
Abstract
Background
The complete sequences of the mitochondrial genomes (mtDNA) of members of the northern and southern genotypes of Anopheles (Nyssorhynchus) darlingi were used for comparative studies to estimate the time to the most recent common ancestor for modern anophelines, to evaluate differentiation within this taxon, and to seek evidence of incipient speciation.
Methods
The mtDNAs were sequenced from mosquitoes from Belize and Brazil and comparative analyses of structure and base composition, among others, were performed. A maximum likelihood approach linked with phylogenetic information was employed to detect evidence of selection and a Bayesian approach was used to date the split between the subgenus Nyssorhynchus and other Anopheles subgenera.
Results
The comparison of mtDNA sequences within the Anopheles darlingi taxon does not provide sufficient resolution to establish different units of speciation within the species. In addition, no evidence of positive selection in any protein-coding gene of the mtDNA was detected, and purifying selection likely is the basis for this lack of diversity. Bayesian analysis supports the conclusion that the most recent ancestor of Nyssorhynchus and Anopheles+Cellia was extant ~94 million years ago.
Conclusion
Analyses of mtDNA genomes of Anopheles darlingi do not provide support for speciation in the taxon. The dates estimated for divergence among the anopheline groups tested is in agreement with the geological split of western Gondwana (95 mya), and provides additional support for explaining the absence of Cellia in the New World, and Nyssorhynchus in the Afro-Eurasian continents.
Similar content being viewed by others


Background
Anopheles darlingi is a major malaria vector in the Americas [1]. Its broad geographical distribution, ranging from southern Mexico to northern Argentina, is coupled with a high morphological, behavioral and genetic diversity [2–8]. Support for considerable genetic variation within this taxon comes from the analysis of polytene chromosomes [6], isozymes [7], as well as nuclear and mitochondrial DNA markers [8–14].
Analysis of the An. darlingi population structure based on the cytochrome-oxidase subunit 1 mitochondrial gene (COI) provided evidence in support of two significant subdivisions among An. darlingi populations from Central (including northwestern Colombia) and South America, with FST values similar to those found among different species [10]. This finding was substantiated by the analysis of microsatellite markers, which revealed limited gene flow between Central America and Brazil and Peru [12]. Also, patterns of variation within the single-copy nuclear white gene, with five fixed non-synonymous mutations detected between the northern and southern An. darlingi populations (hereafter termed northern and southern genotypes) [14], were consistent with the COI studies. Three localities were identified in the Amazon basin where the two genotypes co-occur (Iquitos, Peru; Guayaramerín, Bolivia; Fortuna near Puerto Ayacucho, Venezuela), but no hybrid specimens were detected there.
Recent studies revealed that the distribution of the COI gene variants within South American populations, especially those recovered from Brazil, reflects subdivisions consistent with geographical elements, such as the Amazon River Delta and mountains in southeastern Brazil, as barriers to gene flow [13]. Significant population structure of An. darlingi, identified using microsatellites markers, also was reported in a malaria-endemic region of eastern Amazonian Brazil [11, 12].
Overall, these findings support the hypothesis that An. darlingi is experiencing incipient speciation, with the northern and southern genotypes as the units of speciation [12], although other results appear to counter this hypothesis [9]. Therefore, it is critical to investigate the status of this taxon and elucidate the processes driving divergence between alternative genotypes.
The mtDNA markers are used often for phylogenetic and population genetics analyses because of ease of their amplification and high information content at different evolutionary levels, including variation within and between populations [15, 16]. To date, mtDNA genomes are available for only three anopheline species, belonging to the subgenera Anopheles (Anopheles quadrimaculatus species A) [17] and Cellia (Anopheles gambiae[18] and Anopheles funestus[19]). Because the evolutionary lineage encompassing the subgenus Nyssorhynchus presumably branched prior to the divergence of the subgenera Cellia and Anopheles lineages, mtDNA of Nyssorhynchus species may possess different characteristics, such as altered gene order or different selection pressures, which may affect the utility of mtDNA sequences as markers for evolutionary studies. The purifying selection mechanism under which the mtDNA is assumed to evolve has been questioned recently with some reports describing positive selection acting at the mitochondrial level [20, 21]. Therefore, testing the pattern of evolution of the mtDNA within the postulated incipient species, i.e. to examine whether the mtDNA is evolving under neutrality versus purifying or positive selection, can contribute to the understanding of An. darlingi differentiation.
