
Abstract
Pancreatic cancer ranks fifth as a cause of cancer-related death in the world with an overall 5-year survival rate of less than 1% and a median survival of less than a year after tumour detection. Most of these patients have already metastases at the time of diagnosis. The oncologic strategies such as chemotherapy, radiotherapy, antihormonal modalities or the systemic use of specific monoclonal antibodies have not achieved a significant improvement in the survival of pancreatic cancer patients. Recent studies suggest that alterations in molecular pathways, particularly in growth factor mediated mechanisms, that regulate cell proliferation and differentiation play a pivotal role in the pathogenesis of this cancer. The molecular knowledge regarding changes in the expression of growth factors in pancreatic cancer has the potential to improve diagnostic and therapeutic treatment strategies in the near future.
Similar content being viewed by others


Introduction
Pancreatic cancer most frequently affects men between 60 and 80 years of age. This disease ranks fifth as a cause of cancer-related death in the world with an overall 5-year survival rate of less than 1% and a median survival of approximately 5–6 months after tumour detection [1]. The currently available diagnostic tools usually do not detect early stages of cancer and as a consequence, most of these patients have already metastases at the time of their first visit. Recent studies suggest that alterations in the molecular pathways that regulate cell proliferation and differentiation play a pivotal role in the pathogenesis of this cancer. These changes result in abnormalities in growth factor mediated signaling cascades and cell cycle control. Since the oncologic strategies such as chemotherapy, radiotherapy, antihormonal modalities or the systemic use of monoclonal antibodies have not achieved a significant improvement in the survival of pancreatic cancer patients, great efforts have been undertaken in investigating new treatment approaches that, based on the specific molecular changes of growth factor expression, could lead to novel treatment concepts in the management of pancreatic cancer.
Pathogenesis of pancreatic cancer
The pathogenesis of pancreatic cancer is nowadays described as a model of step-by-step accumulation of genetic and molecular changes leading to defects in cell growth, cell adhesion and integration of epithelial cells. The alterations in pancreatic cancer include changes in the expression of oncogenes, inactivation of tumour suppressor genes and aberrant expression of cyclins (proteins regulating cell cycle).
The K-ras oncogene seems to play the most important role in the pathogenesis of pancreatic cancer, since it is mutated in 90% of pancreatic cancer cases [2–4]. K-ras is a guanyl-nucleotide binding protein with GTPase activity that mediates intracellular signaltransduction of growth factors that bind to tyrosine-kinase receptors. The most common mutations are localized at codons 12, 13 or 61 and render the protein unable to hydrolyze GTP and thereby continually transduce unregulated proliferative signals. Further studies on experimental models of pancreatic cancer and chronic pancreatitis revealed that this mutation appears in the early phase of malignant transformation of ductal epithelial cells [5]. However, the value of the K-ras oncogene as an indicator of malignant transformation of the exocrine pancreas and the possible application of K-ras as a molecular-diagnostic test are biased by the fact that K-ras mutations can be detected in hyperplastic and metaplastic lesions in tissues from normal pancreas or chronic pancreatitis as well [5, 6].
The p53 tumour suppressor gene is the most commonly mutated gene in the carcinogenesis of epithelial cancers: mutations of this gene are present in 40% of pancreatic cancer [7–9]. Point mutations of p53 lead to the accumulation of p53 proteins in the nucleus. However, p53 can be inactivated through MDM2, the expression of this gene is also increased in pancreatic cancer [10]. The most important functions of p53 are the regulation of the cell cycle, e.g. the G1/S transition, and the induction of apoptosis. The activation and progression of the cell cycle is regulated by the activity of cyclins and cyclin-dependent kinases (CDK). WAF1, that is activated by wild type p53, but not by mutated p53, inhibits the activation of cyclin-CDK complex and the phosphorylation and inactivation of Retinoblastoma proteins thereby stopping the progression of cell cycle in phase G1. Cyclin D1 is also overexpressed in pancreatic cancer and this overexpression is associated with a worse prognosis. In a study comparing pancreatic cancer to chronic pancreatitis, mutations of the p53 gene were detected in 47% of pancreatic cancers, but not in any of the chronic pancreatitis cases [9].
