
Abstract
Lung cancer is the number one cancer killer in the United States. This disease is clinically divided into two sub-types, small cell lung cancer, (10–15% of lung cancer cases), and non-small cell lung cancer (NSCLC; 85–90% of cases). Early detection of NSCLC, which is the more common and less aggressive of the two sub-types, has the highest potential for saving lives. As yet, no routine screening method that enables early detection exists, and this is a key factor in the high mortality rate of this disease. Imaging and cytology-based screening strategies have been employed for early detection, and while some are sensitive, none have been demonstrated to reduce lung cancer mortality. However, mortality might be reduced by developing specific molecular markers that can complement imaging techniques. DNA methylation has emerged as a highly promising biomarker and is being actively studied in multiple cancers. The analysis of DNA methylation-based biomarkers is rapidly advancing, and a large number of potential biomarkers have been identified. Here we present a detailed review of the literature, focusing on DNA methylation-based markers developed using primary NSCLC tissue. Viable markers for clinical diagnosis must be detectable in 'remote media' such as blood, sputum, bronchoalveolar lavage, or even exhaled breath condensate. We discuss progress on their detection in such media and the sensitivity and specificity of the molecular marker panels identified to date. Lastly, we look to future advancements that will be made possible with the interrogation of the epigenome.
Similar content being viewed by others

Background
Worldwide lung cancer kills over one million people each year, and as the leading cause of cancer death in men and second leading cause in women, it is a major health problem [1]. This disease is largely smoking-associated. While in developed countries smoking rates are decreasing, the use of tobacco products is increasing in developing countries. In combination with a spike in the number of lung cancer cases in never smokers, this ensures that lung cancer will remain a major health problem [1]. Clinically, lung cancer is divided into two subtypes, small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). SCLC is the more aggressive subtype, and accounts for 10–15% of all cases. The remaining 85–90% of cases are classified as NSCLC, which is further histologically subdivided into four categories; adenocarcinoma (AD), squamous cell carcinoma (SQ), large cell carcinoma (LC) and 'others', for example cancers of neuroendocrine origin.
In the United States lung cancer is the number one cancer killer in both men and women, accounting for over 160,000 deaths each year [2]. Interestingly, it is not the most commonly diagnosed cancer; breast and prostate cancer have a higher incidence. A reason for this disparity is that early detection methods exist for breast and prostate cancer, and these are widely used in the population. As a result, the five-year survival rate is 89 and 99% (respectively) for these cancers, as opposed to a very low 15% for lung cancer [2]. When early stage lung cancer is detected, the survival rate can increase dramatically. For example, one report on detection of early stage cancers using low dose spiral computed tomography (LDSCT) described a ten-year survival rate of 88% [3]. While there is concern that LDSCT leads to overdiagnosis (detection of indolent cancers that would normally not lead to death), it is undisputed that effective early detection of lesions that would otherwise progress to invasive cancer could reduce lung cancer mortality. In an effort to achieve early detection many imaging and cytology-based strategies have been employed, however none have yet been proven effective. Molecular markers would provide an alternative approach and among them, DNA methylation alterations show great promise. Here we present an update of the field of DNA methylation markers for early lung cancer detection.
Early detection of lung cancer
Original early detection methods for lung cancer were focused on screening using chest X-ray and sputum cytology. Randomized controlled trials demonstrated no reduction in mortality using these techniques [4, 5]. The question has been raised as to whether these trials had enough statistical power to determine a mortality benefit [5, 6]. The Prostate, Lung, Colorectal and Ovarian cancer trial currently being conducted by the National Cancer Institute is a larger trial and may conclusively reveal whether chest X-ray screening can reduce mortality [5]. As discussed later, studies of molecular instead of cytological changes in sputum samples appear promising [7].
Following the apparent failure of chest X-ray and sputum cytology as effective screening techniques, attention was focused on a more sensitive imaging method – Low Dose Spiral Computed Tomography (LDSCT). Several trials of LDSCT as a screening tool in high-risk populations have been conducted [8–14]. It is clear that LDSCT is more sensitive than chest X-ray [11, 12], as it can detect non-calcified nodules as small as 1 mm. Such high sensitivity comes with a price. The number of non-calcified nodules detected is far greater than the number of actual cancers. A Mayo Clinic study in 1999 reported that <2.0% of non-calcified nodules detected were actually cancer [15]. This presents two potential problems for LDSCT as an early detection method. Firstly, there is the potential for many false positive results, which would result in low specificity if LDSCT were applied as a lung cancer screening tool. The second problem is that in order to determine which nodules are actually cancer, patients will require follow up procedures (further scans, possibly biopsies or resections). These are costly, invasive, and can result in patient morbidity and mortality. Crestanello et al. report that 9 out of 54 patients underwent surgery for benign nodules [16]. A review of seven studies by Diederich and Wormanns reported that 4–55% of patients had invasive procedures for benign lesions [6].
An increase in survival in LDSCT-screened lung cancer patients has been reported; the IELCAP study reports an 88% 10-year survival [3]. Many argue that the increased survival rate seen is due to an overdiagnosis bias. Using the Yankelevitz criteria of overdiagnosis – a tumor volume doubling time (VDT) of > 400 days [17] – 27% of the detected cancers in a study by Lindell et al. would be considered overdiagnosed [8]. In a review by Jett of a Japanese study, 33% of the cancers detected have a VDT of >400 days [18], and hence would be considered overdiagnosed [15]. Using a predictive model, Bach and colleagues recently examined the combined results of LDSCT screening trials from three centers. They found an excess number of cases diagnosed at each screening point compared to the predicted number, without a decline in the number of advanced cancers being detected. This supports the notion of overdiagnosis in LDSCT screening [19]. The true measure of efficacy of an early detection method is a reduction in mortality. Whether LDSCT screening in high-risk populations decreases lung cancer mortality remains unknown. The answer to this question will hopefully be provided by one of several ongoing randomized controlled trials (for example the US-based National Lung Screening Trial, and in the Netherlands, the Dutch Lung Cancer Screening Trial). The conclusions from such trials will determine the fate of LDSCT as an early detection strategy.
Another imaging-based early detection approach is autofluoresence bronchoscopy (AFB). This distinguishes between tumor and non-tumor tissue based on the tumor-specific change in tissue autofluoresence. AFB has been shown to be effective at detecting preneoplastic lesions and lung cancers [20]. The drawbacks of the method are that it is invasive, it mainly detects centrally located cancers [21], and it is not highly specific [21, 22].
Since imaging techniques have not yet proven effective as an early detection method, a sensitive and specific screening strategy remains to be found. To fill this void, research focus has shifted to molecular approaches. The goal is to identify molecular markers (generally DNA, RNA or protein) that reflect characteristics of lethal tumors, and that can be exploited for early detection of these lesions at the pre-invasive stage. To function as molecular markers in a screening test, these molecules must be detectible in remote media. If molecular markers that allow detection of cancer are identified, they will require complementary highly sensitive imaging methods such as LDSCT to locate the cancer. Identified molecular markers could be potentially targeted by agents to help specifically enhance tumor imaging [23].
DNA methylation
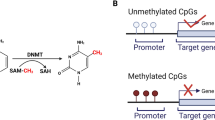
One highly promising molecular biomarker is DNA methylation. This enzymatic addition of a methyl group at the 5-position of the cytosine in a CpG (cytosine-guanine) dinucleotide is a normal process within cells. In cancer, despite a global hypomethylation, one observes hypermethylation in regions of the genome described as CpG islands [24, 25]. These islands are present in almost half of all genes and are frequently promoter-associated [26]. The common occurrence of DNA hypermethylation in all types of cancer makes it an ideal biomarker, one that has been extensively investigated. An advantage of DNA methylation over protein-based markers is that it is readily amplifiable and easily detectable using PCR-based approaches. In addition, contrary to cancer-specific mutations, which could occur anywhere in a gene, cancer-specific DNA hypermethylation occurs in defined regions, usually in or near the promoter of genes. Thus, it is easy to devise targeted probes to measure this molecular alteration. Conveniently, these probes can be readily combined into panels, which is important because no single molecular alteration involved in cancer can be expected to be present in every cancer case. Thus DNA methylation at a single gene would likely allow detection of a subset of cancers. Assembly of a complementary panel of DNA methylation probes would therefore increase sensitivity [27, 28]. Finally, it has been demonstrated that methylated DNA can be isolated from 'remote media' making it well-suited for non-invasive detection [29, 30].
Overview of DNA methylation analysis in NSCLC
In this review, we focus on DNA methylation-based biomarkers for early detection of NSCLC. Because NSCLC is the less aggressive lung cancer subtype, and accounts for 85–90% of all cases, its early detection holds the most promise for saving lives. A plethora of studies describing DNA methylation in non-small-cell lung cancer exist. These studies are summarized in three tables, which, due to their size, are attached to this manuscript as Additional files 1, 2, 3. Each file lists the relevant loci in alphabetical order. Additional file 1 lists information from studies of less than 20 loci. Additional file 2 lists the results of DNA methylation studies of 20 or more loci, or genome wide approaches. Lastly, Additional file 3 discusses loci studied in remote media from cancer patients. The contents of these tables are discussed in more detail below.
Initial DNA methylation studies in NSCLC focused on single loci (or a small number of well known loci) that were selected because of their potential functional role in cancer. The goals of these studies were a) to see if methylation was involved in lung cancer pathogenesis, or b) to determine if methylation of a given gene could be correlated with clinical factors, and hence serve as a prognostic marker. This led to the characterization of the DNA methylation status of many loci in NSCLC (listed in Additional file 1) [31–103]. The information gathered in these studies could be of clinical use for early detection, chemo prevention, diagnosis, treatment or prognosis [104]. Further studies employed panels of 8–19 loci (including these previously reported loci) for DNA methylation profiling [105–116] (see Additional file 1). This profiling was aimed at characterizing methylation status of many loci in NSCLC, or in some cases, at identifying loci with the highest methylation frequency in tumors versus non-tumor tissues, that could potentially be used as DNA methylation-based biomarkers of the disease.
Several loci identified in both types of studies (e.g. APC, CADM1, CDH1, CDH13, CDKN2A/p14(ARF), CDKN2A/p16, DAPK, FHIT, GSTP1, MGMT, MLH1 and RASSF1A) are reported to be methylated multiple independent times in the literature (reviewed in Additional file 1), and there is general consistency in the observed methylation frequency for these loci. Any inconsistencies could have multiple explanations, for example: the use of different techniques to study the methylation status, differences in the population in each study, and a difference in the subtype composition of the NSCLC collection studied.