Different approaches have been used to estimate divergence times within An. darlingi and to determine the most likely scenario for its diversification. Based on a single mitochondrial COI gene, the date of divergence was estimated to the Pleistocene, and the Pleistocene refugium was the main hypothesis to explain current distribution [10, 22]. Recently, a Bayesian approach was employed to estimate divergence within other species complexes, including Anopheles annulipes in Australia and Papua New Guinea [23], and the Neocellia Series in the Oriental region [24]. In addition, Krzywinski et al. [18] calculated divergence dates of mtDNAs using a molecular clock approach with the subgenera Anopheles and Cellia, as well as with Aedes and between An. gambiae and An. funestus. No information exists on the time of divergence of Nyssorhynchus, which is hypothesized to be one of the earliest branching lineages of Anopheles[25]. This study tests the differentiation of the An. darlingi genotypes using the whole mtDNA sequence, analyses patterns of selection within this taxon, and infers a date of Nyssorhynchus divergence from other Anopheles groups.
Methods
Sample origin and DNA extraction
Two resting female mosquitoes were collected near local homes from Manaus, Amazonas State, Brazil (3° 08'S/60° 01' W) in August 2006 and in Central Cayo District of Belize (17° 09'N/88° 36'W) in 2008. The Belize specimens were identified morphologically following the Wilkerson and Strickman [26] illustrated morphological key and the Brazilian mosquito was identified based on both Faran and Linthicum [27] and Forattini [28] entomological keys. Mosquitoes were stored on silica gel in a sealed container or preserved in isopropanol, both at room temperature. DNA was extracted from whole mosquitoes by two different kit extractions, a DNeasy Blood and Tissue kit (Qiagen, Hilden, Germany) and Wizard Genomic DNA Purification kit (Promega, Madison, WI), following the procedures of the manufacturers.
PCR amplification and sequencing of Anopheles darlingi mtDNA
The white gene genotype of each specimen was checked using the PCR-RFLP protocol described in Mirabello [14]. Gene amplification primers for the complete mtDNA were designed using Primer3 0.4.0 [29] based on published An. gambiae, An. quadrimaculatus and An. funestus mtDNA sequences (GenBank Accession NC_002084, NC_000875 and DQ146364). Each fragment overlapped upstream (5') by ~50 base pairs (bp) (see Additional file 1) and was amplified using a Ready-To-Go-PCR bead (Amersham Pharmacia/Biotech, NJ, USA) and run on a PTC-200 thermal cycler (BioRad, Inc.). Amplification conditions included an initial denaturation at 95° for 2 min, 35 cycles of denaturation at 95°C for 1 min, annealing at 58°C for 2 min and extension at 72°C for 1 min, with a final extension step at 72°C for 10 min. Amplification products were purified with the Exo-SAP-IT kit (Amersham Biosciences) and sequenced directly. Sequencing of both strands was performed at Applied Genomic Technologies Core (Wadsworth Center) on an ABI 3700 DNA automated sequencer. To ensure fidelity during amplification, each fragment was sequenced at least three times and a consensus sequence established. The regions with unclear electropherograms, from the 14,000-nucleotide position covering the whole AT-rich region, were cloned with the vector TOPO TA (Invitrogen) according to the manufacturer's recommendations.
Sequence assembly and annotation
Sequencher 3.0 (Gene Codes Corporation) was used for sequence assembly into contigs and for proofreading sequence files. Complete consensus sequences were aligned using the ClustalW application in Bioedit v 7.0 and alignments were annotated in SEQUIN [30]. Identification of transfer RNA genes was conducted using tRNAscan-SE [31]. Protein-coding and ribosomal RNA genes were by comparison with the An. gambiae mtDNA sequences (GenBank accession number NC_002084).
Genomic and evolutionary analyses
Nucleotide composition and the Relative Synonymous Codon Usage (RSU) values were calculated using the MEGA 4.1 program [32]. The GC- and AT-skews were used to determine the base compositional difference and strand asymmetry among the samples analysed [33]. Analyses of sequences also were performed with DnaSP v 4.10.9 [34] and DAMBE v 4.2.13 [35]. AT-rich region analyses and plot profiles were completed with the MacVector software (MacVector, Inc) and mreps v 2.5 [36].
