
Abstract
Background
Metallothionein 3 (MT3) inhibits growth in a variety of cell types. We measured MT3 gene expression by RT-PCR, and DNA methylation in the MT3 promoter by combined bisulphite restriction analysis, in four oesophageal cancer cell lines and the resected oesophagus from 64 patients with oesophageal squamous cell carcinoma (SCC).
Results
MT3 expression was not detected in one of the four oesophageal cell lines. The MT3 promoter was methylated in all of the oesophageal cell lines, but the degree of methylation was greater in the non-expressing cell line. After treatment with 5-aza-2'-deoxycytidine there was a reduction in the degree of methylation, and an increase in MT3 expression, in each of the cell lines (p < 0.01). Methylation was detected in 52% (33 of 64) of primary SCC and 3% (2 of 62) of histologically normal resection margins. MT3 expression was measured in 29 tumours, 17 of which had methylation of MT3. The expression of MT3 was significantly less in the methylated tumours compared to either the unmethylated tumours (p = 0.03), or the matched margin (p = 0.0005). There was not a significant difference in MT3 expression between the tumour and the margin from patients with unmethylated tumour. No correlations were observed between methylation of MT3 and survival time, patient age, gender, smoking or drinking history, tumour stage, volume, or lymph node involvement.
Conclusion
We conclude that MT3 ex pression is frequently down-regulated in oesophageal SCC, by DNA methylation, but that this is not a prognostic indicator.
Similar content being viewed by others


Background
The metallothioneins (MT) are a group of low molecular weight, cysteine-rich intracellular proteins that are involved in maintaining intracellular metal homeostasis by binding transition metals such as zinc and copper. There are 10 functional isoforms of MTs described, which are divided into 4 classes, designated MT1 – 4, on the basis of small differences in protein sequence and charge characteristics [1, 2]. The MTs have been proposed to play an important role in protecting against DNA damage, apoptosis and oxidative stress [3].
Metallothionein 3 (MT3) was first identified as a growth inhibitory factor, expressed in normal brain, which inhibited the survival of neurones in culture and also neurite formation [4]. Subsequent studies using glial [5] or tumour[6–8] cells, stably transfected with MT3, showed that its endogenous over-expression could inhibit cell growth. More recently, down-regulation of MT3 was reported as one of 17 changes in gene expression which was most likely to be associated with metastasis and poor clinical outcome in a range of solid tumours [9]. One mechanism for reducing gene expression is methylation of the CpG island when present in the promoter region of the gene [10]. Methylation of the MT3 promoter has been observed and has been suggested to cause reduced expression in gastric cancer [11].
The levels of MT3 expression, the frequency of MT3 methylation, and correlations between MT3 methylation and clinical parameters, have not been investigated in oesophageal squamous cell carcinoma (SCC). In this study we used combined bisulfite restriction analysis (COBRA) [12] to estimate the frequency of methylation at specific sites within the MT3 promoter in oesophageal cancer cell lines, and investigated the relationship between methylation and its expression by quantitative real-time RT-PCR. We then measured MT3 gene expression and the frequency of MT3 methylation in primary oesophageal SCCs and, when available, the histologically normal, proximal resection margin from those patients.
Results
Methylation analysis of MT3 in normal lymphocytes and oesophageal cell lines
The methylation status of the MT3 CpG island was measured by COBRA within 3 overlapping regions (Figure 1). Complete digestion of the COBRA PCR product indicated methylation at one or more of the BstUI sites within that region of all alleles. Incomplete digestion indicated methylation at one or more of the BstUI sites within that region of some but not all alleles.

Schematic diagram of the MT3 promoter. The diagram was generated by downloading the MT3 CpG island genetic element with the addition of 200 bases upstream from http://genome.ucsc.edu/, using the Human July 2003 Freeze. The location of 5' UTR (□), exon 1 and exon 2 (■) of MT3 (NM_005954) are shown. Each vertical line represents a CpG. The arrows represent the location of the 12 BstUI restriction enzyme sites analysed. The regions amplified for the 3 COBRA PCRs, R1, R2 and R3, are shown.