To further characterize DNA methylation in NSCLC and facilitate the discovery of new markers, more recent studies have employed approaches that analyze large numbers of loci at one time. In these studies, the goal has been to identify DNA methylation-based discriminators of tumor and normal tissues, and tumor subtypes. Some of these approaches were targeted; the loci analyzed were selected based on their relationship to cancer. Other approaches were not designed to interrogate DNA methylation at specific loci, instead they examined the genome in greater depth and identified potentially informative DNA methylation biomarkers based on comparative profiling between tumor and non-tumor cells/tissues. The most promising loci to emerge from these reports are reviewed in Additional file 2. One targeted approach is to use sodium bisulfite-treated DNA (in which unmethylated Cs have been converted to Us) for semi-quantitative real time PCR (MethyLight) to examine methylation levels of multiple loci. Three recent reports described an examination of 27 loci in NSCLC [117], 28 loci in AD [27], and 42 loci in SQ [28] using MethyLight. All three studies described a panel of loci with the ability to sensitively and specifically detect cancer. Using a MALDI-TOF based approach 47 loci were studied in tumor and non-tumor tissues from 96 patients. Six loci (CLEC3B (previously TNA), MGP, RASSF1, SDK2, SERPINB5 and XAGE1A (previously GAGED2)) with statistically significantly higher methylation in tumor samples compared with non-tumor samples were identified [118]. A targeted microarray was used to study the methylation status of 59 loci (245 CpGs) and a set of loci to discriminate SQ (ADPRH (formerly ARH1), GP1BB, RARB and TMEFF2) and AD (CDKN1C, MGMT, TMEFF2) from normal lung was identified [119]. In a similar system, the promoter regions of 288 cancer-related genes were examined. Twenty-eight potential biomarker loci were identified and 5 were further examined in lung cancer tissues, yielding two (PAX3 and PYCARD/ASC) that showed frequent hypermethylation [120]. Restriction landmark genomic scanning (RLGS) allows interrogation of up to 2000 promoter sequences. In a study of 1184 CpG islands Dai et al. discovered 11 genes that are differentially methylated in cancer, two of which are methylated in ≥ 50% of tumors (GNAL and PDX1) [121]. A newer high throughput approach is the Illumina GoldenGate platform, which examines 1505 CpG sites in 807 genes. Recently a panel of loci that detects adenocarcinoma was discovered, of which 8 were further examined by bisulfite genomic sequencing (ASCL2, CDH13, HOXA11, HOXA5, NPY, RUNX3, TERT and TP73) [122]. A study using a large methylation microarray analyzed the promoter regions of 8091 loci, identifying the frequently methylated CIDEB gene [123].
While these approaches can be used to determine the DNA methylation status of large numbers of genes, a non locus-targeted approach that allows unbiased interrogation of DNA methylation in the genome could examine far more loci. This could yield additional biomarkers, as well as new information about general DNA methylation patterns in lung cancer. Using an expression microarray one can identify genes induced in cell lines treated with a DNA methylation inhibitor. Such genes are potential DNA methylation targets. Using this approach, Shames et al. identified 132 tumor-specific methylation candidates, 45 of which were further investigated, revealing seven potential lung cancer markers (ALDH1A3, BNC1, CCNA1, CTSZ, LOX, MSX1 and NRCAM) three of which showed frequent tumor-specific hypermethylation compared to non-tumors [124]. Cortese et al. used a different approach, studying the DNA methylation of genes that are differentially expressed in fetal vs. adult lung. Four loci (FGFR3, LAPTM5, MDK, MEOX2) were identified as aberrantly methylated in lung cancer, one with high frequency [125].
Using a methylated CpG island recovery assay coupled with microarray analysis (MIRA-microarray), Rauch et al. enriched for CpG regions and then hybridized this to a CpG microarray containing 12,192 CpG islands, ≥ 60% of which map to the 5' end of known or putative genes. Multiple highly methylated loci were identified, of which the top 50 were reported [126]. In follow-up studies they identified several loci as markers for SQ lung cancer [127], including HOXA7 and HOXA9 [128]. It is of note that while the non-targeted approaches have the potential to rapidly identify many more biomarkers, the candidate biomarker loci must still be validated in primary tumors using traditional approaches.
In general, there is not a large overlap between the top loci identified in the targeted and non-targeted approaches. Several frequently methylated loci identified in early studies, for example CDKN2A/p16, CDH13, MGMT and RASSF1 remain viable markers when assessed in a larger context, providing support for their role in cancer development/progression [27, 28, 118, 119, 122]. Methylation of genes that are occupied by transcriptionally repressive polycomb group protein in embryonic stem cells, such as members of the HOX and PAX families, was detected by targeted as well as genomic approaches. This reinforces the notion that these genes may be prone to cancer-specific methylation [129]. Further investigation of this group of genes is warranted.
Modest overlap between the top loci from the non-targeted studies is seen. This might be expected as each of these approaches differ in their methods of experimentation, data analysis and ranking of loci as biomarkers. It also indicates that further markers remain to be identified and that development of the optimal panel will require additional studies. Ongoing genome-wide analyses using a multitude of approaches will help solve this issue, but it is important that these analyses be carried out on all histological subtypes of lung cancer. As previously discussed, NSCLC is comprised of four histological sub-groups. The two most common subtypes, adenocarcinoma and squamous cell lung cancer, are quite distinct in both physical location and molecular profile [118, 119, 130–133]. They show differential methylation profiles as reported by Field et al. and Brena et al. [79, 119]. Indeed work in our lab supports the notion of different methylation patterns in SQ and AD [27, 28]. The distinct nature of AD and SQ means an optimal lung cancer methylation panel will probably require markers for both subtypes. Markers for LC and other minor NSCLC groups, such as neuroendocrine cancers, remain to be developed.
DNA methylation in remote media
While using primary tissue to study methylation status is useful to discover potential biomarkers, this material is not non-invasively accessible and is therefore not useful for screening an at-risk population. The ideal system for early diagnosis is material collected in a non-invasive/minimally invasive way that will contain methylated DNA. For this, one looks to remote patient media – blood, naturally produced or induced sputum, exhaled breath-condensate (EBC, non-invasive), and bronchoalveolar lavage (BAL, semi-invasive). Multiple studies show that DNA methylation of certain loci can be detected in blood, sputum and BAL (Additional file 3). A few show that genetic alterations can be detected in EBC, as discussed below, although no published studies of DNA methylation detection in this medium exist.
The ideal remote medium is blood – it can be applied to all patients, both those at minimal and high risk, and is minimally invasive to obtain. It is reported that cancer patients have a higher level of circulating DNA than non-cancer cases [134], and that genetic [135–137], and epigenetic [138] alterations can be detected in said DNA. It is postulated that this DNA is released due to necrotic cell death [139]. Over 25 loci have been reported to be methylated in plasma/serum of NSCLC patients [29, 41, 45, 52, 55, 60, 140–144] (reviewed in Additional file 3). Several studies examined methylation in primary tumor material and corresponding plasma/serum, and in these cases methylation in blood was only seen in patients in which the primary tumor also exhibited methylation [52, 60, 142]. Many of the most promising markers from Additional file 1 and 2 have not yet been investigated in blood.
There are, however, caveats to detection of DNA methylation in blood. It is questioned as to whether there is enough methylated DNA in the blood to efficiently detect tumors at an early enough stage for curative resection. While DNA quantity may be low, ongoing research on more sensitive detection methods may overcome this issue. Another potential problem is that blood as a remote medium is not organ-specific; loci that are methylated in lung cancer may be methylated as well in other cancers, for example TNFRSF10C and D [113] TCF21 [36], RUNX3 [89], APC [145], FBN2 [68]. Thus, methylation of these loci in blood could point to cancer in any one of several organs. The best markers for lung cancer would therefore be ones that show methylation only in lung cancer. Given the recent focus on more genome wide approaches to study methylation in many cancer types, a comparison of DNA methylation profiles across cancer sites should soon be possible. An alternative to this is to complement DNA methylation marker screening with sensitive imaging techniques to identify the cancer site. Another option is to examine remote media that are more lung-specific.
Sputum is produced by increased bronchial secretions, and is commonly found in smokers, hence it can be used to screen high-risk populations. (In former or non-smokers, it is much more difficult to obtain, though it can be induced.) The advantages of sputum as a screening tool include its non-invasive procurement, and the fact that it contains cells from the lungs and lower respiratory tract. However, the material in sputum is from the center of the lungs, and it may not be as useful for the detection of adenocarcinoma, which generally occurs at the periphery. DNA methylation, mutations, and microsatellite alterations have been detected in sputum, indicating it is a useful source of tumor material [7, 29, 146]. Reports of DNA methylation in sputum are summarized in Additional file 3[29, 57, 59, 77, 80, 97, 113, 140, 147, 148]. It has been demonstrated that promoter methylation in sputum increases with cancer risk [29], increases as the time to lung cancer decreases [147], and in the case of CDKN2A/p16 and/or MGMT, can be found in sputum up to 3 years before diagnosis of squamous cell lung cancer [149]. A study by Liu et al. using 50 matched tumor, plasma and sputum samples showed that CDKN2A/p16 hypermethylation is detected in 84% of tumors, and 76% of sputum samples from the same patients, demonstrating that this remote medium is potentially effective in detecting lung cancer [55]. However, whether this detection is applicable to all NSCLC subtypes remains to be determined.
Exhaled breath provides a source of materials that can reflect the disease state of the lungs. Breath condensate, comprised mostly of water vapors, also contains lipids, proteins, DNA and oxidation products – the levels of which may differ between healthy and diseased subjects [150]. Several studies report the utility of EBC in detection of asthma, chronic obstructive pulmonary disease (COPD) and cystic fibrosis [150]. EBC has also been used for NSCLC detection. Carpagnano et al. reported detection of the mitogenic factor endothelin-1 (ET1-1) in EBC of lung cancer patients. In a small study they showed a statistically significant difference in ET-1 levels between healthy controls and NSCLC patients, and between stage I-III and stage IV patients [151]. They have shown similar results when looking at interleukin-6 [152]. While these studies are protein-based, they do demonstrate the promise of EBC for early detection of lung cancer. Thus far, there are no published reports of DNA methylation detection in EBC, although two studies reported collecting sufficient DNA quantities to perform PCR-based assays for microsatellite alterations and p53 mutations [153, 154]. Of concern is the fact that the p53 mutations detected in EBC differ from those found in the primary tumor from the same patient [153, 154]. This raises concern regarding the origin of DNA obtained from EBC (it may also come from cells in the esophagus, throat or mouth) and its utility as a remote medium.
Bronchoalveolar lavage (BAL) is another potential screening material for early detection of lung cancer. While obtaining lavage fluid is not as invasive as a biopsy, it requires bronchoscopy. However, bronchoscopy is routinely performed in suspected lung cancer cases and lavage fluid can be easily obtained during this procedure. An advantage of BAL is that it allows localized harvesting of lung-specific material, so that the fluid can be expected to contain lung cancer cells and/or DNA. Several investigations of DNA methylation in BAL have been conducted [30, 39, 99, 100, 155–160] (Additional file 3). Results vary between studies. De Fraipont showed low levels of DNA methylation in BAL from tumor-bearing patients, indicating that this would not be a good medium for marker detection [157]. In contrast, Topalogu used a panel of loci and detected 68% of their tumor cases by examining DNA methylation in the corresponding BAL from the same patients [39]. Kim et al. also reported a good correlation between methylation in tumors and BAL, ranging from 39–61% for the five loci they analyzed [30]. DNA methylation has also been detected in control BAL from non-neoplastic patients [30, 159, 160]. The detection of DNA methylation in cancer-free patients is cause for concern if presence/absence of DNA methylation is being used as a diagnostic measure of cancer. However, if a quantitative assay to determine DNA methylation levels is applied, then one can determine a cut-off value, above which a sample would be considered positive, as was done by Grote et al. [159] and Schmiemann et al. [160].