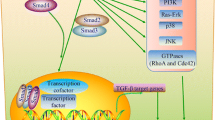
Inactivation of the APC gene is found in approx. 30% of gastric and colorectal cancer. In pancreatic cancer, a Japanese group detected APC mutations in up to 40% of cases but this result could not be confirmed by others [6]. On the contrary, loss of heterozygosity (LOH) of the DCC gene was reported in approx. 60% of pancreatic cancers [11]. Moreover, Hahn et al found LOH at chromosome 18q as well. Further studies of this group led to the identification of the DPC4 gene that is inactivated in approx. 50% of pancreatic cancer [12]. DPC4 belongs to the group of Smad-genes that mediate the signaltransduction of the TGF-β family. Inactivation of Smad-proteins can result in loss of the growth-inhibitory effects of TGF-β1 which plays a central role in the pathogenesis of pancreatic cancer. Thus, pancreatic cancer cell growth, despite TGF-β1 overexpression, may be supported by defects in DPC4 or the TGF-β-recptor II. Furthermore, the loss of Smad4/DPC4-expression along with the inhibition of Smad2/3-expression through the ras-protein can lead to the resistance of epithelial cells against the growth-inhibitory and antiproliferative function of TGF-β1 [13]. Interestingly, Smad4/DPC4 also leads to the induction of p21/WAF1 so that Smad4 can contribute to the inhibition of cell cycle via the activation of WAF1 [14].
The inactivation of the p16 gene on chromosome 9p21 is another essential genetic alteration in pancreatic cancer [15]. This gene encodes the p16-inhibitor of the Cyclin D/CDK-4 complex thereby regulating the coordinated progression of cell cycle [16]. Inactivation of p16 or other genes involved in this signalling pathway can be detected in approx. 90% of pancreatic cancer cases [17]. Mutations of the p16 gene are reported in approx. 40% of cases, deletions in approx. 40% of cases, and loss of transcription due to hypermethylation of the promoter region is responsible for the rest of the cases [14].
Impaired function of DNA repair genes leading to the development of microsatellite instability have been described in colorectal and gastric cancers. Replication error positive cases are found in approx. 30–40% of gastric and colorectal cancer whereas this mechanism seems to play only a minor role in pancreatic cancer [18]. However, Han et al found the same type of replication error in 6 out of 9 ductal pancreatic cancer cases [19].
Growth factors and pancreatic cancer
Growth factors are produced by many different cell types and exert their effects via autocrine and paracrine mechanisms. They function as stimulators or inhibitors of the division, differentiation and migration of cells and are involved in carcinogenesis, in which they influence a variety of functions including cell proliferation, cell invasion, metastasis formation, angiogenesis, local immune system functions and extracellular matrix synthesis. The molecular knowledge acquired regarding changes in the expression of growth factors in pancreatic cancer has the potential to improve diagnostic and therapeutic treatment strategies in the near future [20].
Epidermal Growth Factor Family
In pancreatic cancer, coexpression of the epidermal growth factor (EGF) receptor and EGF and/or transforming growth factor (TGF)-α or amphiregulin results in a worse survival. Furthermore, the upregulation of EGF and the EGF receptor is even more frequent in pancreatic adenocarcinoma with lymph node and distant metastases [21, 22]. A marked increase in the expression of human EGF receptor has proven to be associated with advanced tumour stage and significantly shorter survival [23]. EGF, TGF-α, HB-EGF and amphiregulin significantly enhance the proliferation of human pancreatic cancer cell lines [22]. Dysplastic changes were found in 50% of the TGF-α transgenic mice, and pancreatic cancers were reported in approximately 20% of TGF-α transgenic mice older than 1 year [24].
Fibroblast Growth Factor Family
Fibroblast growth factor (FGF) receptors are expressed by human pancreatic cancer cell lines as well [25]. Glypican-1 seems to be the most important coreceptor for heparin-binding growth factors being overexpressed in a large proportion of pancreatic cancers. The glypican-1 expression occurs predominantly in the cancer cells and in the fibroblasts surrounding the tumour mass. Reduction in glypican-1 expression results in the attenuation of the responsiveness of pancreatic cancer cells to heparin-binding growth factors [27]. In addition, transfection of a truncated FGFR-1 into PANC-1 cells induces the blockade of FGF receptor-dependent pathways and reduces tumour growth in nude mice [28]. FGFs and their receptors are suggested to play a critical role in tumour angiogenesis in a transgenic mouse model of pancreatic β-cell carcinogenesis as well [29].
