ADRP siRNA prior to LPS stimulation of THP-1 cells inhibited the LC3-II/I ratio as determined by western blotting and the percentage of cells with autophagosomes was calculated. Results were presented as mean ± SEM of 3 independent experiments, each in triplicate. *significant difference from LPS treatment P < 0.05; **, P < 0.01.
Discussion
● The data in this study focus on the functional relationships between autophagy and lipid metabolism. We found that autophagy at least partially augmented LPS-induced foam cell formation owing to its effect on the induction of ADRP.
Foam cell formation is a hallmark of the initial stage of atherosclerosis. The activation of macrophages by LPS has been shown to increase fat accumulation (Figure 1). In fact, studies have demonstrated that LPS influences many aspects of macrophage-derived foam cell formation, such as the promotion of LDL oxidation [23], lipid uptake [24], up-regulation of macrophage fatty acid transport and inhibition of cholesterol efflux [25, 26].
Recently, a study has demonstrated that blocking autophagy in cultured preadipocytes decreases the cellular triglyceride content and levels of key adipogenic transcription factors CEBP-α (CCAAT/enhancer-binding protein alpha), CEBP-β (CCAAT/enhancer binding protein beta ) and PPAR-γ [27]. However, little is known about the role of autophagy in atherosclerosis. In this study, we demonstrate that autophagy is activated in LPS-induced macrophage foam cells (Figure 2). Here, we just focus on the autophagy involved in the uptake of lipid and lipid droplet formation in macrophage-derived foam cells. In this work, the activation of autophagy enhanced levels of intracellular cholesterol and triglyceride while inhibition of autophagy could block the accumulation of lipid droplets in macrophages (Figure 3). Consist with these results, an increase rather than a decrease in foam cell formation would be expected in macrophages undergoing autophagy because protein degradation, as well as the decline of protein synthesis, in autophagic cells readily blocks the utilization of lipids for lipid–protein conjugation, which in turn results in the formation of lipid droplets [28].
ADRP is the major macrophage LD coat protein, and its levels are directly related with cellular neural lipid content [29]. Whole-body as well as macrophage specific ADRP deficiency inhibits foam cell formation and protects against atherosclerosis development [29]. Despite ADRP is an absolute LD coat protein, and was also detected in lysosomes in macrophage foam cell, suggesting that ADRP may play an important role in autophagy [30]. Our results also support a possible role for ADRP in LPS-induced autophagic signaling. Overexpression of ADRP leads to augment of autophagy, which has been shown to increase autophagic signaling. Moreover, inhibition of ADRP can suppress the activation of autophagy (Figures 4, 5). Interestingly, ADRP overexpression prevented cholesterol efflux to apolipoprotein A-I (ApoA-I) [31]. One possible explanation is that ADRP may be localized around cytosolic lipid droplets in the macrophages, protecting them from the activity of cholesterol esterases and thereby reducing the availability of free cholesterol (FC) for efflux. Thus, autophagy is activated in response to a lipid challenge. These results indicated that ADRP could protect LDs against the lipolysis mediated by autophagy and lysosomes in the LPS-induced macrophage foam cell.
In conclusion, these data link autophagy and lipid metabolism, two separate pathways that are both activated by LPS. Although further studies are required to establish a correlation between the regulation of autophagy in macrophages and atherosclerosis, our results suggest that the autophagic activity may be involved in the accumulation of lipid droplets in macrophage foam cells. Furthermore, our data indicate that ADRP facilitates autophagic activity in the LPS-induced macrophage foam cells. Summarily, these results are helpful for us to understand the role of autophagy in the development of atherosclerosis.
References
Mori M, Itabe H, Higashi Y, Fujimoto Y, Shiomi M, Yoshizumi M, Ouchi Y, Takano T: Foam cell formation containing lipid droplets enriched with free cholesterol by hyperlipidemic serum. J Lipid Res. 2001, 42: 1771-1781.