The analyses of DNA methylation markers in remote media are still in their early stages, and although many show low sensitivity, the inclusion of more of the recently identified promising markers (Additional file 2) in future studies would likely boost detection of cancer cases. Published data so far supports the continued analysis of these fluids in search of an early detection method that can, at the very least, complement imaging-based screening of at risk subjects.
Selection of DNA methylation-based biomarkers for early detection of NSCLC
While a plethora of loci are reported to serve as potential DNA methylation-based biomarkers for NSCLC, the important question is: Which should be chosen for further evaluation, and eventually for screening of subjects? When performing a screening test there are four potential outcomes. The first two of these, true-positive results (TP, those who test positive and actually have cancer), and true-negative results (TN, those who test negative and do not have cancer), are the desired outcome of a screening test. However, false-negative results (FN, those who have cancer but do not test positive), and false-positive results (FP, those who do not have cancer but test positive), could do serious harm to the screening populations. False negative results have the ramification of delaying diagnosis of the disease, hence endangering patients' lives, while false positive results significantly affect patient quality of life [161]. Sensitivity, defined as TP/(TP+FN), and specificity, defined as TN/(TN+FP), measure the balance of these results in the population. These measures can serve as the selection criteria to determine which potential biomarkers are pursued further. An ideal DNA methylation-based biomarker would be highly sensitive and specific in all populations studied, regardless of age, gender, ethnicity, risk factors and tumor stage. However, given the differences between NSCLC subtypes and smoking and non-smoking associated NSCLC, markers that function accurately in a subset of the population could also be of use. The likelihood of identifying a single marker with 100% sensitivity and specificity is negligible.
The methylation frequency for many loci examined in early studies is quite low in primary tumors (Additional file 1, for example, DAPK 16–47%, p16 23–81%, CDH13 28–48%, and RASSF1A 15–54%). If the methylation frequency is low, sensitivity will suffer as the locus yields too few cases. Even for the more frequently methylated loci listed in Additional file 2, one DNA methylation marker cannot be expected to detect all cases of a particular cancer. The way to address this problem is to study the DNA methylation status of multiple loci (a panel) in a sample population. To ensure high sensitivity individual loci in the panel should be highly penetrant, i.e. have a high frequency in the population, and be complementary, i.e. detect different tumor cases.
While ensuring high sensitivity is important, given very sensitive imaging approaches like LDSCT, the more critical issue in lung cancer screening is high specificity. False-positive results precipitate not only patient anxiety, but also follow up procedures that are invasive, costly, and have associated morbidity and mortality. The incidence for lung cancer in the United States is 79.4/100,000 in men and 52.6/100,000 in women [162]. This shows that less than 0.1% of the population will get lung cancer. Hence, a population-based screening using any marker with a specificity of less that 99.9% will detect more false positive cases than true positive ones. Such a marker therefore cannot function as a screening marker in the population at large. However, in current smokers the risk of lung cancer is greatly increased (incidence of over 230 per 100,000 for both men and women [163]), and the specificity of a marker can be slightly lower when screening is targeted to this high-risk group.
Sensitivity and specificity have been reported for several locus panels when examining methylation in DNA isolated from primary tissue. The area under the curve (AUC) of a receiver operating characteristic (ROC) curve is a measure of the ability of a continuous marker to accurately classify tumor and non-tumor tissue. Such a curve is a plot of sensitivity vs 1 minus specificity values associated with all dichotomous markers that can be formed by varying the value threshold used to designate a marker "positive". An AUC of 1 corresponds to a marker with perfect accuracy, while an AUC of 0.5 corresponds to an uninformative marker. Shivapurkar et al. studied the DNA methylation of 11 loci to distinguish between NSCLC and adjacent non-tumor lung tissue. Using a logistic regression with a binary outcome indicator of tumor and non-tumor lung tissue, and a marker panel as covariates, they demonstrated that a combination of HS3ST2 (3OST2), DAPK and TNFRSF10C (DcR1) gave an ROC curve with an AUC of 0.959 when comparing tumor and adjacent non-tumor lung tissue. This implies that this combination of markers could sensitively and specifically detect lung cancer [113]. Ehrich et al. studied the methylation of 47 loci and developed a panel of 6 that could distinguish cancer from adjacent normal tissue with >95% sensitivity and specificity [118]. Feng et al. developed a panel of 8 loci, of which the presence of methylation of one gene was found in 80% of NSCLC tissues [117]. In an effort to develop markers for specific NSCLC subtypes, Tsou et al. reported a panel of 4 loci with 94% sensitivity and 90% specificity for AD [27], while Anglim et al. reported a panel of 4 loci that with 96.5% sensitivity and 93.3% specificity for SQ lung cancer [28]. Both reports compare DNA methylation in tumor and adjacent non-tumor tissue from the same patients. On a larger scale, Bibikova et al. identified 55 loci that distinguished AD from adjacent non-tumor lung with 100% sensitivity and 92% specificity [122]. These are all encouraging results, implying that DNA methylation detection could serve as a viable early detection biomarker, but these loci must be further validated in larger, racially/ethnicially and gender balanced independent populations in order to ensure equal functionality for all patients. Also, primary tissue would not be the source material tested in screening for early detection, hence, promising loci must be interrogated for their potential to sensitively and specifically detect cancer in remote media.
There are multiple reports of DNA methylation in blood, but not all assess the sensitivity and specificity of the loci. In those that do, it appears that detection in blood is commonly not sensitive [140, 143]. For example, sensitivity ranged from 7–27% for CDH13, CDKN2A/p16, DAPK, GATA5, MGMT, PAX5α, PAX5β and RASSF1A in serum, but is much higher in sputum for the same samples [140]. One way in which investigators have tried to increase sensitivity is by defining a patient positive if a minimum number of loci are methylated. For example, Fujiwara et al. also described a low sensitivity of 49.5% when looking at methylation of at least one of 5 loci in serum (CDKN2A/p16, DAPK, MGMT, RARB and RASSF1A) but specificity was 85% [143]. Recently a report examining the methylation of CDH13, CDKN2A/p16, FHIT, RARB, RASSF1A and ZMYND10 (BLU) in which methylation of any 2 loci in plasma was considered cancer positive showed 73% sensitivity and 82% specificity [45]. This reinforces the notion that a panel of complementary loci is necessary. In an interesting report, Bearzatto et al. showed that combining CDKN2A methylation with microsatellite alterations in plasma increased sensitivity to 62%, and using CDKN2A methylation combined with circulating DNA levels increased specificity to 80%, as opposed to examining CDKN2A methylation alone [164]. While neither of these is ideal as a clinical test, it is of note that the marker panels need not consist solely of DNA methylation-based markers.
Many studies indicate that sputum could be a promising remote medium for early detection. Shivapurkar et al. described a combination of 4 loci, APC, CDKN2A/p16, HS3ST2 (3OST2), and RASSF1A that serve as a good panel for early detection of NSCLC in sputum, with an AUC of 0.8 [113]. Similarly, Li et al. reported a combination of FHIT and HYAL2 with 76% sensitivity and 85% specificity [7]. Wang et al. described MLH1 methylation in sputum to have 60% sensitivity and 86% specificity [77], and Belinsky showed that concomitant methylation of three or more of a panel of 6 loci resulted in 64% sensitivity and specificity [147]. In contrast, Cirincione et al. reported that 3 loci, CDKN2A/p16, RARβ2 and RASSF1A are of limited use in early detection of lung cancer using sputum as a remote medium [59].
Detection of DNA methylation in bronchoalveolar lavage is also documented. Grote et al. published two reports, using either APC or RASSF1A alone for NSCLC detection. In both cases there is low sensitivity (30 and 34% respectively) but high specificity (98.5 and 100% respectively) [156, 158]. Using just CDKN2A/p16, Xie et al. describe a higher sensitivity (64%) than any other reports on DNA methylation in BAL when examining a single locus and a modest specificity (75%) [165]. Grote et al. explored the use of marker combinations in two studies. In the first they used CDKN2A/p16 and RARB2 in combination and showed 69% sensitivity and 87% specificity in their population [159]. In another study they applied a marker panel (APC, CDKN2A/p16, and RASSF1) to detect cancer in 247 patients, and reported 53% sensitivity and, in cases without a previous history of cancer, >99% specificity [160]. It is probable that the inclusion of more highly penetrant markers in such panels would increase sensitivity. This again highlights the need for a panel of markers, and underlines the need to combine molecular markers with imaging techniques.
Conclusion
Lung cancer is responsible for a million cancer deaths per year worldwide, and its detrimental effects will continue to increase. Research focused on biomarker-based early detection has the potential to reduce mortality rates. What will it take to obtain functional DNA methylation markers for early lung cancer detection?
Sullivan-Pepe outlined the five phases of biomarker discovery[166]. The first phase, clinical exploratory, consists of identification of promising markers. Much work on identification of DNA-methylation based markers has already been done, as described in Additional files 1 and 2, and a number of markers has been carried forward to phase two, the clinical detection of established disease (Additional file 3). However, with the advent of new techniques, a thorough evaluation of the epigenome of all types of cancer will soon be possible. The pool of potential DNA methylation markers for lung cancer has by no means been exhausted, and it is expected that additional high penetrance markers will be identified. It will be important to examine DNA methylation in each of the major lung cancer histological subtypes and ensure the functionality of identified markers in lung cancers from both genders and all races/ethnic groups. In addition, given the fact that half of all new lung cancer cases arise in ex-smokers or never smokers [167], and the observed molecular differences between lung cancer from smokers and non-smokers [168], it would be important to ensure representation of lung cancer from never smokers in these marker screens. Standardization of epigenomic assay techniques and data analysis would facilitate comparisons of DNA methylation profiles between cancer types, which may allow the identification of true lung-cancer specific hypermethylation. Ideally, only reproducibly hypermethylated high penetrance DNA methylation markers should be carried forward to the analysis of systematically collected remote media (because remote media are such a valuable resource). The most promising markers can then be tested in retrospective longitudinal studies (phase three), in which materials collected prior to disease onset are examined. Studies of DNA methylation in sputum and BAL collected prior to diagnosis already look promising (e.g. [149, 160]), and results can improve further with the inclusion of new high sensitivity/specificity marker panels. If results are promising, prospective screening studies (phase four) should follow to determine the extent and properties of detected disease and measure the false referral rate. Lastly, case control studies should be done to measure any effect on lung cancer mortality.