To test if positive selection is acting as a force for genotype divergence, the identification of sites with an excess of amino acid replacements with selective pressure over long evolutionary time was determined. We calculated the dN/dS ratio (nonsynonymous and synonymous substitutions) using the entire sequence of each gene in pairwise comparisons to investigate the substitution rates in each protein-encoding gene [37]. In addition, the software PAML 3.15 was used to analyse DNA sequences with maximum likelihood methods in a phylogenetic framework [38]. CODEML is one of the programmes that can detect sites that have been under recurrent positive selection over long periods of time, e.g., to detect any departure from the neutrality theory in the protein sequences of An. darlingi. The selective pressure at the protein level was measured by ω, the ratio of nonsynonymous to synonymous rates dN/dS, with ω < 1, = 1, or >1 indicating conserved, neutral or adaptive evolution, respectively [39]. The measure of this ratio provides a sensitive measure of selective pressure at the amino acid level. Codon-based likelihood analysis was conducted under the random-sites model: Model M0 (one ratio) assumes constant selective pressure across codon sites and over time. Models M1a (neutral), M2a (selection), M7 (β) and M8 (β & ω) of variable selective pressure across codon sites were used to estimate selective pressure and to test for positive selection. Model M0 is the most simple and can be used to check that parameter estimates in more complex models are consistent. M0 (model = 0 NSsites = 0) was run to provide initial branch lengths, and the initial values of this tree were then used to run the other models. Because selection can act differently in some species, another approach, the branch-specific model, was also tested [40, 41] to detect any evidence of positive selection in any branch of the designated tree. For this analysis the whole protein-encoding genes were used in a combined file, setting the An. darlingi branch as the background one (ω = 1 fixed), and allowing ω to vary in the other anopheline branches of the tree topology. In both approaches, the tree topology for each protein obtained in PAUP [42] was used as the topology to be tested under maximum likelihood.
Phylogenetic analysis
In addition to An. darlingi mtDNA, the following sequences were included in the analysis: An. quadrimaculatus species A (GenBank L_04272), An. funestus (Genbank NC_008070), An. gambiae (NC_002084), Aedes albopictus (GenBank NC_006817) and Aedes aegypti (GenBank NC_010241). Drosophila yakuba (Genbank accession number NC_001322.1) was selected as the out-group [43].
Three different alignments were defined to test the phylogeny of the samples; one with the whole mtDNA sequence, the second with all the protein-encoding gene sequences concatenated and a third new data set with only 1st and 2nd positions of these protein-encoding genes to minimize homoplasy in the 3rd codon position [44]. The software Modeltest v. 3.7 [45] was used to determine the nucleotide substitution model and the alignments followed a GTR+I+γ model (although with different rates) and were used for the subsequent phylogenetic analysis. Three different approaches were followed to reconstruct the phylogeny for posterior analyses, maximum parsimony (MP), maximum likelihood (ML) and Bayesian inference (BI). MP and ML were conducted with PAUP v 4.0b10 [42] and bootstrap values calculated with 1,000 replicates. The BI was performed employing MrBayes v 3.1.1. [46], with two simultaneous runs of 5,000,000 generations, discarding the first 25% as burn-in.
Estimation of divergence date
Culicidae fossil records are not common, and while most of them are compressed, some specimens have been detected in amber inclusions [47]. Anopheles (Nyssorhynchus) dominicanus is the oldest fossil record of the Anophelinae subfamily in the New World, with controversy in the proposed age ranging from 15-20 million years ago (mya) based on foraminifera [48] and 30-45 mya based on coccoliths [49]. The deficient Diptera fossil record offers only an age estimate for the divergence between Drosophila and Anopheles at ~259.9 mya [50], and this deep external calibration might not provide reliable estimation dates, especially for the youngest nodes and for intraspecific divergences within the An. darlingi taxa [51]. Alternatively, Brower [52] estimated that the standard arthropod mtDNA sequence divergence is 2.3% per million years based on calculations across five arthropod genera. The limitation of applying this information to our data is that it may underestimate divergences older than 3.25 million years, because of mutation saturation [53].
A Bayesian Markov Chain Monte Carlo approach, (MCMC) available in the software BEAST v.1.4.6 [54] was performed to infer the topology and the node ages by estimating the Bayesian posterior distribution of the divergence. Following the recommendations of the authors, the uncorrelated relaxed lognormal clock was applied to estimate how clock-like our data were. The ucld.stdev parameter was checked, and in all partitions, the value was close to 0, indicating that the data were clock-like and that the rate of heterogeneity among species was low. Therefore, an uncorrelated lognormal relaxed clock was followed [55] and defined the rate prior to have a normal distribution with a mean of 0.1. The SRD06 model for the partition in the protein-encoding genes was selected, as it seems to provide a better fit for protein-coding nucleotide data [54]. This partition was run using the GTR with gamma and invariant sites model, the best fit with our nucleotide substitution detected by Modeltest. The Yule tree prior is the most suitable for trees describing the relationships among individuals from different species and it was selected to calculate the divergence time between taxa. Final analyses consisted of three separate MCMC runs of 20 million generations sampled every 1000 generations. Tracer v1.4 software was used to confirm the adequate mixing of the MCMC chains of the three separate runs and to check that the effective sample sizes of all the parameters were greater than 500. LogCombiner v1.4.7 was employed for combining the separate runs into one file. Finally, the maximum clade credibility tree was calculated with the mean node branch using TreeAnnotator v1.4.7. FigTree v1.2.1 was employed for visualization of the node branches.