Transforming Growth Factor-β
Cancer cells are resistant to the growth-suppressive effects of transforming growth factor(TGF)-β [30]. The overexpression of TGF-β isoforms is associated with a poor prognosis in pancreatic cancer [31]. The inactivation of Smad4 which is part of the TGF-β signalling pathway is common (up to 42%) in this cancer [32]. According to in situ hybridization analyses, Smad6 and Smad7 (TGF-β signaling inhibitors) are also overexpressed in the cancer cells within the tumour mass. Following transfections with Smad6 and Smad7, a complete blockade of the growth-inhibitory effects of TGF-β occurred in the TGF-β-responsive pancreatic cancer cell line COLO-357 and in nude mice [33, 34]. Thus, pancreatic cancer cells might have multiple mechanisms to avoid the suppressive effect of TGF-β and thereby can retain the capability to express metastasis-promoting genes.
Vascular Endothelial Growth Factor
Vascular endothelial growth factor (VEGF) plays an important role in tumour angiogenesis. Its two shorter isoforms (VEGF121 and VEGF165) induce angiogenic effects by binding to the specific transmembrane tyrosine kinase receptors KDR/flk-1 and flt-1, respectively, which are expressed selectively on endothelial cells. A significant correlation was observed between VEGF positivity and microvascular density (MVD) [35]. Moreover, VEGF expression was found to be associated with K-ras mutation [36]. Several groups demonstrated that patients with pancreatic cancer showing moderate or high VEGF expression have significantly shorter survival than patients with low or without any VEGF expression [35–38]. Luo et al, reported that stable transfection of an anti-sense VEGF(189) into PANC-1 pancreatic cancer cells resulted in decreased VEGF expression and secretion, a decreased capacity of the resultant conditioned medium to enhance endothelial cell proliferation and a significant attenuation of tumour cell proliferation in vitro, and when injected into athymic nude mice, the antisense-VEGF(189)-expressing cells exhibited an 80% decrease in tumour growth compared to control cells [39].
Platelet-Derived Growth Factor Family
Platelet derived growth factors (PDGF) are proteins consisting of A and/or B chains [40]. Through disulphide bonds they can form the three isoforms PDGF-AA, -AB-, -BB [41]. Two PDGF receptors have been cloned and characterized, all of which possess tyrosine-kinase activity. PDGFs have several biological functions, including their mitogenic and chemoattractant role towards fibroblasts, monocytes and endothelial cells. Furthermore, they play an important role in wound healing and repair [42, 43]. With regard to the formation of tumors, they have been shown to stimulate the production of stromal tissue in malignant tumors, thereby potentially contributing to the development and progression of human cancers [44, 45]. The expression of PDGFs is induced by TGF-β1 in pancreatic cancer cells and both PDGF receptors are overexpressed in this malignancy.
Nerve Growth Factor
The tumourous infiltration of pancreatic nerves is a well-known phenomenon causing severe pain in the back which is often the first clinical manifestation of pancreatic cancer. The involvement of the extrapancreatic nerve plexus leads to retropancreatic tumour extension, inhibits curative resection, enhances local recurrence and reduces the prognosis of pancreatic cancer patients [46]. The expression of nerve growth factor (NGF) and TrkA (the high-affinity receptor for NGF) is significantly higher in pancreatic cancer than in normal pancreas, and this overexpression is associated with more frequent perineural invasion and a higher degree of pain [47, 48].
Future perspectives in the treatment of pancreatic cancer
Pancreatic cancer obtains a significant growth advantage through autocrine and paracrine mechanisms of several growth factors, therefore novel therapeutic modalities are designed focusing on abrogating signaling of these ligands. Hereby we summarize the most promising therapeutic approaches executed on human cancer cell lines or in animal models of pancreatic cancer.
Epidermal Growth Factor
Nude mice were treated with a novel orally given EGF-R tyrosine kinase inhibitor, PKI 166 (4-(R)-phenethylamino-6-(hydroxyl)phenyl-7H-pyrrolo [2.3-d]-pyrimidine), 7 days after orthotopic injection of L3.6pl human pancreatic cancer cells. The volume of pancreatic cancers was reduced by 59% in mice treated with gemcitabine, by 45% in mice treated with PKI 166, and by 85% in those treated with both drugs. The combination therapy also significantly inhibited lymph node and liver metastasis formation, which led to a significant improvement in overall survival. EGF-R activation was significantly blocked by therapy with PKI 166 and was associated with a significant decrease in tumour cell production of VEGF and IL-8, which correlated with a significant reduction in angiogenesis, and an increase in apoptotic tumour and endothelial cells [49–51].