If a strong DNA methylation marker panel were developed, the manner in which it would be applied would depend on its sensitivity and specificity. It is unlikely that DNA methylation markers, or any molecular markers for that matter, would be used on their own. Instead, we envision that they will be applied in concert with high-resolution imaging. In the near future, the prospect of genome-wide interrogation of DNA methylation in lung cancer is extremely exciting. The resulting information may provide not only new candidate markers for early detection, but also for monitoring response to therapy and recurrence. In addition, methylation information could be linked to pathobiology and clinical characteristics, potentially providing indicators for treatment and prognosis. Much work remains to be done, but using epigenomics while building on the experience and materials obtained from prior studies, we are well armed to make non-invasive testing for early lung cancer detection a reality.
References
Molina JR, Yang P, Cassivi SD, Schild SE, Adjei AA: Non-small cell lung cancer: epidemiology, risk factors, treatment, and survivorship. Mayo Clin Proc. 2008, 83: 584-594.
Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ: Cancer statistics, 2007. CA Cancer J Clin. 2007, 57: 43-66.
Henschke CI, Yankelevitz DF, Libby DM, Pasmantier MW, Smith JP, Miettinen OS: Survival of patients with stage I lung cancer detected on CT screening. N Engl J Med. 2006, 355: 1763-1771. 10.1056/NEJMoa060476
Gavelli G, Giampalma E: Sensitivity and specificity of chest X-ray screening for lung cancer: review article. Cancer. 2000, 89: 2453-2456. 10.1002/1097-0142(20001201)89:11+<2453::AID-CNCR21>3.0.CO;2-M
Bach PB, Kelley MJ, Tate RC, McCrory DC: Screening for lung cancer: a review of the current literature. Chest. 2003, 123: 72S-82S. 10.1378/chest.123.1_suppl.72S
Diederich S, Wormanns D: Impact of low-dose CT on lung cancer screening. Lung Cancer. 2004, 45 (Suppl 2): S13-19. 10.1016/j.lungcan.2004.07.997
Li R, Todd NW, Qiu Q, Fan T, Zhao RY, Rodgers WH, Fang HB, Katz RL, Stass SA, Jiang F: Genetic deletions in sputum as diagnostic markers for early detection of stage I non-small cell lung cancer. Clin Cancer Res. 2007, 13: 482-487. 10.1158/1078-0432.CCR-06-1593
Lindell RM, Hartman TE, Swensen SJ, Jett JR, Midthun DE, Tazelaar HD, Mandrekar JN: Five-year lung cancer screening experience: CT appearance, growth rate, location, and histologic features of 61 lung cancers. Radiology. 2007, 242: 555-562. 10.1148/radiol.2422052090
Bastarrika G, Garcia-Velloso MJ, Lozano MD, Montes U, Torre W, Spiteri N, Campo A, Seijo L, Alcaide AB, Pueyo J, Cano D, Vivas I, Cosin O, Dominguez P, Serra P, Richter JA, Montuenga L, Zulueta JJ: Early lung cancer detection using spiral computed tomography and positron emission tomography. Am J Respir Crit Care Med. 2005, 171: 1378-1383. 10.1164/rccm.200411-1479OC
Carter D, Vazquez M, Flieder DB, Brambilla E, Gazdar A, Noguchi M, Travis WD, Kramer A, Yip R, Yankelevitz DF, Henschke CI: Comparison of pathologic findings of baseline and annual repeat cancers diagnosed on CT screening. Lung Cancer. 2007, 56: 193-199. 10.1016/j.lungcan.2006.12.001
Gohagan J, Marcus P, Fagerstrom R, Pinsky P, Kramer B, Prorok P: Baseline findings of a randomized feasibility trial of lung cancer screening with spiral CT scan vs chest radiograph: the Lung Screening Study of the National Cancer Institute. Chest. 2004, 126: 114-121. 10.1378/chest.126.1.114
Gohagan JK, Marcus PM, Fagerstrom RM, Pinsky PF, Kramer BS, Prorok PC, Ascher S, Bailey W, Brewer B, Church T, Engelhard D, Ford M, Fouad M, Freedman M, Gelmann E, Gierada D, Hocking W, Inampudi S, Irons B, Johnson CC, Jones A, Kucera G, Kvale P, Lappe K, Manor W, Moore A, Nath H, Neff S, Oken M, Plunkett M: Final results of the Lung Screening Study, a randomized feasibility study of spiral CT versus chest X-ray screening for lung cancer. Lung Cancer. 2005, 47: 9-15. 10.1016/j.lungcan.2004.06.007
Sone S, Nakayama T, Honda T, Tsushima K, Li F, Haniuda M, Takahashi Y, Hanaoka T, Takayama F, Koizumi T, Kubo K, Yamanda T, Kondo R, Fushimi H, Suzuki T: CT findings of early-stage small cell lung cancer in a low-dose CT screening programme. Lung Cancer. 2007, 56: 207-215. 10.1016/j.lungcan.2006.12.014
van Klaveren RJ, Habbema JDF, Pedersen JH, de Koning HJ, Oudkerk M, Hoogsteden HC: Lung cancer screening by low-dose spiral computed tomography. Eur Respir J. 2001, 18: 857-866. 10.1183/09031936.01.00076701
Jett JR: Limitations of screening for lung cancer with low-dose spiral computed tomography. Clin Cancer Res. 2005, 11: 4988s-4992s. 10.1158/1078-0432.CCR-05-9000
Crestanello JA, Allen MS, Jett JR, Cassivi SD, Nichols FC, Swensen SJ, Deschamps C, Pairolero PC: Thoracic surgical operations in patients enrolled in a computed tomographic screening trial. J Thorac Cardiovasc Surg. 2004, 128: 254-259. 10.1016/j.jtcvs.2004.02.017
Yankelevitz DF, Kostis WJ, Henschke CI, Heelan RT, Libby DM, Pasmantier MW, Smith JP: Overdiagnosis in chest radiographic screening for lung carcinoma: frequency. Cancer. 2003, 97: 1271-1275. 10.1002/cncr.11185
Hasegawa M, Sone S, Takashima S, Li F, Yang ZG, Maruyama Y, Watanabe T: Growth rate of small lung cancers detected on mass CT screening. Br J Radiol. 2000, 73: 1252-1259.
Bach PB, Jett JR, Pastorino U, Tockman MS, Swensen SJ, Begg CB: Computed tomography screening and lung cancer outcomes. Jama. 2007, 297: 953-961. 10.1001/jama.297.9.953
Feller-Kopman D, Lunn W, Ernst A: Autofluorescence bronchoscopy and endobronchial ultrasound: a practical review. Ann Thorac Surg. 2005, 80: 2395-2401. 10.1016/j.athoracsur.2005.04.084
McWilliams A, MacAulay C, Gazdar AF, Lam S: Innovative molecular and imaging approaches for the detection of lung cancer and its precursor lesions. Oncogene. 2002, 21: 6949-6959. 10.1038/sj.onc.1205831
Haussinger K, Becker H, Stanzel F, Kreuzer A, Schmidt B, Strausz J, Cavaliere S, Herth F, Kohlhaufl M, Muller KM, Huber RM, Pichlmeier U, Bolliger ChT: Autofluorescence bronchoscopy with white light bronchoscopy compared with white light bronchoscopy alone for the detection of precancerous lesions: a European randomised controlled multicentre trial. Thorax. 2005, 60: 496-503. 10.1136/thx.2005.041475
Weissleder R, Pittet MJ: Imaging in the era of molecular oncology. Nature. 2008, 452: 580-589. 10.1038/nature06917
Rideout WM, Eversole-Cire P, Spruck CH, Hustad CM, Coetzee GA, Gonzales FA, Jones PA: Progressive increases in the methylation status and heterochromatinization of the myoD CpG island during oncogenic transformation. Mol Cell Biol. 1994, 14: 6143-6152.
Takai D, Jones PA: The CpG island searcher: a new WWW resource. In Silico Biol. 2003, 3: 235-240.
Suzuki MM, Bird A: DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet. 2008, 9: 465-476. 10.1038/nrg2341
Tsou JA, Galler JS, Siegmund KD, Laird PW, Turla S, Cozen W, Hagen JA, Koss MN, Laird-Offringa IA: Identification of a panel of sensitive and specific DNA methylation markers for lung adenocarcinoma. Mol Cancer. 2007, 6: 70- 10.1186/1476-4598-6-70
Anglim PP, Galler JS, Koss MN, Hagen JA, Turla S, Campan M, Weisenberger DJ, Laird PW, Siegmund KD, Laird-Offringa IA: Identification of a panel of sensitive and specific DNA methylation markers for squamous cell lung cancer. Mol Cancer. 2008, 7: 62- 10.1186/1476-4598-7-62
Belinsky SA, Klinge DM, Dekker JD, Smith MW, Bocklage TJ, Gilliland FD, Crowell RE, Karp DD, Stidley CA, Picchi MA: Gene promoter methylation in plasma and sputum increases with lung cancer risk. Clin Cancer Res. 2005, 11: 6505-6511. 10.1158/1078-0432.CCR-05-0625
Kim H, Kwon YM, Kim JS, Lee H, Park JH, Shim YM, Han J, Park J, Kim DH: Tumor-specific methylation in bronchial lavage for the early detection of non-small-cell lung cancer. J Clin Oncol. 2004, 22: 2363-2370. 10.1200/JCO.2004.10.077
Dai Z, Popkie AP, Zhu WG, Timmers CD, Raval A, Tannehill-Gregg S, Morrison CD, Auer H, Kratzke RA, Niehans G, Amatschek S, Sommergruber W, Leone GW, Rosol T, Otterson GA, Plass C: Bone morphogenetic protein 3B silencing in non-small-cell lung cancer. Oncogene. 2004, 23: 3521-3529. 10.1038/sj.onc.1207441
Marsit CJ, Kim DH, Liu M, Hinds PW, Wiencke JK, Nelson HH, Kelsey KT: Hypermethylation of RASSF1A and BLU tumor suppressor genes in non-small cell lung cancer: implications for tobacco smoking during adolescence. Int J Cancer. 2005, 114: 219-223. 10.1002/ijc.20714
Toyooka KO, Toyooka S, Virmani AK, Sathyanarayana UG, Euhus DM, Gilcrease M, Minna JD, Gazdar AF: Loss of expression and aberrant methylation of the CDH13 (H-cadherin) gene in breast and lung carcinomas. Cancer Res. 2001, 61: 4556-4560.
Toyooka S, Toyooka KO, Miyajima K, Reddy JL, Toyota M, Sathyanarayana UG, Padar A, Tockman MS, Lam S, Shivapurkar N, Gazdar AF: Epigenetic down-regulation of death-associated protein kinase in lung cancers. Clin Cancer Res. 2003, 9: 3034-3041.
Virmani AK, Rathi A, Sathyanarayana UG, Padar A, Huang CX, Cunnigham HT, Farinas AJ, Milchgrub S, Euhus DM, Gilcrease M, Herman J, Minna JD, Gazdar AF: Aberrant methylation of the adenomatous polyposis coli (APC) gene promoter 1A in breast and lung carcinomas. Clin Cancer Res. 2001, 7: 1998-2004.