Results and Discussion
Genome organization and structure
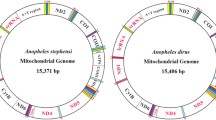
The complete mtDNAs of An. darlingi are 15,385 bp and 15,386 bp in length for the southern and northern genotypes, respectively. The typical 37 genes in animal mtDNA, comprising 13 protein-encoding genes, two rRNA genes (12S rRNA and 16S rRNA), 22 tRNA genes and a control region are found in both genomes (Figure 1 and Table 1). DNA sequences were deposited in GenBank under accession number GQ918272 (northern genotype) and GQ918273 (southern genotype). Genes are encoded on both strands (Table 1) with some open-reading frames overlapping adjacent genes (ATP8/ATP6 and NAD4/NAD4L as reported in other insects [56]), resulting in a compact genome. The An. darlingi gene arrangement structure is highly conserved when compared with the mitochondrion of the other three sequenced anopheline species.

Schematic representation of the organization of the mtDNA of Anopheles darlingi. A) Genes encoded by the L-strand are shaded. tRNA genes are designated by single-letter amino acid codes; L1, L2, S1 and S2 indicate tRNA- Leu (TAA), tRNA-Leu (TAG), tRNA- Ser (GCT) and tRNA- Ser (TGA). B) Graphical representation of the A+T and G+C content across the whole mtDNA. X-axis values represent the length of the genome and the Y-axis represents the percentage.
The nucleotide composition of the An. darlingi southern genotype is as follows: the L-strand has a GC content of 20.73% and 79.27% AT (A = 35.06%, T = 44.21%, C = 9.23% and G = 11.5%) and the H-strand has A = 44.5%, C = 16.12%, G = 7.98%, T = 31.4%. The composition of both strands of the northern genotype was not significantly different from the southern genotype. The mtDNAs of both An. darlingi genotypes are biased towards a high A+T content (Table 2). The overall AT content in the whole mitochondrion sequence was 78.2% and 78.1% in northern and southern, reaching 93.68% and 93.67% in the AT rich region, respectively (Figure 1). This result is similar to that obtained with other anophelines, although it is slightly lower than in the other species included in the study (Table 2). The base composition, measured by the G-skew and T-skew, i.e. asymmetry in nucleotide composition, showed both skews (-0.144 and -0.141, for the G skew) and (-0.028 for the T-skew), indicating the preference for these two nucleotides, a widespread characteristic of animal mtDNAs [57].
Transfer RNA and ribosomal RNA genes
The mtDNA of An. darlingi includes 22 tRNA genes with anticodons representing 20 different amino acids, identical to other anophelines (Table 1).
The lengths of the 12S and 16S rRNA genes are 793 bp and 1329 bp, respectively, identical in both genotypes, and are encoded on the L-strand. The ends of those genes were assumed to be extended to the boundaries of the flanking genes [16]. As in other anophelines, the lrRNA gene is flanked by the tRNALeuand tRNAVal, while the srRNA gene is between tRNAValand the control region. Their A+T contents were 82.54-82.39% for lrRNA and 79.95-79.70% for srRNA, which are within the range of other insects.
Protein-coding genes
Thirteen protein-coding genes could be identified in the mtDNA of An. darlingi, similar to those of other Anophelinae [17–19]. Start codons in all the protein-coding genes are conserved and follow the ATN rule described previously in Anopheles[18], except for the COI gene, which has a TCG start codon characteristic of Anopheles[17, 18, 58]. Eight genes (NAD2, ATP8, ATP6, NAD3, NAD6, CYTB, NAD4L, NAD1) use the complete stop codon TAA (Table 1), whereas five genes have incomplete stop codons, TA_(NAD4, NAD5) and T_(COI, COII, COIII), which could be completed as a result of postranscriptional polyadenylation [59].
The codon usage of both genotypes of An. darlingi is nearly identical (Table 3), and the total number of non-stop codons is 3733 in both. This latter value is similar in the family Anophelinae, ranging from 3715 in An. quadrimaculatus to 3733 in An. gambiae (Figure 2).

Codon distribution in anopheline mtDNA. The Y-axis represents the contribution of each of the codons to the total. Numbers to the left refer to the total number of codons. Amino acids names are indicated on the X-axis. N: Anopheles darlingi northern genotype; S: Anopheles darlingi southern genotype.
The sequence identity within the An. darlingi taxon showed a high similarity across the whole genome (Table 4), with the lowest value (98.5) for the COII gene. Moreover, the sequence of the protein-encoding genes is conserved in Anophelinae, while more differences are found in the Control Region (CR) [60].