COBRA was performed on the bisulphite modified lymphocyte DNA from 19 different normal donors without any known cancers or other genetic abnormalities. In each of these donor lymphocytes samples an average of two percent (± 1% SD) of molecules were methylated at site 12 in the R1 region, located in intron 1. There was no evidence of methylation at any of the other sites analysed by COBRA in the R1 and the R2 regions (data not shown).
COBRA was performed on the oesophageal cancer cell lines OE19, OE21, OE33 and TE7, cultured with or without the DNA demethylation drug 5-aza-2'-deoxycytine (aza-dC) (Figure 2). Complete digestion of each of the 3 regions was observed for OE33. For TE7, incomplete digestion was observed in the R1 and R2 regions, whilst complete digestion was observed in R3 region. Incomplete digestion was observed in each of the 3 regions for OE21. For OE19, no digestion was observed in the R2 region, but incomplete digestion was observed in the R1 and R3 regions. The frequency of methylation at any BstUI site within each of the regions, R1, R2 or R3, was estimated in the oesophageal cell lines (Table 1). The frequency of methylation in each region varied from cell line to cell line, and the frequency of methylation in a given cell line varied from region to region. We then estimated the frequency of methylation at each BstUI site within a region (Table 2). We observed that the frequency of methylation at each BstUI site within a region varied for a given cell line and varied between cell lines.

The MT3 COBRA on regions 1 (A), 2 (B) and 3 (C) for the oesophageal cell lines with or without treatment with Aza-dC. The COBRA was performed on the oesophageal cell lines following 72 to 96 hrs of treatment with either 1 μM aza-dC (+) or vehicle (-). M, molecular weight marker.
Re-expression of the MT3 after 5-aza-2'-deoxycytine treatment
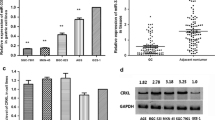
To determine the relationship between methylation and transcription of the MT3 gene, we measured the expression of MT3 by RT-PCR in 4 oesophageal cell lines, OE19, OE21, OE33 and TE7, cultured in the presence or absence of aza-dC (Figure 3). MT3 expression was undetectable in the completely methylated cell line OE33. Low levels of MT3 were measured in the partially methylated cell lines OE19, OE21 and TE7. Demethylation treatment with aza-dC reduced the frequency of methylation both within and between different regions for all cell lines provided that the intial frequency of methylation was ≥ 10 %. Demethylation treatment did not completely demethylate all the cells in culture. The expression of MT3 was significantly increased following aza-dC treatment in each of the oesophageal cell lines (p < 0.01). The increase in MT3 expression induced by aza-dC was significantly less in the OE33 cell line compared to the OE19, OE21 or TE7 (p = 0.02).

The expression of MT3 in oesophageal cell lines. A) MT3 expression in the oesophageal cell lines grown for 72 to 96 h with either 1 μM aza-dC (■) or vehicle (□). Results are the average of triplicate values ± SD, normalised to that of PBGD. B) Representative example of gel electrophoresis of the MT3 and PBGD RT-PCR products for the cell lines following treatment with either 1 μM aza-dC (+) or vehicle (-).
Methylation and expression of MT3 in oesophageal SCC
The frequency of MT3 methylation was measured in resected primary tumour and, when available, the proximal resection margin from 64 patients with oesophageal SCC. From each sample the R1 and R2 regions of the MT3 CpG island were amplified by PCR and then analysed for methylation by COBRA. As with the lymphocyte samples, low levels of methylation at BstUI site 12 in the R1 region were observed in all the histologically normal margins and the tumour samples from these patients. Thus, this ubiquitous low level of methylation was not considered in the subsequent analysis. Methylation of the R1 region was observed in 33 (52%) and of the R2 region in 10 (16%) of the tumour samples. All samples that were methylated in the R2 region were also methylated in the R1 region. No correlations were observed between patient age, gender, smoking or drinking history, tumour, volume, or lymph node involvement and the methylation of MT3 (Table 3). Methylation of the R2 region was observed in none of the 62 proximal resection margin tissues available, whilst methylation of the R1 region was observed in 2 (3%).