Smith LT, Lin M, Brena RM, Lang JC, Schuller DE, Otterson GA, Morrison CD, Smiraglia DJ, Plass C: Epigenetic regulation of the tumor suppressor gene TCF21 on 6q23-q24 in lung and head and neck cancer. Proc Natl Acad Sci USA. 2006, 103: 982-987. 10.1073/pnas.0510171102
Tessema M, Willink R, Do K, Yu YY, Yu W, Machida EO, Brock M, Van Neste L, Stidley CA, Baylin SB, Belinsky SA: Promoter methylation of genes in and around the candidate lung cancer susceptibility locus 6q23-25. Cancer Res. 2008, 68: 1707-1714. 10.1158/0008-5472.CAN-07-6325
Harden SV, Tokumaru Y, Westra WH, Goodman S, Ahrendt SA, Yang SC, Sidransky D: Gene promoter hypermethylation in tumors and lymph nodes of stage I lung cancer patients. Clin Cancer Res. 2003, 9: 1370-1375.
Topaloglu O, Hoque MO, Tokumaru Y, Lee J, Ratovitski E, Sidransky D, Moon CS: Detection of promoter hypermethylation of multiple genes in the tumor and bronchoalveolar lavage of patients with lung cancer. Clin Cancer Res. 2004, 10: 2284-2288. 10.1158/1078-0432.CCR-1111-3
Brabender J, Usadel H, Danenberg KD, Metzger R, Schneider PM, Lord RV, Wickramasinghe K, Lum CE, Park J, Salonga D, Singer J, Sidransky D, Holscher AH, Meltzer SJ, Danenberg PV: Adenomatous polyposis coli gene promoter hypermethylation in non-small cell lung cancer is associated with survival. Oncogene. 2001, 20: 3528-3532. 10.1038/sj.onc.1204455
Usadel H, Brabender J, Danenberg KD, Jeronimo C, Harden S, Engles J, Danenberg PV, Yang S, Sidransky D: Quantitative adenomatous polyposis coli promoter methylation analysis in tumor tissue, serum, and plasma DNA of patients with lung cancer. Cancer Res. 2002, 62: 371-375.
Nagatake M, Osada H, Kondo M, Uchida K, Nishio M, Shimokata K, Takahashi T, Takahashi T: Aberrant hypermethylation at the bcl-2 locus at 18q21 in human lung cancers. Cancer Res. 1996, 56: 1886-1891.
Agathanggelou A, Dallol A, Zochbauer-Muller S, Morrissey C, Honorio S, Hesson L, Martinsson T, Fong KM, Kuo MJ, Yuen PW, Maher ER, Minna JD, Latif F: Epigenetic inactivation of the candidate 3p21.3 suppressor gene BLU in human cancers. Oncogene. 2003, 22: 1580-1588. 10.1038/sj.onc.1206243
Ito M, Ito G, Kondo M, Uchiyama M, Fukui T, Mori S, Yoshioka H, Ueda Y, Shimokata K, Sekido Y: Frequent inactivation of RASSF1A, BLU, and SEMA3B on 3p21.3 by promoter hypermethylation and allele loss in non-small cell lung cancer. Cancer Lett. 2005, 225: 131-139. 10.1016/j.canlet.2004.10.041
Hsu HS, Chen TP, Hung CH, Wen CK, Lin RK, Lee HC, Wang YC: Characterization of a multiple epigenetic marker panel for lung cancer detection and risk assessment in plasma. Cancer. 2007, 110: 2019-2026. 10.1002/cncr.23001
Lee MN, Tseng RC, Hsu HS, Chen JY, Tzao C, Ho WL, Wang YC: Epigenetic inactivation of the chromosomal stability control genes BRCA1, BRCA2, and XRCC5 in non-small cell lung cancer. Clin Cancer Res. 2007, 13: 832-838. 10.1158/1078-0432.CCR-05-2694
Esteller M, Corn PG, Baylin SB, Herman JG: A gene hypermethylation profile of human cancer. Cancer Res. 2001, 61: 3225-3229.
Zhong S, Fields CR, Su N, Pan YX, Robertson KD: Pharmacologic inhibition of epigenetic modifications, coupled with gene expression profiling, reveals novel targets of aberrant DNA methylation and histone deacetylation in lung cancer. Oncogene. 2007, 26: 2621-2634. 10.1038/sj.onc.1210041
Nakata S, Sugio K, Uramoto H, Oyama T, Hanagiri T, Morita M, Yasumoto K: The methylation status and protein expression of CDH1, p16(INK4A), and fragile histidine triad in nonsmall cell lung carcinoma: epigenetic silencing, clinical features, and prognostic significance. Cancer. 2006, 106: 2190-2199. 10.1002/cncr.21870
Kim JS, Han J, Shim YM, Park J, Kim DH: Aberrant methylation of H-cadherin (CDH13) promoter is associated with tumor progression in primary nonsmall cell lung carcinoma. Cancer. 2005, 104: 1825-1833. 10.1002/cncr.21409
Sato M, Mori Y, Sakurada A, Fujimura S, Horii A: The H-cadherin (CDH13) gene is inactivated in human lung cancer. Hum Genet. 1998, 103: 96-101. 10.1007/s004390050790
Ulivi P, Zoli W, Calistri D, Fabbri F, Tesei A, Rosetti M, Mengozzi M, Amadori D: p16INK4A and CDH13 hypermethylation in tumor and serum of non-small cell lung cancer patients. J Cell Physiol. 2006, 206: 611-615. 10.1002/jcp.20503
Jarmalaite S, Kannio A, Anttila S, Lazutka JR, Husgafvel-Pursiainen K: Aberrant p16 promoter methylation in smokers and former smokers with nonsmall cell lung cancer. Int J Cancer. 2003, 106: 913-918. 10.1002/ijc.11322
Furonaka O, Takeshima Y, Awaya H, Ishida H, Kohno N, Inai K: Aberrant methylation of p14(ARF), p15(INK4b) and p16(INK4a) genes and location of the primary site in pulmonary squamous cell carcinoma. Pathol Int. 2004, 54: 549-555. 10.1111/j.1440-1827.2004.01663.x
Liu Y, An Q, Li L, Zhang D, Huang J, Feng X, Cheng S, Gao Y: Hypermethylation of p16INK4a in Chinese lung cancer patients: biological and clinical implications. Carcinogenesis. 2003, 24: 1897-1901. 10.1093/carcin/bgg169
Liu Y, Lan Q, Siegfried JM, Luketich JD, Keohavong P: Aberrant promoter methylation of p16 and MGMT genes in lung tumors from smoking and never-smoking lung cancer patients. Neoplasia. 2006, 8: 46-51. 10.1593/neo.05586
Belinsky SA, Nikula KJ, Palmisano WA, Michels R, Saccomanno G, Gabrielson E, Baylin SB, Herman JG: Aberrant methylation of p16(INK4a) is an early event in lung cancer and a potential biomarker for early diagnosis. Proc Natl Acad Sci USA. 1998, 95: 11891-11896. 10.1073/pnas.95.20.11891
Breuer RH, Snijders PJ, Sutedja GT, Sewalt RG, Otte AP, Postmus PE, Meijer CJ, Raaphorst FM, Smit EF: Expression of the p16(INK4a) gene product, methylation of the p16(INK4a) promoter region and expression of the polycomb-group gene BMI-1 in squamous cell lung carcinoma and premalignant endobronchial lesions. Lung Cancer. 2005, 48: 299-306. 10.1016/j.lungcan.2004.11.026
Cirincione R, Lintas C, Conte D, Mariani L, Roz L, Vignola AM, Pastorino U, Sozzi G: Methylation profile in tumor and sputum samples of lung cancer patients detected by spiral computed tomography: a nested case-control study. Int J Cancer. 2006, 118: 1248-1253. 10.1002/ijc.21473
Fromont-Hankard G, Philippe-Chomette P, Delezoide AL, Nessmann C, Aigrain Y, Peuchmaur M: Glial cell-derived neurotrophic factor expression in normal human lung and congenital cystic adenomatoid malformation. Arch Pathol Lab Med. 2002, 126: 432-436.
Mizuno K, Osada H, Konishi H, Tatematsu Y, Yatabe Y, Mitsudomi T, Fujii Y, Takahashi T: Aberrant hypermethylation of the CHFR prophase checkpoint gene in human lung cancers. Oncogene. 2002, 21: 2328-2333. 10.1038/sj.onc.1205402
Zhang P, Wang J, Gao W, Yuan BZ, Rogers J, Reed E: CHK2 kinase expression is down-regulated due to promoter methylation in non-small cell lung cancer. Mol Cancer. 2004, 3: 14- 10.1186/1476-4598-3-14
Yano M, Toyooka S, Tsukuda K, Dote H, Ouchida M, Hanabata T, Aoe M, Date H, Gazdar AF, Shimizu N: Aberrant promoter methylation of human DAB2 interactive protein (hDAB2IP) gene in lung cancers. Int J Cancer. 2005, 113: 59-66. 10.1002/ijc.20531
Liu Y, Gao W, Siegfried JM, Weissfeld JL, Luketich JD, Keohavong P: Promoter methylation of RASSF1A and DAPK and mutations of K-ras, p53, and EGFR in lung tumors from smokers and never-smokers. BMC Cancer. 2007, 7: 74- 10.1186/1471-2407-7-74
Luxen S, Belinsky SA, Knaus UG: Silencing of DUOX NADPH oxidases by promoter hypermethylation in lung cancer. Cancer Res. 2008, 68: 1037-1045. 10.1158/0008-5472.CAN-07-5782
Yue W, Dacic S, Sun Q, Landreneau R, Guo M, Zhou W, Siegfried JM, Yu J, Zhang L: Frequent inactivation of RAMP2, EFEMP1 and Dutt1 in lung cancer by promoter hypermethylation. Clin Cancer Res. 2007, 13: 4336-4344. 10.1158/1078-0432.CCR-07-0015
Tai KY, Shiah SG, Shieh YS, Kao YR, Chi CY, Huang E, Lee HS, Chang LC, Yang PC, Wu CW: DNA methylation and histone modification regulate silencing of epithelial cell adhesion molecule for tumor invasion and progression. Oncogene. 2007, 26: 3989-3997. 10.1038/sj.onc.1210176
Chen H, Suzuki M, Nakamura Y, Ohira M, Ando S, Iida T, Nakajima T, Nakagawara A, Kimura H: Aberrant methylation of FBN2 in human non-small cell lung cancer. Lung Cancer. 2005, 50: 43-49. 10.1016/j.lungcan.2005.04.013
Tzao C, Tsai HY, Chen JT, Chen CY, Wang YC: 5'CpG island hypermethylation and aberrant transcript splicing both contribute to the inactivation of the FHIT gene in resected non-small cell lung cancer. Eur J Cancer. 2004, 40: 2175-2183. 10.1016/j.ejca.2004.06.022
Kim JS, Kim H, Shim YM, Han J, Park J, Kim DH: Aberrant methylation of the FHIT gene in chronic smokers with early stage squamous cell carcinoma of the lung. Carcinogenesis. 2004, 25: 2165-2171. 10.1093/carcin/bgh217
Guo M, Akiyama Y, House MG, Hooker CM, Heath E, Gabrielson E, Yang SC, Han Y, Baylin SB, Herman JG, Brock MV: Hypermethylation of the GATA genes in lung cancer. Clin Cancer Res. 2004, 10: 7917-7924. 10.1158/1078-0432.CCR-04-1140
Irimia M, Fraga MF, Sanchez-Cespedes M, Esteller M: CpG island promoter hypermethylation of the Ras-effector gene NORE1A occurs in the context of a wild-type K-ras in lung cancer. Oncogene. 2004, 23: 8695-8699. 10.1038/sj.onc.1207914
Shigematsu H, Suzuki M, Takahashi T, Miyajima K, Toyooka S, Shivapurkar N, Tomlinson GE, Mastrangelo D, Pass HI, Brambilla E, Sathyanarayana UG, Czerniak B, Fujisawa T, Shimizu N, Gazdar AF: Aberrant methylation of HIN-1 (high in normal-1) is a frequent event in many human malignancies. Int J Cancer. 2005, 113: 600-604. 10.1002/ijc.20622
Takai D, Yagi Y, Wakazono K, Ohishi N, Morita Y, Sugimura T, Ushijima T: Silencing of HTR1B and reduced expression of EDN1 in human lung cancers, revealed by methylation-sensitive representational difference analysis. Oncogene. 2001, 20: 7505-7513. 10.1038/sj.onc.1204940
Dunn JR, Panutsopulos D, Shaw MW, Heighway J, Dormer R, Salmo EN, Watson SG, Field JK, Liloglou T: METH-2 silencing and promoter hypermethylation in NSCLC. Br J Cancer. 2004, 91: 1149-1154.