Non-coding regions
The mtDNA of An. darlingi contains five intergenic spacers. There are two spacers with only two nucleotides, while two others are formed by nine and 17 nucleotides. The A+T region, flanked by 12sRNA and tRNAIle, has 574 and 573 nucleotides (northern and southern, respectively), and is presumed to be the origin of DNA replication of the organelle, as reported in other insects [61]. High levels of sequence divergence were found in the CRs among anopheline species, even when low diversity was detected in other genes (Table 4). This is consistent with the CR variation observed among dipteran mtDNAs [60]. Four motifs have been described in arthropod CRs: long sequence of thymines, tandem repeat sequences, a sub-region of even higher A+T content and stem-loop structures [62]. The CR of the An. darlingi genotypes showed only 15 variable sites among them, including nine indels (five insertions in the northern genotype). This is surprisingly small variation, even lower than within the An. gambiae complex, where 52 variable positions in CR were described [63]. The anopheline CR does not have the distinct domains seen in other insects [64]. In addition, the T-stretch structure, hypothesized as the regulation of transcription of the mtDNA, is located in position 142 (17 bp of extension) and this structure is a commonly observed in the other anophelines analysed as well in other Diptera, and in different insect orders [60, 64, 65].
Phylogenetic analyses
The relationships among Anopheles inferred using the MP, ML and Bayesian approaches are consistent with earlier reports based on mitochondrial and nuclear genes [25]. Nyssorhynchus was inferred as a sister taxon to Anopheles + Cellia with high bootstrap support for each node. Inconsistent topology was obtained only in the case of rDNA sequences, which grouped Anopheles with Nyssorhynchus, likely due to non-independence of substitutions within loop regions in rDNA genes. Thus, the tree topology was consistent and employed in posterior estimation of parameters in tests of selection.
No evidence of positive selection in the mtDNA
The Likelihood Ratio Test did not detect any sites under positive selection in any mtDNA gene. Parameter estimations of the variable ω were derived under different models for the 13 protein-encoding genes (Table 5). Analyses conducted with all the partitions were consistent with the conclusion that there is no positive selection acting on the protein-encoding genes of An. darlingi mtDNA, and that those are evolving by purifying selection. Even with models that allow variable selective pressure among sites (M2), ω values are distant from 1 and no indication of different selection intensities was detected. Under M8, the ω was >1 in three genes, NAD4, NAD6 and COIII, with less than one site with this pattern, although the comparison between models was insignificant. This could be an indication of a lack of power of the analysis, perhaps as a result of the few sequences that were available, the low divergence between them, or the lack of power of the Likelihood Ratio Test [39]. Concerns about small sample size and sequence divergence have been discussed [66]. Recently, it has been established that reasonable statistical power can be achieved with up to five genomes with divergence similar to those observed among some bacterial strains [67]. In addition, a recent study based on molecular evolution in mosquito immunity-related genes used this approach with similar sample size and with reliable results [68].
Estimates of time of most recent common ancestor
The analyses performed with all mtDNA genes pushed back the split between Culicinae (subgenus Stegomyia) and Anophelinae (genus Anopheles) to 190 mya, in the Early Jurassic. The Anopheles radiation took place during the Early Cretaceous (130 mya), and the most recent ancestor of the Anopheles and Cellia subgenera is estimated at 107 mya. Finally, the split within the Cellia subgenus (An. gambiae and An. funestus) was estimated at 82 mya. Alternatively, the analysis of the protein-coding genes provides earlier divergence dates, and although they correspond to the same geological periods, the nodes are dated more recently in agreement with an older origin of the mosquitoes in the Jurassic [69, 70]. A recent study based on six nuclear genes and morphological characters provides further support for a Jurassic origin of mosquitoes although the estimated divergence between Anopheles and Cellia was more recent (~43 mya) than the dates derived from our study [71]. The different evolutionary histories of the molecular markers (mtDNA vs nDNA), besides incomplete species sampling, may explain the apparent incongruence between the dates.
The inclusion of an additional calibration point of the divergence of Anophelinae and Culicinae estimated to 120 mya in the Early Cretaceous [72] was incorporated in the analysis, providing a node age divergence of the Nyssorhynchus-Anopheles of 79 mya. The process of separation between South America and Africa began in the Permian and the last connection by land was estimated to exist ~95 mya. In addition, the split between Anopheles-Cellia took place 58 mya (Figure 3). Altogether, these data provide estimates for the radiation of the subgenus Cellia concordant with the loss of land bridges between Africa-Europe and Europe-North America (through Greenland), that may explain the current distribution of the Cellia subgenus, absent in the Americas. Anopheles distribution across the American continent, in agreement with the radiation dates derived from this study, is sustained from a latitudinal migration and subsequent radiation from the north to the south. The large confidence intervals of both analyses include congruent dates that fit with the above explanation, although these estimates should be read carefully, since the use of few calibration points could provide weak estimates of divergence [73].