MT3 gene expression was analysed by quantitative real-time RT-PCR in 29 of the primary tumours and their matched non-cancerous proximal resection margins for which RNA was available. MT3 was methylated in 17 of these tumours, and unmethylated in 12. The MT3 expression was normalized to that of ACTB. The expression of MT3 was significantly less in the tumours in which the promoter was methylated compared to those in which it was unmethylated (Figure 4, p = 0.03). MT3 expression in methylated tumours was significantly less than that of the matched margin (p = 0.0005). There was not a statistically significant difference in expression between the tumour and matched margin from patients in whom the tumour was unmethylated.

MT3 expression in patient-matched tumour and non-cancerous proximal resection margin from patients with SCC of the oesophagus. MT3 expression in the oesophageal tissue from patients with either methylated tumour (○) or umethylated tumour (●). Results are the average of triplicate values, normalised to that of ACTB. The grey bars represent the median expression for each group. The black lines link expression values of tissues from the same patient.
Survival data was available for 41 patients, 19 with an unmethylated tumour, 22 methylated. There was no difference between these 2 groups with respect to the time since operation (unmethylated: range 3 – 22 months, median 20 months; methylated: range 1 – 22 months, median 21 months). There were 5 deaths in the unmethylated and 4 in the methylated group (P = 0.7). There was no difference in the survival time following operation, with deaths in the unmethylated group occurring 3, 13, 13, 17 and 18 months post-operatively, compared to 1, 4, 8 and 22 months in the methylated group.
Discussion
In this study we investigated the expression of MT3 and its control by DNA methylation in oesophageal SCC. We measured methylation of MT3 by COBRA in three separate regions of its promoter, and gene expression by quantitative real-time RT-PCR. In the oesophageal cell line OE33, which had complete methylation at all the CpG sites analysed, there was also complete transcriptional silencing of the MT3 gene. In a further 3 cell oesophageal cancer cell lines, each of which had partial methylation, there was a partial but not complete reduction in gene expression. Treatment of each of these cell lines with aza-dC, which reduced methylation, resulted in an increase in gene expression.
In those the cell lines which had partial methylation, the amount of methylation could vary between contiguous CpGs in the same cell line, and could vary in particular CpGs between cell lines. Thus, a particular CpG which might be unmethylated in one cell line could be partially or completely methylated in other cell lines expressing similar levels of MT3. This is consistent with the reported variability of methylation of the p16 CpG island in primary human mammary epithelial cells during escape from growth arrest [13], and the O6-methylguanine-DNA methyltransferase gene in human cell lines [14]. The significance of this finding is that methods for the measurement of DNA methylation such as methylation specific PCR (MSP) which analyse methylation at one or two CpGs only may misrepresent the methylation pattern of a region. In addition these methods would mislead if they probed CpGs with low levels of methylation in all tissues analysed, such as the site 12 in intron 1 in our analysis of MT3. This was methylated in normal lymphocytes and normal oesophageal tissue, as well oesophageal SCC, even though other CpGs were unmethylated and gene expression was high.
In order to show the relationship between methylation within the regions studied and MT3 gene expression, we incubated cultures of the oesophageal cell lines for approximately two cell divisions with aza-dC, a potent inhibitor of DNA methyltransferase. Because the rate of division varied for each of the cell lines, we determined the length of time required for each of the cell lines to divide twice in the presence of aza-dC using PKH-26 labelling [15]. This stable red fluorescent dye inserts into the cytoplasmic membrane, and is distributed equally amongst each of the daughter cells at the time of cell division, such that the mean fluorescence intensity of the cell population is halved with each cell division. As expected, treatment with aza-dC for two cell divisions did not completely demethylate the cell lines. This is because it is only newly synthesised strands which are demethylated, so if in a cell a particular region of DNA is completely methylated on both alleles, after 2 divisions only 75% of the alleles will be demethylated. We found that there was a significant reduction in the amount of methylation at each of the methylated sites analysed following drug treatment. Interestingly, the amount of the reduction varied between different CpG sites within a cell line, and varied from cell line to cell line at a given site. The reasons for this cell line and regional variation in the extent of demethylation are unknown.