Glass CK, Witztum JL: Atherosclerosis: the road ahead. Cell. 2001, 104: 503-516. 10.1016/S0092-8674(01)00238-0
Mizushima N, Ohsumi Y, Yoshimori T: Autophagosome formation in mammalian cells. Cell Struct Funct. 2002, 27: 421-429. 10.1247/csf.27.421
Klionsky DJ, Abeliovich H, Agostinis P, Agrawal DK, Aliev G, Askew DS, Baba M, Baehrecke EH, Bahr BA, Ballabio A: Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008, 4: 151-175.
Yang Z, Klionsky DJ: Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010, 22: 124-131. 10.1016/j.ceb.2009.11.014
Yuan H, Perry CN, Huang C, Iwai-Kanai E, Carreira RS, Glembotski CC, Gottlieb RA: LPS-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. Am J Physiol Heart Circ Physiol. 2009, 296: H470-H479.
Ostos MA, Recalde D, Zakin MM, Scott-Algara D: Implication of natural killer T cells in atherosclerosis development during a LPS-induced chronic inflammation. FEBS Lett. 2002, 519: 23-29. 10.1016/S0014-5793(02)02692-3
Feingold KR, Kazemi MR, Magra AL, McDonald CM, Chui LG, Shigenaga JK, Patzek SM, Chan ZW, Londos C, Grunfeld C: ADRP/ADFP and Mal1 expression are increased in macrophages treated with TLR agonists. Atherosclerosis. 2010, 209: 81-88. 10.1016/j.atherosclerosis.2009.08.042
Waltz P, Carchman EH, Young AC, Rao J, Rosengart MR, Kaczorowski D, Zuckerbraun BS: Lipopolysaccaride induces autophagic signaling in macrophages via a TLR4, heme oxygenase-1 dependent pathway. Autophagy. 2011, 7: 315-320. 10.4161/auto.7.3.14044
Martinet W, De Meyer GR: Autophagy in atherosclerosis. Curr Atheroscler Rep. 2008, 10: 216-223. 10.1007/s11883-008-0034-y
Martinet W, De Meyer GR: Autophagy in atherosclerosis: a cell survival and death phenomenon with therapeutic potential. Circ Res. 2009, 104: 304-317. 10.1161/CIRCRESAHA.108.188318
Liu Y, Schiff M, Czymmek K, Talloczy Z, Levine B, Dinesh-Kumar SP: Autophagy regulates programmed cell death during the plant innate immune response. Cell. 2005, 121: 567-577. 10.1016/j.cell.2005.03.007
Ogawa M, Yoshimori T, Suzuki T, Sagara H, Mizushima N, Sasakawa C: Escape of intracellular Shigella from autophagy. Science. 2005, 307: 727-731. 10.1126/science.1106036
Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V: Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004, 119: 753-766. 10.1016/j.cell.2004.11.038
Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T: LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000, 19: 5720-5728. 10.1093/emboj/19.21.5720
Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF: Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012, 15: 534-544. 10.1016/j.cmet.2012.02.011
Singh R, Kaushik S, Wang Y, Xiang Y, Novak I, Komatsu M, Tanaka K, Cuervo AM, Czaja MJ: Autophagy regulates lipid metabolism. Nature. 2009, 458: 1131-1135. 10.1038/nature07976
Zhang Y, Goldman S, Baerga R, Zhao Y, Komatsu M, Jin S: Adipose-specific deletion of autophagy-related gene 7 (atg7) in mice reveals a role in adipogenesis. Proc Natl Acad Sci USA. 2009, 106: 19860-19865. 10.1073/pnas.0906048106
Xu J, Dang Y, Ren YR, Liu JO: Cholesterol trafficking is required for mTOR activation in endothelial cells. Proc Natl Acad Sci USA. 2010, 107: 4764-4769. 10.1073/pnas.0910872107
Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD: ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci USA. 2010, 107: 4153-4158. 10.1073/pnas.0913860107
Larigauderie G, Cuaz-Perolin C, Younes AB, Furman C, Lasselin C, Copin C, Jaye M, Fruchart JC, Rouis M: Adipophilin increases triglyceride storage in human macrophages by stimulation of biosynthesis and inhibition of beta-oxidation. FEBS J. 2006, 273: 3498-3510. 10.1111/j.1742-4658.2006.05357.x