Sathyanarayana UG, Toyooka S, Padar A, Takahashi T, Brambilla E, Minna JD, Gazdar AF: Epigenetic inactivation of laminin-5-encoding genes in lung cancers. Clin Cancer Res. 2003, 9: 2665-2672.
Wang YC, Lu YP, Tseng RC, Lin RK, Chang JW, Chen JT, Shih CM, Chen CY: Inactivation of hMLH1 and hMSH2 by promoter methylation in primary non-small cell lung tumors and matched sputum samples. J Clin Invest. 2003, 111: 887-895.
Nishioka M, Kohno T, Tani M, Yanaihara N, Tomizawa Y, Otsuka A, Sasaki S, Kobayashi K, Niki T, Maeshima A, Sekido Y, Minna JD, Sone S, Yokota J: MYO18B, a candidate tumor suppressor gene at chromosome 22q12.1, deleted, mutated, and methylated in human lung cancer. Proc Natl Acad Sci USA. 2002, 99: 12269-12274. 10.1073/pnas.192445899
Brena RM, Morrison C, Liyanarachchi S, Jarjoura D, Davuluri RV, Otterson GA, Reisman D, Glaros S, Rush LJ, Plass C: Aberrant DNA methylation of OLIG1, a novel prognostic factor in non-small cell lung cancer. PLoS Med. 2007, 4: e108- 10.1371/journal.pmed.0040108
Palmisano WA, Crume KP, Grimes MJ, Winters SA, Toyota M, Esteller M, Joste N, Baylin SB, Belinsky SA: Aberrant promoter methylation of the transcription factor genes PAX5 alpha and beta in human cancers. Cancer Res. 2003, 63: 4620-4625.
Gery S, Komatsu N, Kawamata N, Miller CW, Desmond J, Virk RK, Marchevsky A, McKenna R, Taguchi H, Koeffler HP: Epigenetic silencing of the candidate tumor suppressor gene Per1 in non-small cell lung cancer. Clin Cancer Res. 2007, 13: 1399-1404. 10.1158/1078-0432.CCR-06-1730
Xu L, Jain RK: Down-regulation of placenta growth factor by promoter hypermethylation in human lung and colon carcinoma. Mol Cancer Res. 2007, 5: 873-880. 10.1158/1541-7786.MCR-06-0141
Cooper WN, Dickinson RE, Dallol A, Grigorieva EV, Pavlova TV, Hesson LB, Bieche I, Broggini M, Maher ER, Zabarovsky ER, Clark GJ, Latif F: Epigenetic regulation of the ras effector/tumour suppressor RASSF2 in breast and lung cancer. Oncogene. 2008, 27: 1805-1811. 10.1038/sj.onc.1210805
Kaira K, Sunaga N, Tomizawa Y, Yanagitani N, Ishizuka T, Saito R, Nakajima T, Mori M: Epigenetic inactivation of the RAS-effector gene RASSF2 in lung cancers. Int J Oncol. 2007, 31: 169-173.
Choi N, Son DS, Song I, Lee HS, Lim YS, Song MS, Lim DS, Lee J, Kim H, Kim J: RASSF1A is not appropriate as an early detection marker or a prognostic marker for non-small cell lung cancer. Int J Cancer. 2005, 115: 575-581. 10.1002/ijc.20916
Wang J, Walsh G, Liu DD, Lee JJ, Mao L: Expression of Delta DNMT3B variants and its association with promoter methylation of p16 and RASSF1A in primary non-small cell lung cancer. Cancer Res. 2006, 66: 8361-8366. 10.1158/0008-5472.CAN-06-2031
Burbee DG, Forgacs E, Zochbauer-Muller S, Shivakumar L, Fong K, Gao B, Randle D, Kondo M, Virmani A, Bader S, Sekido Y, Latif F, Milchgrub S, Toyooka S, Gazdar AF, Lerman MI, Zabarovsky E, White M, Minna JD: Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J Natl Cancer Inst. 2001, 93: 691-699. 10.1093/jnci/93.9.691
Chang HC, Cho CY, Hung WC: Downregulation of RECK by promoter methylation correlates with lymph node metastasis in non-small cell lung cancer. Cancer Sci. 2007, 98: 169-173. 10.1111/j.1349-7006.2006.00367.x
Li QL, Kim HR, Kim WJ, Choi JK, Lee YH, Kim HM, Li LS, Kim H, Chang J, Ito Y, Youl Lee K, Bae SC: Transcriptional silencing of the RUNX3 gene by CpG hypermethylation is associated with lung cancer. Biochem Biophys Res Commun. 2004, 314: 223-228. 10.1016/j.bbrc.2003.12.079
He B, You L, Uematsu K, Zang K, Xu Z, Lee AY, Costello JF, McCormick F, Jablons DM: SOCS-3 is frequently silenced by hypermethylation and suppresses cell growth in human lung cancer. Proc Natl Acad Sci USA. 2003, 100: 14133-14138. 10.1073/pnas.2232790100
Suzuki M, Shigematsu H, Nakajima T, Kubo R, Motohashi S, Sekine Y, Shibuya K, Iizasa T, Hiroshima K, Nakatani Y, Gazdar AF, Fujisawa T: Synchronous alterations of Wnt and epidermal growth factor receptor signaling pathways through aberrant methylation and mutation in non small cell lung cancer. Clin Cancer Res. 2007, 13: 6087-6092. 10.1158/1078-0432.CCR-07-0591
Fukui T, Kondo M, Ito G, Maeda O, Sato N, Yoshioka H, Yokoi K, Ueda Y, Shimokata K, Sekido Y: Transcriptional silencing of secreted frizzled related protein 1 (SFRP 1) by promoter hypermethylation in non-small-cell lung cancer. Oncogene. 2005, 24: 6323-6327. 10.1038/sj.onc.1208777
Xu XL, Wu LC, Du F, Davis A, Peyton M, Tomizawa Y, Maitra A, Tomlinson G, Gazdar AF, Weissman BE, Bowcock AM, Baer R, Minna JD: Inactivation of human SRBC, located within the 11p15.5-p15.4 tumor suppressor region, in breast and lung cancers. Cancer Res. 2001, 61: 7943-7949.
Kuramochi M, Fukuhara H, Nobukuni T, Kanbe T, Maruyama T, Ghosh HP, Pletcher M, Isomura M, Onizuka M, Kitamura T, Sekiya T, Reeves RH, Murakami Y: TSLC1 is a tumor-suppressor gene in human non-small-cell lung cancer. Nat Genet. 2001, 27: 427-430. 10.1038/86934
Fukami T, Fukuhara H, Kuramochi M, Maruyama T, Isogai K, Sakamoto M, Takamoto S, Murakami Y: Promoter methylation of the TSLC1 gene in advanced lung tumors and various cancer cell lines. Int J Cancer. 2003, 107: 53-59. 10.1002/ijc.11348
Kikuchi S, Yamada D, Fukami T, Maruyama T, Ito A, Asamura H, Matsuno Y, Onizuka M, Murakami Y: Hypermethylation of the TSLC1/IGSF4 promoter is associated with tobacco smoking and a poor prognosis in primary nonsmall cell lung carcinoma. Cancer. 2006, 106: 1751-1758. 10.1002/cncr.21800
Shivapurkar N, Stastny V, Xie Y, Prinsen C, Frenkel E, Czerniak B, Thunnissen FB, Minna JD, Gazdar AF: Differential methylation of a short CpG-rich sequence within exon 1 of TCF21 gene: a promising cancer biomarker assay. Cancer Epidemiol Biomarkers Prev. 2008, 17: 995-1000. 10.1158/1055-9965.EPI-07-2808
Wang G, Hu X, Lu C, Su C, Luo S, Luo ZW: Promoter-hypermethylation associated defective expression of E-cadherin in primary non-small cell lung cancer. Lung Cancer. 2008.
Chan EC, Lam SY, Tsang KW, Lam B, Ho JC, Fu KH, Lam WK, Kwong YL: Aberrant promoter methylation in Chinese patients with non-small cell lung cancer: patterns in primary tumors and potential diagnostic application in bronchoalevolar lavage. Clin Cancer Res. 2002, 8: 3741-3746.
Guo M, House MG, Hooker C, Han Y, Heath E, Gabrielson E, Yang SC, Baylin SB, Herman JG, Brock MV: Promoter hypermethylation of resected bronchial margins: a field defect of changes?. Clin Cancer Res. 2004, 10: 5131-5136. 10.1158/1078-0432.CCR-03-0763
Shivapurkar N, Toyooka S, Toyooka KO, Reddy J, Miyajima K, Suzuki M, Shigematsu H, Takahashi T, Parikh G, Pass HI, Chaudhary PM, Gazdar AF: Aberrant methylation of trail decoy receptor genes is frequent in multiple tumor types. Int J Cancer. 2004, 109: 786-792. 10.1002/ijc.20041
Brabender J, Usadel H, Metzger R, Schneider PM, Park J, Salonga D, Tsao-Wei DD, Groshen S, Lord RV, Takebe N, Schneider S, Holscher AH, Danenberg KD, Danenberg PV: Quantitative O(6)-methylguanine DNA methyltransferase methylation analysis in curatively resected non-small cell lung cancer: associations with clinical outcome. Clin Cancer Res. 2003, 9: 223-227.