Previous studies have shown that the methylation of only a subset of available CpG sites within a region can be sufficient to reduce transcription [16–18, 13], with a greater reduction in transcription as the density of CpG methylation increases [19]. Complete methylation of all the sites in the regions which we studied correlated with lack of any gene expression, while methylation of only a subset of sites correlated with some MT3 expression, and the further reduction in methylation resulting from demethylation with aza-dC always correlated with an increase in MT3 expression. In this study we evaluated MT3 expression only by RT-PCR, because an antibody specific for the MT3 protein was not commercially available. However, previous studies have shown that when the MT3 transcript was able to be detected the protein was also detectable [20, 21, 7, 8].
Our interest in MT3 came from a report that solid tumours with a specific pattern of expression of 17 genes, including down-regulation of MT3, were most likely to be associated with metastasis and poor outcome [9]. Oesophageal SCC is an aggressive disease with a 5 year survival of about 20%, death most commonly due to secondaries. Molecular markers which assist in predicting metasases might help to tailor treatment options better. MT3 was first identified as a growth inhibitory factor which decreased the survival of neuronal cultures [4]. Subsequent studies using stably transfected cell lines showed that up-regulation of MT3 was growth inhibitory in some, but not all, cell lines [6–8].
The reported role of MT3 in carcinogenesis is unclear. In gastric carcinomas MT3 expression was found to be markedly reduced, but there was no indication of any relationship to outcome in this report [22]. In contrast, levels of MT3 protein were shown to be elevated in bladder [20] and breast carcinomas [21]. This elevated expression was a poor prognostic indicator, being associated with increased tumour stage in bladder carcinoma, and poor disease outcome in some breast carcinomas. In our series of 64 patients with oesophageal SCC we found a degree of methylation in 52% of primary tumours and 3% of the histologically normal proximal resection margin. We observed that MT3 expression was frequently down-regulated in primary oesophageal SCC when compared to normal mucosa, and significant down-regulation was most commonly observed in those tumours that were methylated for MT3, and not unmethylated tumours. Interestingly, 3 tumours which had methylated DNA also had normal expression of MT3. This might reflect a heterozygous pattern of methylation, with only one allele methylated, or a mixed tumour cell population containing some cells which were methylated and some which were not. The techniques used in this study could not distinguish between these possibilities. Also, 3 patients with unmethylated DNA had low levels of MT3 expression, perhaps reflecting mutation or other change in the tumour. We found no relationship between methylation status and survival in the subset of patients for whom data were obtainable, suggesting that in these patients there would also be no relationship between gene expression and survival. Methylation and reduced expression of MT3 was also not associated with tumour stage or tumour size. Thus a change in MT3 expression by itself does not appear to favour increased tumour growth, and methylations status is not associated with survival, in patients with oesophageal SCC.
Conclusion
We have shown that the MT3 promoter has a heterogeneous pattern of methylation in oeosphageal cancer cell lines, and is associated with a reduction in the gene expression. In resected oesophageal SCC tissue MT3 expression was frequently down-regulated, most commonly in those tumours in which the promoter region was methylated. Methylation or down-regulation of MT3 did not correlate with and patient age, gender, smoking or drinking history, tumour stage, volume, lymph node involvement, or patient survival. MT3 methylation does not show promise as a prognostic marker in patients with oesophageal SCC.
Methods
Cell lines and tissue samples
The oesophageal cancer cell lines OE19, OE21, OE33 and TE7 [23] were each cultured in RPMI 1640 supplemented with 10% foetal bovine serum at 37°C with 5% CO2. Whole blood was obtained from 19 normal donors, none of whom had known cancers or genetic abnormalities. Primary tumour and, when available, non-cancerous proximal resection margin from 64 consecutive patients undergoing oesophagectomy for SCC at the Department of Thoracic Surgery, Fourth Hospital, Hebei Medical University, were collected into RNAlater (Ambion, Austin, TX). Patient gender, age at the time of operation, smoking and drinking history and tumour type, stage, volume, and conventional histopathology were recorded. Survival data were available for 41 patients. The study complied with the appropriate institutional guidelines.