The study of incipient species has been of paramount importance in the understanding of speciation in different organisms [74, 75]. Even though there is strong evidence for diverging genotypes in An. darlingi, its mtDNA does not sustain this level of variance. The persistence of permeable barriers that permit exchange of genetic information within genotypes could preclude the detection of the differentiation. Questions focused on reproductive isolation among the An. darlingi genotypes need to be addressed, as well as assortative mating features between co-existing populations. Taxa with a recent divergence time are expected to share ancestral variation in high proportions, which may confound the reconstruction of their historical relationships. Furthermore, it is likely that young species, with a recent divergence process, have fixed differences only at genes directly responsible for speciation. The major African malaria vector, An. gambiae, has incomplete barriers to gene flow, where divergent selection is acting on local regions of the genome, and gradually increasing the divergence of the populations until gene flow disappears between them [76, 77]. Further support for this form of divergence was provided with the "speciation island model", which suggests small regions with high differentiation in the genome of this taxon [77, 78].
The current studies focused on deciphering the complete genome of An. darlingi will provide new elements and tools to further evaluate the speciation of this taxon. Subsequently, the potential identification of genes affecting speciation should improve the surveillance and management of malaria programs in the Americas and help to elucidate the different patterns of malaria transmission involving An. darlingi.
In addition, the radiation of anophelinae species underscores the significance of these studies together with the origin of malaria parasites. The link between the anophelines in the New World and malaria parasites is still controversial [79–81]. If human Plasmodium parasites were transferred to the Americas by European colonialists in post-Colombian times, then the contact between neotropical malaria vectors and protozoan is very recent. This supports the hypothesis that the susceptibility of mosquitoes to malaria infection is a basal characteristic and further investigations should be conducted to elucidate the refractoriness to this parasite in non-malaria vector species.
Conclusions
The lack of conclusive evidence of speciation at the mtDNA level demands further investigation of other markers, such as nuclear genes that can reflect differences in ecological preferences within An. darlingi. The divergence dates estimated for the subgenus Nyssorhynchus provide valuable information, which, although there is a need to be cautious, because the constraints of the method gave an approximation of the radiation of this group. Finally, it seems that purifying selection is predominant at the mitochondrial level, and the differences observed within the taxon may not be a consequence of that force.
Abbreviations
- The abbreviations used in this work were:
-
COI, COII, COIII: cytochrome oxidase subunit I, II, and III protein genes
- CYTB :
-
cytochrome b gene
- ATP6 :
-
ATP8: ATP synthase subunit 6 and 8 genes
- NAD1 :
-
NAD2, NAD3, NAD4, NAD4L, NAD5, NAD6: NADH dehydrogenase subunit 1-6, 4L genes.
References
World Health Organization: World Malaria Report 2008. 2008, WHO. Geneva, HO/HTM/GMP/2008.1
Linthicum KJ: A revision of the Argyritarsis section of the subgenus Nyssorhynchus of Anopheles (Diptera: Culicidae). Mosq Syst. 1988, 20: 98-271.
Charlwood JD: Biological variation in Anopheles darlingi Root. Mem Inst Oswaldo Cruz. 1996, 91: 391-398.
Lounibos LP, Conn JE: Malaria vector heterogeneity in South America. American Entomologist. 2000, 46: 238-249.
Zimmerman RH, Galardo AK, Lounibos LP, Arruda M, Wirtz R: Bloodmeal hosts of Anopheles species (Diptera: Culicidae) in a malaria-endemic area of the Brazilian Amazon. J Med Entomol. 2006, 43: 947-956. 10.1603/0022-2585(2006)43[947:BHOASD]2.0.CO;2.
Tadei WP, Santos JMM, Rabbani MG: Biologia de anofelinos amazônicos. V. Polimorfismo cromossômico de Anopheles darlingi Root (Diptera: Culicidae). Acta Amazonica. 1982, 12: 353-369.
Steiner WWM, Narang S, Kitzmiller JB, Swofford DL: Genetic divergence and evolution in Neotropical Anopheles (subgenus Nyssorhynchus). Recent developments in the genetics of insect disease vectors. Edited by: Steiner WWM, Tabachnick WJ, Rai KS, Narang S. 1982, Stipes Champaign, Illinois, 523-551.
Malafronte RS, Marrelli MT, Marinotti O: Analysis of ITS2 DNA sequences from Brazilian Anopheles darlingi (Diptera: Culicidae). J Med Entomol. 1999, 36: 631-634.