By blocking the conversion of 3-Hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) to mevalonate, HMG-CoA reductase inhibitors (i.e. fluvastatin and lovstatin) inhibit the synthesis of other products derived from this metabolite. EGF induced a dose-dependent increase of PANC-1 cell invasion in vitro. Treatment of PANC-1 cells with fluvastatin markedly attenuated EGF-induced translocation of RhoA from the cytosol to the membrane fraction and actin stress fiber assembly, whereas it did not inhibit the tyrosine phosphorylation of EGF receptor and c-erbB-2 [52].
Vascular Endothelial Growth Factor
In pancreatic cancer, VEGF expression and the degree of MVD, which are closely correlated, are reliable markers of early tumour recurrence after resection. The soluble form of flt-1 VEGF receptor inhibits VEGF activity. PANC-1 and PK-8 were utilized as lower- and higher-VEGF-producing cell lines, respectively. The in vitro proliferation of cancer cells infected with adenovirus vectors encoding soluble flt-1 (Adsflt) and control vectors (AdLacZ) exhibited no difference. Cancer cells were inoculated in severe combined immunodeficient (SCID) mice in order to evaluate the in vivo tumour growth suppression. Adsflt, AdLacZ, or vehicle was injected directly into the tumours. The tumour growth and the MVD of the Adsflt-treated group was significantly inhibited both in PANC-1 cells and PK-8 cells. Apoptosis index increased and tumour angiogenesis decreased in the Adsflt group in contrast to groups of wild-type cells and AdLacZ-infected cells [53, 54].
Injection of 100 mg/kg of SU6668 (inhibitor of the receptor-tyrosine kinase activity of VEGF, FGF, and PDGF) markedly suppressed tumour growth and decreased tumour blood flow in CFPAC human pancreatic carcinoma cells and additionally extended survival in tumour-bearing mice compared to control. Daily SU6668 administration and a single dose of 15 Gy of X-irradiation was significantly more effective than either treatment alone in suppressing tumour growth [55].
Platelet-Derived Growth Factor
The recent introducton of the signal transduction inhibitor imatinib mesylate (formerly ST1571) has had an important influence on the practice of oncology as well as on the process of drug development [56]. Imatinib mesylate was originally designed to specifically target chronic myeloid leukemia (CML) by blocking bcr-abl oncoprotein [57]. Having obtained larger amount of experience, imatinib mesylate proved to have a striking activity against gastrointestinal stromal tumours (GISTs) as well by blocking c-kit [58]. PDGF was one of the first polypeptide growth factors identified that signals through a cell surface tyrosine kinase receptor to stimulate several cellular functions including growth, proliferation, and differentiation in various cancers such as prostate and pancreatic cancer [59]. Since imatinib mesylate inhibits the PDGF-R kinase activity, it could possibly be a potent therapeutical approach for pancreatic cancer as well.
Hepatocyte Growth Factor
Hepatocyte growth factor (HGF) enhances tumour invasion and metastasis through tumour-stromal interactions. NK4 is a four-kringle fragment of HGF and acts as an HGF-antagonist and an angiogenesis inhibitor. NK4 blocked the conversion of orthotopic pancreatic tumours from in situ carcinoma to invading cancers during days 3–14, when administration of NK4 was started from the third day after orthotopic injection of SUIT-2 human pancreatic cancer cells into nude mice. NK4 therapy, started on day 10 when cancer cells were already invading surrounding tissues, suppressed tumour growth, peritoneal dissemination, and ascites accumulation resulting in the improvement of survival rates. The antitumour effects of NK4 correlated with the reduction of MVD [60].
Conclusion
Molecular alterations in cellular pathways that regulate normal cell growth and differentiation are associated with the development of pancreatic cancer. Recently developed assays for the detection of such genetic markers might become useful diagnostic tools. The pharmacological manipulation of growth-factor mediated signaling cascades and the molecular inactivation of growth regulatory pathways using specifically designed tyrosine kinase inhibitors or other gene targeting techniques are beginning to be applied in the treatment of this cancer. This should lead to improvements in diagnostic and therapeutic strategies and subsequently result in a better prognosis of patients with pancreatic cancer.
Authors contributions
Authors MJ, BN and ME wrote the manuscript, authors PM and ME discussed the manuscript with the other authors and finalized the version submitted for publication. All authors read and approved the final manuscript.