Demethylation of oesophageal cell lines by 5-aza-2'-deoxycytine treatment
Re-expression studies for MT3 were performed on each of the oesophageal cancer cell lines. To determine the culture conditions required to achieve at least 2 cell divisions in the presence or absence of 5-aza-2'-deoxycytine (aza-dC), cells, labelled with PKH-26 (Sigma-Aldrich, Sydney, NSW, Australia), were analysed by flow cytometry as described previously [15]. The cells were treated for 72 (OE21, OE33 and TE7) or 96 (OE19) hr with culture medium containing either vehicle or 1 μM aza-dC (Sigma-Aldrich). The medium was then replaced with fresh medium not containing aza-dC, and the cells were incubated for a further 24 hr before harvesting.
Preparation of bisulphite modified DNA
Genomic DNA was isolated from normal donor lymphocytes, cultured cells, and RNAlater stabilized tissues. Lymphocytes were isolated from whole blood using Ficoll-Paque (Pharmacia Biotech, Uppsala, Sweden). The adherent cell lines were harvested by detaching the cells from culture flasks using trypsin/EDTA. RNAlater stabilized tissues were homogenised using disposable pestles (Edwards Instruments, Narellan, NSW, Australia). Next, the samples of lymphocytes, cultured cells, or RNAlater stabilised tissues were digested for 3 days with proteinase K and sodium dodecyl sulfate in TES, pH 8. The protein was removed by NaCl precipitation. The DNA was precipitated with ethanol, and resuspended in TE, pH 8. The DNA was bisulphite modified using a modification of the method previously described [24]. Briefly, 2 μg of genomic DNA was denatured by treatment with NaOH at 37°C for 15 min. The DNA was modified using a mixture of 5 M sodium bisulfite (Sigma-Aldrich) and 0.72 μM hydroquinone (Sigma-Aldrich) for 4 hr at 56°C. The samples were purified using Rapid PCR Purification System (Marligen Biosciences, Ijamsville, MD), desulphonated with NaOH for 15 min at 37°C, precipitated with ethanol and sodium acetate, and resuspended in 50 μl of TE, pH 8.
Methylation analysis of the MT3 promoter by COBRA
Bisulphite modified DNA was amplified using primers that amplified 3 overlapping regions, designated R1, R2 and R3 (Figure 1). The primers did not discriminate between methylated and unmethylated alleles. The primers and PCR conditions were specific for bisulphite modified DNA, and did not amplify unmodified DNA. All COBRA PCRs were performed in a volume of 50 μl containing 2 μl of bisulphite modified DNA, 2 mM MgCl2, 0.2 mM dNTPs, 0.2 μM of forward and reverse primer, and 0.5 U of HotStar Taq in 1 × PCR Buffer (Qiagen, Hilden, Germany). The reactions were incubated in an Eppendorf Mastercycler at 95°C for 15 min; then 45 cycles of 94°C for 1 min, 52, 54 or 60°C for 1 min, and 72°C for 1 min; and a 4 min final extension at 72°C (Table 4). The PCR products (5 μl) were digested with 5 units of BstUI restriction enzyme (New England Biolabs, Beverly, MA) in a final volume of 10 μl, for 4 hr at 60°C. BstUI specifically digests at CGCG sites that are retained after bisulphite modification when CpGs are methylated. The unmethylated cytosines are deaminated to uracil by the bisulphite reaction, then amplified as thymine in the PCR reaction, and so are not digested by BstUI. The digested PCR products were resolved on 2% agarose gels and stained with ethidium bromide. The intensity of each band was quantified and converted to nanograms of DNA using Kodak ID Image Analysis Software (Kodak, Rochester, NY). For each gel, 250 ng of the molecular weight marker pUC19/Hpa II (GeneWorks, Adelaide, SA, Australia) was used as a standard to determine DNA fragment size and mass. Restriction maps for each digest were used to determine the length of all possible fragments, and the number of molecules in each band were determined by multiplying the mass of DNA in each band by its' fragment length. The frequency of methylation at each BstUI site was then estimated by dividing the number of molecules for each fragment by the total number of molecules in the digest.