Manguin S, Wilkerson RC, Conn JE, Rubio-Palis Y, Danoff-Burg JA, Roberts DR: Population structure of the primary malaria vector in South America, Anopheles darlingi, using isozyme, random amplified polymorphic DNA, internal transcribed spacer 2, and morphologic markers. Am J Trop Med Hyg. 1999, 60: 364-376.
Mirabello L, Conn JE: Molecular population genetics of the malaria vector Anopheles darlingi in Central and South America. Heredity. 2006, 96: 311-321. 10.1038/sj.hdy.6800805.
Conn JE, Vineis JH, Bollback JP, Onyabe DY, Wilkerson RC, Povoa MM: Population structure of the malaria vector Anopheles darlingi in a malaria-endemic region of eastern Amazonian Brazil. Am J Trop Med Hyg. 2006, 74: 798-806.
Mirabello L, Vineis JH, Yanoviak SP, Scarpassa VM, Povoa MM, Padilla N, Achee NL, Conn JE: Microsatellite data suggest significant population structure and differentiation within the malaria vector Anopheles darlingi in Central and South America. BMC Ecol. 2008, 8: 3-10.1186/1472-6785-8-3.
Pedro P, Sallum MAM: Spatial expansion and population structure of the neotropical malaria vector, Anopheles darlingi. Biol J Linnean Soc. 2009, 97: 854-866. 10.1111/j.1095-8312.2009.01226.x.
Mirabello L: Molecular population genetics of the malaria vector Anopheles darlingi throughout Central and South America using mitochondrial, nuclear, and microsatellite markers. PhD thesis. 2007, State University of New York at Albany, Biomedical Sciences Department
Boore JL, Collins TM, Stanton D, Daehler LL, Brown WM: Deducing the pattern of arthropod phylogeny from mitochondrial DNA rearrangements. Nature. 1995, 376: 163-165. 10.1038/376163a0.
Boore JL, Macey JR, Medina M: Sequencing and comparing whole mitochondrial genomes of animals. Methods Enzymol. 2005, 395: 311-348. full_text.
Mitchell SE, Cockburn AF, Seawright JA: The mitochondrial genome of Anopheles quadrimaculatus species A: complete nucleotide sequence and gene organization. Genome. 1993, 36: 1058-1073. 10.1139/g93-141.
Beard CB, Hamm DM, Collins FH: The mitochondrial genome of the mosquito Anopheles gambiae: DNA sequence, genome organization, and comparisons with mitochondrial sequences of other insects. Insect Mol Biol. 1993, 2: 103-124. 10.1111/j.1365-2583.1993.tb00131.x.
Krzywinski J, Grushko OG, Besansky NJ: Analysis of the complete mitochondrial DNA from Anopheles funestus: an improved dipteran mitochondrial genome annotation and a temporal dimension of mosquito evolution. Mol Phylogenet Evol. 2006, 39: 417-423. 10.1016/j.ympev.2006.01.006.
Bazin E, Glemin S, Galtier N: Population size does not influence mitochondrial genetic diversity in animals. Science. 2006, 312: 570-572. 10.1126/science.1122033.
Meiklejohn CD, Montooth KL, Rand DM: Positive and negative selection on the mitochondrial genome. Trends Genet. 2007, 23: 259-63. 10.1016/j.tig.2007.03.008.
Conn JE, Mirabello L: The biogeography and population genetics of neotropical vector species. Heredity. 2007, 99: 245-256. 10.1038/sj.hdy.6801002.
Foley DH, Wilkerson RC, Cooper RD, Volovsek ME, Bryan JH: A molecular phylogeny of Anopheles annulipes (Diptera: Culicidae) sensu lato: the most species-rich anopheline complex. Mol Phylogenet Evol. 2007, 43: 283-297. 10.1016/j.ympev.2006.10.008.
Morgan K, O'Loughlin SM, Mun-Yik F, Linton YM, Somboon P, Min S, Htun PT, Nambanya S, Weerasinghe I, Sochantha T, Prakash A, Walton C: Molecular phylogenetics and biogeography of the Neocellia Series of Anopheles mosquitoes in the oriental region. Mol Phylogenet Evol. 2009, 52: 588-601. 10.1016/j.ympev.2009.01.022.
Krzywinski J, Besansky NJ: Molecular systematics of Anopheles: from subgenera to subpopulations. Annu Rev Entomol. 2003, 48: 111-139. 10.1146/annurev.ento.48.091801.112647.
Wilkerson RC, Strickman D: Illustrated key to the female Anophelinae mosquitoes of Central America and Mexico. J Am Mosq Control Assoc. 1990, 6: 7-34.