Abbreviations
- CDK:
-
cyclin dependent kinase
- LOH:
-
loss of heterozygosity
- TGF:
-
transforming growth factor. EGF-R, epithelial growth factor receptor
- HMG-CoA:
-
3-Hydroxy-3-methylglutaryl-coenzyme A
- VEGF-R:
-
vascular endothelial growth factor receptor
- MVD:
-
microvascular density
- PDGF-R:
-
platelet derived growth factor receptor
- HGF:
-
hepatocyte growth factor receptor
References
Urrutia R, DiMagno EP: Pancreatic cancer: cellular and molecular mechanisms. In: Encyclopedia of Cancer. Edited by: Bertino JR. 1996, Academic Press Editor-in-Chief, 1201-1211.
Almoguera C, Shibata D, Forrester K, Martin J, Arnheim N, Perucho M: Most human carcinomas of the exocrine pancreas contain mutant c-K-ras genes. Cell. 1988, 53: 549-554.
Grünewald K, Lyons J, Fröhlich A, Feichtinger H, Weger RA, Schwab G, Janssen JW, Bartram CR: High frequency of Ki-ras codon 12 mutations in pancreatic adenocarcinomas. Int J Cancer. 1989, 43: 1037-1041.
Ebert MP, Hoffmann J, Schneider-Stock R, Kasper HU, Schulz HU, Lippert H, Roessner A, Malfertheiner P: Analysis of K-ras gene mutations in rare pancreatic and ampullary tumours. Eur J Gastroenterol Hepatol. 1998, 10: 1025-1029.
Lüttges J, Schlehe B, Menke MA, Vogel I, Henne-Bruns D, Kloppel G: The K-ras mutation pattern in pancreatic ductal adenocarcinoma usually is identical to that in associated normal, hyperplastic, and metaplastic ductal epithelium. Cancer. 1999, 85: 1703-17. 10.1002/(SICI)1097-0142(19990415)85:8<1703::AID-CNCR9>3.3.CO;2-I.
Bramhall SR: The use of molecular technology in the differentiation of pancreatic cancer and chronic pancreatitis. Int J Pancreatol. 1998, 23: 83-100. 10.1385/IJGC:23:2:83.
Barton CM, Staddon SL, Hughes CM, Hall PA, O'Sullivan C, Kloppel G, Theis B, Russell RC, Neoptolemos J, Williamson RC: Abnormalities of the p53 tumour suppressor gene in human pancreatic cancer. Br J Cancer. 1991, 64: 1076-1082.
Kalthoff H, Schmiegel W, Roeder C, Kasche D, Schmidt A, Lauer G, Thiele HG, Honold G, Pantel K, Riethmuller G: p53 and K-RAS alterations in pancreatic epithelial cell lesions. Oncogene. 1993, 8: 289-298.
Casey G, Yamanaka Y, Friess H, Kobrin MS, Lopez ME, Büchler M, Beger HG, Korc M: p53 mutations are common in pancreatic cancer and are absent in chronic pancreatitis. Cancer Lett. 1993, 69: 151-160.
Ebert M, Yokoyama M, Kobrin MS, Friess H, Büchler MW, Korc M: Increased MDM2 expression and immunoreactivity in human pancreatic ductal adenocarcinoma. Int J Oncol. 1994, 5: 1279-1284.
Simon B, Weinel R, Hohne M, Watz J, Schmidt J, Kortner G, Arnold R: Frequent alterations of the tumour suppressor genes p53 and DCC in human pancreatic carcinoma. Gastroenterology. 1994, 106: 1645-1651.
Hahn SA, Schutte M, Hoque AT, Moskaluk CA, da Costa LT, Rozenblum E, Weinstein CL, Fischer A, Yeo CL, Hruban RH, Kern SE: DPC4, a candidate tumour suppressor gene at human chromosome 18q21.1. Science. 1996, 271: 350-353.
Calonge MJ, Massague J: Smad4/DPC4 silencing and hyperactive Ras jointly disrupt transforming growth factor-beta antiproliferative responses in colon cancer cells. Biol Chem. 1999, 274: J33637-33643. 10.1074/jbc.274.47.33637.
Hahn SA, Bartsch D, Schroers A, Galehdari H, Becker M, Ramaswamy A, Schwarte-Waldhoff I, Maschek H, Schmiegel W: Mutations of the DPC4/Smad4 gene in biliary tract carcinoma. Cancer Res. 1998, 58: 1124-1126.
Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, Weinstein CL, Hruban RH, Yeo CJ, Kern SE: Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994, 8: 27-32.
Perugini RA, McDade TP, Vittimberga FJ, Callery MP: The molecular and cellular biology of pancreatic cancer. Crit Rev Eukaryot Gene Expr. 1998, 8: 377-393.
Mangray S, King TC: Molecular pathobiology of pancreatic adenocarcinoma. Front Biosci. 1998, 3: D1148-1160.
Myeroff LL, Parsons R, Kim SJ, Hedrick L, Cho KR, Orth K, Mathis M, Kinzler KW, Lutterbaugh J, Park K: A transforming growth factor beta receptor type II gene mutation common in colon and gastric but rare in endometrial cancers with microsatellite instability. Cancer Res. 1995, 55: 5545-5547.
Han HJ, Yanagisawa A, Kato Y, Park JG, Nakamura Y: Genetic instability in pancreatic cancer and poorly differentiated type of gastric cancer. Cancer Res. 1993, 53: 5087-5089.
Shi X, Friess H, Kleeff J, Ozawa F, Büchler MW: Pancreatic cancer: Factors regulating tumour development, maintenance and metastasis. Pancreatology. 2001, 1: 517-524. 10.1159/000055854.
Yamanaka Y, Friess H, Kobrin MS, Büchler M, Beger HG, Korc M: Coexpression of epidermal growth factor receptor and ligands in human pancreatic cancer is associated with enhanced tumour aggressiveness. Anticancer Res. 1993, 13: 565-569.
Ebert M, Yokoyama M, Kobrin MS, Friess H, Lopez ME, Büchler MW, Johnson GR, Korc M: Induction and expression of amphiregulin in human pancreatic cancer. Cancer Res. 1994, 54: 3959-3962.
Friess H, Yamanaka Y, Kobrin MS, Do DA, Büchler MW, Korc M: Enhanced erbB-3 expression in human pancreatic cancer correlates with tumour progression. Clin Cancer Res. 1995, 1: 1413-420.
Wagner M, Luhrs H, Kloppel G, Adler G, Schmid RM: Malignant transformation of duct-like cells originating from acini in transforming growth factor transgenic mice. Gastroenterology. 1998, 115: 1254-1262.
Balaz P, Friess H, Büchler MW: Growth Factors in pancreatic health and disease. Pancreatology. 2001, 1: 343-355. 10.1159/000055833.
Friess H, Kobrin MS, Korc M: Acidic and basic fibroblast growth factors and their receptors are expressed in human pancreas. Pancreas. 1992, 7: 737-741.
Kleeff J, Ishiwata T, Kumbasar A, Friess H, Büchler MW, Lopez ME, Korc M: The cell surface heparan sulfate proteoglycan glypican-1 regulates growth factor action in pancreatic carcinoma cells and is overexpressed in human pancreatic cancer. J Clin Invest. 1998, 102: 1662-1673.
Wagner M, Lopez ME, Cahn M, Korc M: Suppression of fibroblast growth factor receptor signaling inhibits pancreatic cancer growth in vitro and in vivo. Gastroenterology. 1998, 114: 798-807.
Compagni A, Wilgenbus P, Impagnatiello MA, Cotten M, Christofori G: Fibroblast growth factors are required for efficient tumour angiogenesis. Cancer Res. 2000, 60: 7163-7169.
Kleeff J, Korc M: Up-regulation of transforming growth factor (TGF)-beta receptors by TGF-beta1 in COLO-357 cells. J Biol Ch. 1998, 273: 7495-7500. 10.1074/jbc.273.13.7495.
Friess H, Yamanaka Y, Büchler MW, Ebert M, Beger HG, Gold LI, Korc M: Enhanced expression of transforming growth factor beta isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology. 1993, 105: 1846-1856.
Jonson T, Albrechtsson E, Axelson J, Heidenblad M, Gorunova L, Johansson B, Höglund M: Altered expression of TGFB receptors and mitogenic effects of TGFB in pancreatic carcinomas. Int J Oncol. 2001, 19: 71-81.
Kleeff J, Maruyama H, Friess H, Büchler MW, Falb D, Korc M: Smad6 suppresses TGF-beta-induced growth inhibition in COLO-357 pancreatic cancer cells and is overexpressed in pancreatic cancer. Biochem Biophys Res Commun. 1999, 255: 268-273. 10.1006/bbrc.1999.0171.