Analysis of MT3 expression by quantitative real-time RT-PCR
Cell line RNA was isolated using the RNeasy kit (Qiagen) with an on-column DNase I digestion. Tissue RNA was isolated using Trizol (Invitrogen, Mount Waverly, VIC, Australia) and treated with DNase I (Ambion). The cDNA was synthesised from 2 μg of RNA using an M-MLV kit (Invitrogen). Quantitative real-time RT-PCR was performed on a RotorGene 2000 PCR thermocycler (Corbett Research, Sydney, NSW, Australia). Triplicate reactions were done using the appropriate PCR primers (Table 4) and the Quantitect Sybr-Green PCR mix (Qiagen). The PCR products were electrophoresed on 1.5 % agarose gels and stained with ethidium bromide. The expression of MT3 was normalized to that of either ACTB or PBGD [25].
Data analysis
Statistical analysis of expression of MT3 before and after aza-dC treatment for each cell line was performed using unpaired T-tests. Comparisons of the levels of MT3 between cell lines with or without aza-dC treatment were performed using ANOVA with Tukey-Kramer multiple comparisons test. The comparison of the volume of methylated and unmethylated tumours was performed using a Kruskal-Wallis Test. The comparison of patients with or without positive lymph nodes and methylation status was performed using a Chi-squared test. The Mann-Whitney test was used to compare MT3 expression in methylated and unmethylated tumour. Comparisons of MT3 expression in tumour and margin were performed using Wilcoxon matched-pairs sign-rank tests. Survival data were analysed using Fisher's Exact Test. All statistics were performed using InStat version 3.0a (GraphPad Software, San Diego, CA).
Abbreviations
- aza-dC:
-
5-aza-2'-deoxycytidine
- COBRA:
-
combined bisulphite restriction analysis
- MT3:
-
metallothionein 3
- RT-PCR:
-
reverse transcriptase-polymerase chain reaction
- SCC:
-
squamous cell carcinoma.
References
Hamer DH: Metallothionein. Annu Rev Biochem. 1986, 55: 913-951.
Kagi JH, Hunziker P: Mammalian metallothionein. Biol Trace Elem Res. 1989, 21: 111-118.
You HJ, Oh DH, Choi CY, Lee DG, Hahm KS, Moon AR, Jeong HG: Protective effect of metallothionein-III on DNA damage in response to reactive oxygen species. Biochim Biophys Acta. 2002, 1573 (1): 33-38.
Uchida Y, Takio K, Titani K, Ihara Y, Tomonaga M: The growth inhibitory factor that is deficient in the Alzheimer's disease brain is a 68 amino acid metallothionein-like protein. Neuron. 1991, 7 (2): 337-347. 10.1016/0896-6273(91)90272-2
Amoureux MC, Wurch T, Pauwels PJ: Modulation of metallothionein-III mRNA content and growth rate of rat C6-glial cells by transfection with human 5-HT1D receptor genes. Biochem Biophys Res Commun. 1995, 214 (2): 639-645. 10.1006/bbrc.1995.2334
Palmiter RD: Constitutive expression of metallothionein-III (MT-III), but not MT-I, inhibits growth when cells become zinc deficient. Toxicol Appl Pharmacol. 1995, 135 (1): 139-146. 10.1006/taap.1995.1216
Dutta R, Sens DA, Somji S, Sens MA, Garrett SH: Metallothionein isoform 3 expression inhibits cell growth and increases drug resistance of PC-3 prostate cancer cells. Prostate. 2002, 52 (2): 89-97. 10.1002/pros.10097
Gurel V, Sens DA, Somji S, Garrett SH, Nath J, Sens MA: Stable transfection and overexpression of metallothionein isoform 3 inhibits the growth of MCF-7 and Hs578T cells but not that of T-47D or MDA-MB-231 cells. Breast Cancer Res Treat. 2003, 80 (2): 181-191. 10.1023/A:1024520801262
Ramaswamy S, Ross KN, Lander ES, Golub TR: A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003, 33 (1): 49-54. 10.1038/ng1060
Jones PA, Laird PW: Cancer epigenetics comes of age. Nat Genet. 1999, 21 (2): 163-167. 10.1038/5947
Deng D, El-Rifai W, Ji J, Zhu B, Trampont P, Li J, Smith MF, Powel SM: Hypermethylation of metallothionein-3 CpG island in gastric carcinoma. Carcinogenesis. 2003, 24 (1): 25-29. 10.1093/carcin/24.1.25
Xiong Z, Laird PW: COBRA: a sensitive and quantitative DNA methylation assay. Nucleic Acids Res. 1997, 25 (12): 2532-2534. 10.1093/nar/25.12.2532
Wong DJ, Foster SA, Galloway DA, Reid BJ: Progressive region-specific de novo methylation of the p16 CpG island in primary human mammary epithelial cell strains during escape from M(0) growth arrest. Mol Cell Biol. 1999, 19 (8): 5642-5651.