Furonaka O, Takeshima Y, Awaya H, Kushitani K, Kohno N, Inai K: Aberrant methylation and loss of expression of O-methylguanine-DNA methyltransferase in pulmonary squamous cell carcinoma and adenocarcinoma. Pathol Int. 2005, 55: 303-309. 10.1111/j.1440-1827.2005.01830.x
Fong KM, Sekido Y, Gazdar AF, Minna JD: Lung cancer. 9: Molecular biology of lung cancer: clinical implications. Thorax. 2003, 58: 892-900. 10.1136/thorax.58.10.892
Suzuki M, Shigematsu H, Iizasa T, Hiroshima K, Nakatani Y, Minna JD, Gazdar AF, Fujisawa T: Exclusive mutation in epidermal growth factor receptor gene, HER-2, and KRAS, and synchronous methylation of nonsmall cell lung cancer. Cancer. 2006, 106: 2200-2207. 10.1002/cncr.21853
Tang M, Torres-Lanzas J, Lopez-Rios F, Esteller M, Sanchez-Cespedes M: Wnt signaling promoter hypermethylation distinguishes lung primary adenocarcinomas from colorectal metastasis to the lung. Int J Cancer. 2006, 119: 2603-2606. 10.1002/ijc.22211
Tsou JA, Shen LY, Siegmund KD, Long TI, Laird PW, Seneviratne CK, Koss MN, Pass HI, Hagen JA, Laird-Offringa IA: Distinct DNA methylation profiles in malignant mesothelioma, lung adenocarcinoma, and non-tumor lung. Lung Cancer. 2005, 47: 193-204. 10.1016/j.lungcan.2004.08.003
Wang Y, Zhang D, Zheng W, Luo J, Bai Y, Lu Z: Multiple gene methylation of nonsmall cell lung cancers evaluated with 3-dimensional microarray. Cancer. 2008, 112: 1325-1336. 10.1002/cncr.23312
Kim DS, Cha SI, Lee JH, Lee YM, Choi JE, Kim MJ, Lim JS, Lee EB, Kim CH, Park TI, Jung TH, Park JY: Aberrant DNA methylation profiles of non-small cell lung cancers in a Korean population. Lung Cancer. 2007, 58: 1-6. 10.1016/j.lungcan.2007.04.008
Yanagawa N, Tamura G, Oizumi H, Takahashi N, Shimazaki Y, Motoyama T: Promoter hypermethylation of tumor suppressor and tumor-related genes in non-small cell lung cancers. Cancer Sci. 2003, 94: 589-592. 10.1111/j.1349-7006.2003.tb01487.x
Sano A, Kage H, Sugimoto K, Kitagawa H, Aki N, Goto A, Fukayama M, Nakajima J, Takamoto S, Nagase T, Yatomi Y, Ohishi N, Takai D: A second-generation profiling system for quantitative methylation analysis of multiple gene promoters: application to lung cancer. Oncogene. 2007, 26: 6518-6525. 10.1038/sj.onc.1210483
Zochbauer-Muller S, Fong KM, Virmani AK, Geradts J, Gazdar AF, Minna JD: Aberrant promoter methylation of multiple genes in non-small cell lung cancers. Cancer Res. 2001, 61: 249-255.
Shivapurkar N, Stastny V, Suzuki M, Wistuba II, Li L, Zheng Y, Feng Z, Hol B, Prinsen C, Thunnissen FB, Gazdar AF: Application of a methylation gene panel by quantitative PCR for lung cancers. Cancer Lett. 2007, 247: 56-71. 10.1016/j.canlet.2006.03.020
Safar AM, Spencer H, Su X, Coffey M, Cooney CA, Ratnasinghe LD, Hutchins LF, Fan CY: Methylation profiling of archived non-small cell lung cancer: a promising prognostic system. Clin Cancer Res. 2005, 11: 4400-4405. 10.1158/1078-0432.CCR-04-2378
Marsit CJ, Houseman EA, Christensen BC, Eddy K, Bueno R, Sugarbaker DJ, Nelson HH, Karagas MR, Kelsey KT: Examination of a CpG island methylator phenotype and implications of methylation profiles in solid tumors. Cancer Res. 2006, 66: 10621-10629. 10.1158/0008-5472.CAN-06-1687
Dammann R, Strunnikova M, Schagdarsurengin U, Rastetter M, Papritz M, Hattenhorst UE, Hofmann HS, Silber RE, Burdach S, Hansen G: CpG island methylation and expression of tumour-associated genes in lung carcinoma. Eur J Cancer. 2005, 41: 1223-1236. 10.1016/j.ejca.2005.02.020
Feng Q, Hawes SE, Stern JE, Wiens L, Lu H, Dong ZM, Jordan CD, Kiviat NB, Vesselle H: DNA methylation in tumor and matched normal tissues from non-small cell lung cancer patients. Cancer Epidemiol Biomarkers Prev. 2008, 17: 645-654. 10.1158/1055-9965.EPI-07-2518
Ehrich M, Field JK, Liloglou T, Xinarianos G, Oeth P, Nelson MR, Cantor CR, Boom van den D: Cytosine methylation profiles as a molecular marker in non-small cell lung cancer. Cancer Res. 2006, 66: 10911-10918. 10.1158/0008-5472.CAN-06-0400
Field JK, Liloglou T, Warrak S, Burger M, Becker E, Berlin K, Nimmrich I, Maier S: Methylation discriminators in NSCLC identified by a microarray based approach. Int J Oncol. 2005, 27: 105-111.
Fukasawa M, Kimura M, Morita S, Matsubara K, Yamanaka S, Endo C, Sakurada A, Sato M, Kondo T, Horii A, Sasaki H, Hatada I: Microarray analysis of promoter methylation in lung cancers. J Hum Genet. 2006, 51: 368-374. 10.1007/s10038-005-0355-4
Dai Z, Lakshmanan RR, Zhu WG, Smiraglia DJ, Rush LJ, Fruhwald MC, Brena RM, Li B, Wright FA, Ross P, Otterson GA, Plass C: Global methylation profiling of lung cancer identifies novel methylated genes. Neoplasia. 2001, 3: 314-323. 10.1038/sj.neo.7900162
Bibikova M, Lin Z, Zhou L, Chudin E, Garcia EW, Wu B, Doucet D, Thomas NJ, Wang Y, Vollmer E, Goldmann T, Seifart C, Jiang W, Barker DL, Chee MS, Floros J, Fan JB: High-throughput DNA methylation profiling using universal bead arrays. Genome Res. 2006, 16: 383-393. 10.1101/gr.4410706
Hatada I, Fukasawa M, Kimura M, Morita S, Yamada K, Yoshikawa T, Yamanaka S, Endo C, Sakurada A, Sato M, Kondo T, Horii A, Ushijima T, Sasaki H: Genome-wide profiling of promoter methylation in human. Oncogene. 2006, 25: 3059-3064. 10.1038/sj.onc.1209331
Shames DS, Girard L, Gao B, Sato M, Lewis CM, Shivapurkar N, Jiang A, Perou CM, Kim YH, Pollack JR, Fong KM, Lam CL, Wong M, Shyr Y, Nanda R, Olopade OI, Gerald W, Euhus DM, Shay JW, Gazdar AF, Minna JD: A genome-wide screen for promoter methylation in lung cancer identifies novel methylation markers for multiple malignancies. PLoS Med. 2006, 3: e486- 10.1371/journal.pmed.0030486
Cortese R, Hartmann O, Berlin K, Eckhardt F: Correlative gene expression and DNA methylation profiling in lung development nominate new biomarkers in lung cancer. Int J Biochem Cell Biol. 2008, 40: 1494-1508. 10.1016/j.biocel.2007.11.018
Rauch T, Li H, Wu X, Pfeifer GP: MIRA-assisted microarray analysis, a new technology for the determination of DNA methylation patterns, identifies frequent methylation of homeodomain-containing genes in lung cancer cells. Cancer Res. 2006, 66: 7939-7947. 10.1158/0008-5472.CAN-06-1888
Rauch TA, Zhong X, Wu X, Wang M, Kernstine KH, Wang Z, Riggs AD, Pfeifer GP: High-resolution mapping of DNA hypermethylation and hypomethylation in lung cancer. Proc Natl Acad Sci USA. 2008, 105: 252-257. 10.1073/pnas.0710735105
Rauch T, Wang Z, Zhang X, Zhong X, Wu X, Lau SK, Kernstine KH, Riggs AD, Pfeifer GP: Homeobox gene methylation in lung cancer studied by genome-wide analysis with a microarray-based methylated CpG island recovery assay. Proc Natl Acad Sci USA. 2007, 104: 5527-5532. 10.1073/pnas.0701059104
Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, Laird PW: Epigenetic stem cell signature in cancer. Nat Genet. 2007, 39: 157-158. 10.1038/ng1941
Zhou W, Heist RS, Liu G, Neuberg DS, Asomaning K, Su L, Wain JC, Lynch TJ, Giovannucci E, Christiani DC: Polymorphisms of vitamin D receptor and survival in early-stage non-small cell lung cancer patients. Cancer Epidemiol Biomarkers Prev. 2006, 15: 2239-2245. 10.1158/1055-9965.EPI-06-0023
Vischioni B, Oudejans JJ, Vos W, Rodriguez JA, Giaccone G: Frequent overexpression of aurora B kinase, a novel drug target, in non-small cell lung carcinoma patients. Mol Cancer Ther. 2006, 5: 2905-2913. 10.1158/1535-7163.MCT-06-0301
Tam IY, Chung LP, Suen WS, Wang E, Wong MC, Ho KK, Lam WK, Chiu SW, Girard L, Minna JD, Gazdar AF, Wong MP: Distinct epidermal growth factor receptor and KRAS mutation patterns in non-small cell lung cancer patients with different tobacco exposure and clinicopathologic features. Clin Cancer Res. 2006, 12: 1647-1653. 10.1158/1078-0432.CCR-05-1981
Raponi M, Zhang Y, Yu J, Chen G, Lee G, Taylor JM, Macdonald J, Thomas D, Moskaluk C, Wang Y, Beer DG: Gene expression signatures for predicting prognosis of squamous cell and adenocarcinomas of the lung. Cancer Res. 2006, 66: 7466-7472. 10.1158/0008-5472.CAN-06-1191
Shapiro B, Chakrabarty M, Cohn EM, Leon SA: Determination of circulating DNA levels in patients with benign or malignant gastrointestinal disease. Cancer. 1983, 51: 2116-2120. 10.1002/1097-0142(19830601)51:11<2116::AID-CNCR2820511127>3.0.CO;2-S
Hagiwara N, Mechanic LE, Trivers GE, Cawley HL, Taga M, Bowman ED, Kumamoto K, He P, Bernard M, Doja S, Miyashita M, Tajiri T, Sasajima K, Nomura T, Makino H, Takahashi K, Hussain SP, Harris CC: Quantitative detection of p53 mutations in plasma DNA from tobacco smokers. Cancer Res. 2006, 66: 8309-8317. 10.1158/0008-5472.CAN-06-0991
Sozzi G, Musso K, Ratcliffe C, Goldstraw P, Pierotti MA, Pastorino U: Detection of microsatellite alterations in plasma DNA of non-small cell lung cancer patients: a prospect for early diagnosis. Clin Cancer Res. 1999, 5: 2689-2692.