Kleeff J, Ishiwata T, Maruyama H, Friess H, Truong P, Büchler MW, Falb D, Korc M: The TGF-beta-signaling inhibitor Smad7 enhances tumourigenicity in pancreatic cancer. Onocogene. 1999, 18: 5363-5372. 10.1038/sj.onc.1202909.
Seo Y, Baba H, Fukuda T, Takashima M, Sugimachi K: High expression of vascular endothelial growth factor is associated with liver metastasis and a poor prognosis for patients with ductal pancreatic adenocarcinoma. Cancer. 2000, 88: 2239-2245. 10.1002/(SICI)1097-0142(20000515)88:10<2239::AID-CNCR6>3.0.CO;2-V.
Ikeda N, Nakajima Y, Sho M, Adachi M, Huang CL, Iki K, Kanehiro H, Hisanaga M, Nakano H, Miyake M: The association of K-ras gene mutation and vascular endothelial growth factor gene expression in pancreatic carcinoma. Cancer. 2001, 92: 488-499. 10.1002/1097-0142(20010801)92:3<488::AID-CNCR1347>3.0.CO;2-F.
Ikeda N, Adachi M, Taki T, Huang C, Hashida H, Takabayashi A, Sho M, Nakajima Y, Kanehiro H, Hisanaga M, Nakano H, Miyake M: Prognostic significance of angiogenesis in human pancreatic cancer. Br J Cancer. 1999, 79: 1553-1563. 10.1038/sj.bjc.6690248.
Knoll MR, Rudnitzki D, Sturm J, Manegold BC, Post S, Jaeger TM: Correlation of postoperative survival and angiogenic growth factors in pancreatic carcinoma. Hepatogastroenterology. 2001, 48: 1162-1165.
Luo J, Guo P, Matsuda K, Truong N, Lee A, Chun C, Cheng SY, Korc M: Pancreatic cancer cell-derived vascular endothelial growth factor is biologically active in vitro and enhances tumourigenicity in vivo. Int J Cancer. 2001, 92: 361-369. 10.1002/ijc.1202.
Ebert M, Yokoyama M, Friess H, Kobrin MS, Büchler MW, Korc M: Induction of platelet derived growth factor A and B chains and overexpression of their receptors in human pancreatic cancer. Int J Cancer. 1995, 62: 529-535.
Westermark B, Heldin CH: Platelet derived growth factor in autocrine transformation. Cancer Res. 1991, 51: 5087-5092.
Siegbahn A, Hammacher A, Westermark B, Heldin CH: Differential effects of the various isoforms of platelet-derived growth factor on chemotaxis of fibroblasts, monocytes, and granulocytes. J Clin Invest. 1990, 85: 916-920.
Kundra V, Escobedo JA, Kazlauskas A, Kim HK, Rhee SG, Williams LT, Zetter BR: Regulation of chemotaxis by the platelet-derived growth factor receptor-A. Nature. 1994, 367: 474-476. 10.1038/367474a0.
Chaudry A, Papanicolau V, Timber K, Heldin CH, Funa K: Expression of platelet-derived growth factor and ist receptors in neuroendocrine tumors of the digestive system. Cancer Res. 1992, 52: 1006-1012.
Lindmark G, Sundberg C, Glimelius B, Pithman L, Rubin K, Gerdin B: Stromal expression of platelet-derived growth factor B-chain in colorectal cancer. Lab Invest. 1993, 69: 682-689.
Zhou Z, Friess H, Wang L, Bogardus T, Korc M, Kleeff J, Büchler MW: Nerve growth factor exerts differential effects on the growth of human pancreatic cancer cells. Clin Cancer Res. 2001, 7: 105-112.
Zhou Z, Friess H, diMola FF, Zimmermann A, Graber HU, Korc M, Büchler MW: Nerve growth factor expression correlates with perineural invasion and pain in human pancreatic cancer. J Clin Oncol. 1999, 17: 2419-2428.
Schneider MB, Standop J, Ulrich A, Wittel U, Friess H, Andren-Sandberg A, Pour PM: Expression of nerve growth factors in pancreatic neural tissue and pancreatic cancer. J Histochem Cytochem. 2001, 49: 1205-1210.
Bruns CJ, Solorzano CC, Harbison MT, Ozawa S, Tsan R, Fan D, Abbruzzese J, Traxler P, Buchdunger E, Radinsky R, Fidler IJ: Blockade of the Epidermal Growth Factor Receptor Signaling by a Novel Tyrosine Kinase Inhibitor Leads to Apoptosis of Endothelial Cells and Therapy of Human Pancreatic Carcinoma. Cancer Res. 2000, 60: 2926-2935.