Listenberger LL, Ostermeyer-Fay AG, Goldberg EB, Brown WJ, Brown DA: Adipocyte differentiation-related protein reduces the lipid droplet association of adipose triglyceride lipase and slows triacylglycerol turnover. J Lipid Res. 2007, 48: 2751-2761. 10.1194/jlr.M700359-JLR200
Memon RA, Staprans I, Noor M, Holleran WM, Uchida Y, Moser AH, Feingold KR, Grunfeld C: Infection and inflammation induce LDL oxidation in vivo. Arterioscler Thromb Vasc Biol. 2000, 20: 1536-1542. 10.1161/01.ATV.20.6.1536
Choi SH, Harkewicz R, Lee JH, Boullier A, Almazan F, Li AC, Witztum JL, Bae YS, Miller YI: Lipoprotein accumulation in macrophages via toll-like receptor-4-dependent fluid phase uptake. Circ Res. 2009, 104: 1355-1363. 10.1161/CIRCRESAHA.108.192880
Miller YI, Choi SH, Fang L, Harkewicz R: Toll-like receptor-4 and lipoprotein accumulation in macrophages. Trends Cardiovasc Med. 2009, 19: 227-232. 10.1016/j.tcm.2010.02.001
Khovidhunkit W, Moser AH, Shigenaga JK, Grunfeld C, Feingold KR: Endotoxin down-regulates ABCG5 and ABCG8 in mouse liver and ABCA1 and ABCG1 in J774 murine macrophages: differential role of LXR. J Lipid Res. 2003, 44: 1728-1736. 10.1194/jlr.M300100-JLR200
Singh R, Xiang Y, Wang Y, Baikati K, Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ, Czaja MJ: Autophagy regulates adipose mass and differentiation in mice. J Clin Invest. 2009, 119: 3329-3339.
Martinet W, Verheye S, De Meyer GR: Selective depletion of macrophages in atherosclerotic plaques via macrophage-specific initiation of cell death. Trends Cardiovasc Med. 2007, 17: 69-75. 10.1016/j.tcm.2006.12.004
Paul A, Chang BH, Li L, Yechoor VK, Chan L: Deficiency of adipose differentiation-related protein impairs foam cell formation and protects against atherosclerosis. Circ Res. 2008, 102: 1492-1501. 10.1161/CIRCRESAHA.107.168070
Ouimet M, Franklin V, Mak E, Liao X, Tabas I, Marcel YL: Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011, 13: 655-667. 10.1016/j.cmet.2011.03.023
Larigauderie G, Furman C, Jaye M, Lasselin C, Copin C, Fruchart JC, Castro G, Rouis M: Adipophilin enhances lipid accumulation and prevents lipid efflux from THP-1 macrophages: potential role in atherogenesis. Arterioscler Thromb Vasc Biol. 2004, 24: 504-510. 10.1161/01.ATV.0000115638.27381.97
Acknowledgements
This study was supported by National Natural Science Foundation of China (NSFC) (81070248, 81070249, 31171132, 81100612) and the fundings from 211 projects of China. The authors gratefully acknowledge the technical assistance from Dr. Linkang Zhou (Tsinghua University, China). We also thank Dr. Rui’an Wang (Fourth Military Medical Univeristy, China) for very favorable discussion and comments to improve the quality of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
There are no competing interest in this study.
Authors’ contributions
XYF and YY carried out the study design, data collection and analysis, and drafted the manuscript. CW and JF carried out the autophagy assays. ZYY and XMZ participated in the real-time PCR experiment. WS and PZH constructed ADRP-siRNA and pCMV5-ADRP plasmids. PFZ participated in the measurement of intracellular cholesterol and triglyceride levels in foam cells. JY conceived of the study, and participated in its design and coordination and helped to draft the manuscript. All authors read and approved the final manuscript.
Xuyang Feng, Yuan Yuan contributed equally to this work.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Feng, X., Yuan, Y., Wang, C. et al. Autophagy involved in lipopolysaccharide-induced foam cell formation is mediated by adipose differentiation-related protein. Lipids Health Dis 13, 10 (2014). https://doi.org/10.1186/1476-511X-13-10
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-511X-13-10