Khan S, Coulson JM, Woll PJ: Genetic abnormalities in plasma DNA of patients with lung cancer and other respiratory diseases. Int J Cancer. 2004, 110: 891-895. 10.1002/ijc.20156
Laird PW, Jaenisch R: The role of DNA methylation in cancer genetic and epigenetics. Annu Rev Genet. 1996, 30: 441-464. 10.1146/annurev.genet.30.1.441
Widschwendter M, Menon U: Circulating methylated DNA: a new generation of tumor markers. Clin Cancer Res. 2006, 12: 7205-7208. 10.1158/1078-0432.CCR-06-2531
Belinsky SA, Grimes MJ, Casas E, Stidley CA, Franklin WA, Bocklage TJ, Johnson DH, Schiller JH: Predicting gene promoter methylation in non-small-cell lung cancer by evaluating sputum and serum. Br J Cancer. 2007, 96: 1278-1283. 10.1038/sj.bjc.6603721
Di Vinci A, Gelvi I, Banelli B, Casciano I, Allemanni G, Romani M: Meth-DOP-PCR: an assay for the methylation profiling of trace amounts of DNA extracted from bodily fluids. Lab Invest. 2006, 86: 297-303. 10.1038/labinvest.3700384
Esteller M, Sanchez-Cespedes M, Rosell R, Sidransky D, Baylin SB, Herman JG: Detection of aberrant promoter hypermethylation of tumor suppressor genes in serum DNA from non-small cell lung cancer patients. Cancer Res. 1999, 59: 67-70.
Fujiwara K, Fujimoto N, Tabata M, Nishii K, Matsuo K, Hotta K, Kozuki T, Aoe M, Kiura K, Ueoka H, Tanimoto M: Identification of epigenetic aberrant promoter methylation in serum DNA is useful for early detection of lung cancer. Clin Cancer Res. 2005, 11: 1219-1225. 10.1158/1078-0432.CCR-04-2363
Wang Y, Yu Z, Wang T, Zhang J, Hong L, Chen L: Identification of epigenetic aberrant promoter methylation of RASSF1A in serum DNA and its clinicopathological significance in lung cancer. Lung Cancer. 2007, 56: 289-294. 10.1016/j.lungcan.2006.12.007
Virmani AK, Tsou JA, Siegmund KD, Shen LY, Long TI, Laird PW, Gazdar AF, Laird-Offringa IA: Hierarchical clustering of lung cancer cell lines using DNA methylation markers. Cancer Epidemiol Biomarkers Prev. 2002, 11: 291-297.
Miozzo M, Sozzi G, Musso K, Pilotti S, Incarbone M, Pastorino U, Pierotti MA: Microsatellite alterations in bronchial and sputum specimens of lung cancer patients. Cancer Res. 1996, 56: 2285-2288.
Belinsky SA, Liechty KC, Gentry FD, Wolf HJ, Rogers J, Vu K, Haney J, Kennedy TC, Hirsch FR, Miller Y, Franklin WA, Herman JG, Baylin SB, Bunn PA, Byers T: Promoter hypermethylation of multiple genes in sputum precedes lung cancer incidence in a high-risk cohort. Cancer Res. 2006, 66: 3338-3344. 10.1158/0008-5472.CAN-05-3408
Olaussen KA, Soria JC, Park YW, Kim HJ, Kim SH, Ro JY, Andre F, Jang SJ: Assessing abnormal gene promoter methylation in paraffin-embedded sputum from patients with NSCLC. Eur J Cancer. 2005, 41: 2112-2119. 10.1016/j.ejca.2005.06.013
Palmisano WA, Divine KK, Saccomanno G, Gilliland FD, Baylin SB, Herman JG, Belinsky SA: Predicting lung cancer by detecting aberrant promoter methylation in sputum. Cancer Res. 2000, 60: 5954-5958.
Hunt J: Exhaled breath condensate: an evolving tool for noninvasive evaluation of lung disease. J Allergy Clin Immunol. 2002, 110: 28-34. 10.1067/mai.2002.124966
Carpagnano GE, Foschino-Barbaro MP, Resta O, Gramiccioni E, Carpagnano F: Endothelin-1 is increased in the breath condensate of patients with non-small-cell lung cancer. Oncology. 2004, 66: 180-184. 10.1159/000077992
Carpagnano GE, Resta O, Foschino-Barbaro MP, Gramiccioni E, Carpagnano F: Interleukin-6 is increased in breath condensate of patients with non-small cell lung cancer. Int J Biol Markers. 2002, 17: 141-145.
Carpagnano GE, Foschino-Barbaro MP, Mule G, Resta O, Tommasi S, Mangia A, Carpagnano F, Stea G, Susca A, Di Gioia G, De Lena M, Paradiso A: 3p microsatellite alterations in exhaled breath condensate from patients with non-small cell lung cancer. Am J Respir Crit Care Med. 2005, 172: 738-744. 10.1164/rccm.200503-439OC
Gessner C, Kuhn H, Toepfer K, Hammerschmidt S, Schauer J, Wirtz H: Detection of p53 gene mutations in exhaled breath condensate of non-small cell lung cancer patients. Lung Cancer. 2004, 43: 215-222. 10.1016/j.lungcan.2003.08.034
Ahrendt SA, Chow JT, Xu LH, Yang SC, Eisenberger CF, Esteller M, Herman JG, Wu L, Decker PA, Jen J, Sidransky D: Molecular detection of tumor cells in bronchoalveolar lavage fluid from patients with early stage lung cancer. J Natl Cancer Inst. 1999, 91: 332-339. 10.1093/jnci/91.4.332
Grote HJ, Schmiemann V, Kiel S, Bocking A, Kappes R, Gabbert HE, Sarbia M: Aberrant methylation of the adenomatous polyposis coli promoter 1A in bronchial aspirates from patients with suspected lung cancer. Int J Cancer. 2004, 110: 751-755. 10.1002/ijc.20196
de Fraipont F, Moro-Sibilot D, Michelland S, Brambilla E, Brambilla C, Favrot MC: Promoter methylation of genes in bronchial lavages: a marker for early diagnosis of primary and relapsing non-small cell lung cancer?. Lung Cancer. 2005, 50: 199-209. 10.1016/j.lungcan.2005.05.019
Grote HJ, Schmiemann V, Geddert H, Bocking A, Kappes R, Gabbert HE, Sarbia M: Methylation of RAS association domain family protein 1A as a biomarker of lung cancer. Cancer. 2006, 108: 129-134. 10.1002/cncr.21717
Grote HJ, Schmiemann V, Geddert H, Rohr UP, Kappes R, Gabbert HE, Bocking A: Aberrant promoter methylation of p16(INK4a), RARB2 and SEMA3B in bronchial aspirates from patients with suspected lung cancer. Int J Cancer. 2005, 116: 720-725. 10.1002/ijc.21090
Schmiemann V, Bocking A, Kazimirek M, Onofre AS, Gabbert HE, Kappes R, Gerharz CD, Grote HJ: Methylation assay for the diagnosis of lung cancer on bronchial aspirates: a cohort study. Clin Cancer Res. 2005, 11: 7728-7734. 10.1158/1078-0432.CCR-05-0999
Kramer B, Gohagan J, Prorok P: Cancer Screening: Theory and Practice. 1999, Marcel Dekker.
Cancer of the Lung and Bronchus. http://seer.cancer.gov/statfacts/html/lungb.html
Bain C, Feskanich D, Speizer FE, Thun M, Hertzmark E, Rosner BA, Colditz GA: Lung cancer rates in men and women with comparable histories of smoking. J Natl Cancer Inst. 2004, 96: 826-834.
Bearzatto A, Conte D, Frattini M, Zaffaroni N, Andriani F, Balestra D, Tavecchio L, Daidone MG, Sozzi G: p16(INK4A) Hypermethylation detected by fluorescent methylation-specific PCR in plasmas from non-small cell lung cancer. Clin Cancer Res. 2002, 8: 3782-3787.
Xie GS, Hou AR, Li LY, Gao YN, Cheng SJ: Aberrant p16 promoter hypermethylation in bronchial mucosae as a biomarker for the early detection of lung cancer. Chin Med J (Engl). 2006, 119: 1469-1472.
Pepe MS, Etzioni R, Feng Z, Potter JD, Thompson ML, Thornquist M, Winget M, Yasui Y: Phases of biomarker development for early detection of cancer. J Natl Cancer Inst. 2001, 93: 1054-1061. 10.1093/jnci/93.14.1054
American Cancer Society. http://www.cancer.org
Sun S, Schiller JH, Gazdar AF: Lung cancer in never smokers–a different disease. Nat Rev Cancer. 2007, 7: 778-790. 10.1038/nrc2190
Acknowledgements
The authors thank Laird-Offringa lab members for critical comments on the manuscript. Grant support for IALO includes: National Institutes of Health/National Cancer Institute R21 CA102247, R01 CA119029 and R01 CA120869, Whittier Foundation Translational Research Grant, a STOP Cancer award, a Joan's Legacy/Thomas Labrecque Foundation grant, and generous gifts from the Kazan, McClain, Abrams, Fernandez, Lyons & Farrise Foundation, the Canary Foundation, Paul and Michelle Zygielbaum, and Conya and Wallace Pembroke. None of the funding agencies played any role in the collection, analysis, interpretation of the data, writing of the manuscript, nor the decision to publish. The content is solely the responsibility of the authors and does not represent the official views of the funding agencies.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
PPA was involved in drafting the manuscript and generation of tables. TAA was involved in reviewing and editing the manuscript. IALO mentored PPA, and revised manuscript drafts. All authors reviewed and commented on the manuscript during its drafting and approved the final version.
Electronic supplementary material
12943_2008_372_MOESM1_ESM.doc
Additional file 1: Alphabetical list of all loci reported to be methylated in NSCLC in studies on single loci/small panels of loci. This table lists loci that were described to be methylated in studies of single loci, or small panels of loci, and describes in what fraction of samples the locus was methylated, from which source material the DNA was extracted, whether a specific NSCLC subtype was investigated, and the bibliography number for the reference. (DOC 664 KB)
12943_2008_372_MOESM2_ESM.doc
Additional file 2: Alphabetical list of selected loci of interest from studies using targeted or genome-wide approaches to examine DNA methylation at more than 20 loci. This table lists the loci of interest that were identified using approaches that examine many loci, the method used to identify them, the details concerning how many loci were examined, the fraction of tissues found to be methylated (where applicable), and the bibliography number for the reference. (DOC 200 KB)
12943_2008_372_MOESM3_ESM.doc
Additional file 3: Alphabetical list of genes for which DNA methylation status has been examined in remote media. This table lists loci for which DNA methylation status has been examined in remote media, the fraction of samples methylated, the remote medium used, the detection method used, and the bibliography number for the reference. (DOC 326 KB)
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Anglim, P.P., Alonzo, T.A. & Laird-Offringa, I.A. DNA methylation-based biomarkers for early detection of non-small cell lung cancer: an update. Mol Cancer 7, 81 (2008). https://doi.org/10.1186/1476-4598-7-81
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-4598-7-81