Qian XC, Brent TP: Methylation hot spots in the 5' flanking region denote silencing of the O6-methylguanine-DNA methyltransferase gene. Cancer Res. 1997, 57 (17): 3672-3677.
Kinugasa S, Smith E, Drew PA, Watson DI, Jamieson GG: Aspirin and indomethacin for the prevention of experimental port-site metastases. Surg Endosc. 2004, 18 (5): 834-838.
Boyes J, Bird A: Repression of genes by DNA methylation depends on CpG density and promoter strength: evidence for involvement of a methyl-CpG binding protein. Embo J. 1992, 11 (1): 327-333.
Kass SU, Goddard JP, Adams RL: Inactive chromatin spreads from a focus of methylation. Mol Cell Biol. 1993, 13 (12): 7372-7379.
Gonzalgo ML, Hayashida T, Bender CM, Pao MM, Tsai YC, Gonzales FA, Nguyen HD, Nguyen TT, Jones PA: The role of DNA methylation in expression of the p19/p16 locus in human bladder cancer cell lines. Cancer Res. 1998, 58 (6): 1245-1252.
Hsieh CL: Dependence of transcriptional repression on CpG methylation density. Mol Cell Biol. 1994, 14 (8): 5487-5494.
Sens MA, Somji S, Lamm DL, Garrett SH, Slovinsky F, Todd JH, Sens DA: Metallothionein isoform 3 as a potential biomarker for human bladder cancer. Environ Health Perspect. 2000, 108 (5): 413-418.
Sens MA, Somji S, Garrett SH, Beall CL, Sens DA: Metallothionein isoform 3 overexpression is associated with breast cancers having a poor prognosis. Am J Pathol. 2001, 159 (1): 21-26.
El-Rifai W, Frierson HFJ, Harper JC, Powell SM, Knuutila S: Expression profiling of gastric adenocarcinoma using cDNA array. Int J Cancer. 2001, 92 (6): 832-838. 10.1002/ijc.1264
Nishihira T, Hashimoto Y, Katayama M, Mori S, Kuroki T: Molecular and cellular features of esophageal cancer cells. J Cancer Res Clin Oncol. 1993, 119 (8): 441-449. 10.1007/BF01215923
Smith E, Bianco-Miotto T, Drew P, Watson D: Method for optimizing methylation-specific PCR. Biotechniques. 2003, 35 (1): 32-33.
Hussey DJ, Moore S, Nicola M, Dobrovic A: Fusion of the NUP98 gene with the LEDGF/p52 gene defines a recurrent acute myeloid leukemia translocation. BMC Genet. 2001, 2 (1): 20- 10.1186/1471-2156-2-20
Acknowledgements
Supported by grants from the National Health and Medical Research Council of Australia. Dr. Z. Q. Tian was supported by a scholarship from The International Society for Diseases of the Esophagus. The authors thank Dr. D. Hussey and Dr. T. Bianco-Miotto for their contributions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
ES isolated the DNA and RNA, performed the bisulphite modifications, COBRAs and RT-PCRs, designed the study, performed the statistical analysis and coordinated and drafted the manuscript. GCM aided with the RT-PCRs. ZQT and JFL collected the oesophagectomy material and clinical data. NJD performed the cell culture and edited the manuscript. AR confirmed the pathology. PD, DIW and GGJ supervised the work and edited the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Smith, E., Drew, P.A., Tian, ZQ. et al. Metallothionien 3 expression is frequently down-regulated in oesophageal squamous cell carcinoma by DNA methylation. Mol Cancer 4, 42 (2005). https://doi.org/10.1186/1476-4598-4-42
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-4598-4-42