Bruns CJ, Harbison MT, Davis DW, Portera CA, Tsan R, McConkey DJ, Evans DB, Abbruzzese JL, Hicklin DJ, Radinsky R: Epidermal growth factor receptor blockade with C225 plus gemcitabine results in regression of human pancreatic carcinoma growing orthotopically in nude mice by antiangiogenic mechanisms. Clin Cancer Res. 2000, 6: 1936-1948.
Solorzano CC, Baker CH, Tsan R, Traxler P, Cohen P, Buchdunger E, Killion JJ, Fidler IJ: Optimization for the Blockade of Epidermal Growth Factor Receptor Signaling for Therapy of Human Pancreatic Carcinoma. Clin Cancer Res. 2001, 7: 2563-2572.
Kusama T, Mukai M, Iwasaki T, Tatsuta M, Matsumoto Y, Akedo H, Nakamura H: Inhibition of Epidermal Growth Factor-induced RhoA Translocation and Invasion of Human Pancreatic Cancer Cells by 3-Hydroxy-3-methylglutaryl-coenzyme A Reductase Inhibitors. Cancer Res. 2001, 61: 4885-4891.
Hoshida T, Sunamura M, Duda DG, Egawa S, Miyazaki S, Shineha R, Hamada H, Ohtani H, Satomi S, Matsuno S: Gene Therapy for Pancreatic Cancer Using an Adenovirus Vector Encoding Soluble flt-1 Vascular Endothelial Growth Factor Receptor. Pancreas. 2002, 25: 111-121. 10.1097/00006676-200208000-00001.
Niedergethmann M, Hildenbrand R, Wostbrock B, Hartel M, Sturm JW, Richter A, Post S: High Expression of Vascular Endothelial Growth Factor Predicts Early Recurrence and Poor Prognosis after Curative Resection for Ductal Adenocarcinoma of the Pancreas. Pancreas. 2002, 25: 122-129. 10.1097/00006676-200208000-00002.
Griffin RJ, Williams BW, Wild R, Cherrington JM, Park H, Song CW: Simultaneous Inhibition of the Receptor Kinase Activity of Vascular Endothelial, Fibroblast, and Platelet-derived Growth Factors Suppresses Tumour Growth and Enhances Tumour Radiation Response. Cancer Res. 2002, 62: 1702-1706.
Griffin J: The biology of signal transduction inhibition: basic science to novel therapies. Semin Oncol. 2001, 28 (suppl 17): 3-8. 10.1053/sonc.2001.29182.
Druker BJ, Sawyers CL, Kantarjian H, Resta DJ, Reese SF, Ford JM, Capdeville R, Talpaz M: Activity of a specific inhibitor of the bcr-abl tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Phliadelphia chromosome. N Engl J Med. 2001, 344: 1038-1042. 10.1056/NEJM200104053441402.
Van Oosterom AT, Judson I, Verweij J, Stroobants S, diPaola ED, Dimitrijevic S, Martens M, Webb A, Sciot R, Van Glabbeke M, Silberman S, Nielsen OS: Safety and efficacy of imatinib (STI571) in metastatic gastrointestinal stromal tumours: a phase I study. Lancet. 2001, 358: 1421-1423. 10.1016/S0140-6736(01)06535-7.
George D: Platelet-derived growth factor receptors: a therapeutic target in solid tumours. Semin Oncol. 2001, 28 (suppl 17): 27-33. 10.1053/sonc.2001.29185.
Tomioka D, Maehara N, Kuba K, Mizumoto K, Tanaka M, Matsumoto K, Nakamura T: Inhibition of Growth, Invasion, and Metastasis of Human Pancreatic Carcinoma Cells by NK4 in an Orthotopic Mouse Model. Cancer Res. 2001, 61: 7518-7524.
Acknowledgements
This work was supported by a grant from the DFG awarded to M.E. (Eb 187/4-1) and a scholarship to M.J. by the EAGE. M. E. is also supported by the Heisenberg Programme of the DFG (Eb 187/5-1).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Juhász, M., Nitsche, B., Malfertheiner, P. et al. Implications of growth factor alterations in the treatment of pancreatic cancer. Mol Cancer 2, 5 (2003). https://doi.org/10.1186/1476-4598-2-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1476-4598-2-5