Faran ME, Linthicum KJ: A handbook of Amazonian species of Anopheles (Nyssorhynchus) (Diptera: Culicidae). Mosq Syst. 1981, 8: 1-107.
Forattini OP: Entomologia Médica. 1962, São Paulo: Universidade São Paulo, I:
Rozen S, Skaletsky H: Primer3 on the www for general users and for biologist programmers. Methods Mol Biol. 2000, 132: 365-386.
Sequin. [http://www.ncbi.nlm.nih.gov/Sequin/sequin.hlp.html]
Lowe TM, Eddy SR: tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25: 955-64. 10.1093/nar/25.5.955.
Kumar S, Tamura K, Nei M: MEGA3: Integrated software for Molecular Evolutionary Genetics Analysis and sequence alignment. Brief Bioinform. 2004, 5: 150-163. 10.1093/bib/5.2.150.
Perna NT, Kocher TD: Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol. 1995, 41: 353-358. 10.1007/BF01215182.
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R: DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003, 19: 2496-2497. 10.1093/bioinformatics/btg359.
Xia X, Xie Z: DAMBE: software package for data analysis in molecular biology and evolution. J Hered. 2001, 92: 371-373. 10.1093/jhered/92.4.371.
Kolpakov R, Bana G, Kucherov G: mreps: Efficient and flexible detection of tandem repeats in DNA. Nucleic Acids Res. 2003, 31: 3672-3678. 10.1093/nar/gkg617.
Nei M, Gojobori T: Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986, 3: 418-426.
Yang Z: PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997, 13: 555-556.
Yang Z, Nielsen R: Codon-substitution models for detecting molecular adaptation at individual sites along specific lineages. Mol Biol Evol. 2002, 19: 908-917.
Yang Z, Wong WSW, Nielsen R: Bayes empirical Bayes inference of amino acid sites under positive selection. Mol Biol Evol. 2005, 22: 1107-1118. 10.1093/molbev/msi097.
Zhang J, Nielsen R, Yang Z: Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol Biol Evol. 2005, 22: 2472-2479. 10.1093/molbev/msi237.
Swofford DL: PAUP* version 4.0b10. Phylogenetic Analysis Using Parsimony (*and Other Methods). 2000, Sinauer Associates, Inc, Sunderland, Massachusetts, USA
Clary DO, Wolstenholme DR: The mitochondrial DNA molecular of Drosophila yakuba: nucleotide sequence, gene organization, and genetic code. J Mol Evol. 1985, 22: 252-271. 10.1007/BF02099755.
Swofford DL, Olsen GJ, Waddell PJ, Hillis DM: Phylogenetic inference. Molecular systematics. Edited by: Hillis DM, Moritz C, Mable BK. 1996, Sinauer, Sunderland, Mass, 407-514. 2
Posada D, Crandall KA: MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998, 14: 817-818. 10.1093/bioinformatics/14.9.817.
Ronquist F, Huelsenbeck JP: MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics. 2003, 19: 1572-1574. 10.1093/bioinformatics/btg180.
Zavortink TJ, Poinar GO: Anopheles (Nyssorhynchus) dominicanus sp. n. (Diptera: Culicidae) from Dominican amber. Ann Entomol Soc Am. 2000, 93: 1230-1235. 10.1603/0013-8746(2000)093[1230:ANDSND]2.0.CO;2.
Iturralde-Vincent MA, MacPhee RDE: Age and paleogeographical origin of Dominican amber. Science. 1996, 273: 1850-1852. 10.1126/science.273.5283.1850.
Schlee D: Das Bernstein-Kabinett. Sttutg Beitr Naturk C. 1990, 28: 100-
Gaunt MW, Miles MA: An insect molecular clock dates the origin of the insects and accords with palaeontological and biogeographic landmarks. Mol Biol Evol. 2002, 19: 748-761.
Ho SY, Larson G: Molecular clocks: when times are a-changin'. Trends Genet. 2006, 22 (2): 79-83. 10.1016/j.tig.2005.11.006.
Brower AV: Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial DNA evolution. Proc Natl Acad Sci USA. 1994, 91: 6491-6495. 10.1073/pnas.91.14.6491.
DeSalle R, Templeton A, Mori I, Pletscher S, Johnston JS: Temporal and spatial heterogeneity of mtDNA polymorphisms in natural populations of Drosophila mercatorum. Genetics. 1987, 116: 215-23.
Drummond AJ, Rambaut A: BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol. 2007, 7: 214-10.1186/1471-2148-7-214.
Drummond AJ, Ho SY, Phillips MJ, Rambaut A: Relaxed phylogenetics and dating with confidence. PLoS Biol. 2006, 4: e88-10.1371/journal.pbio.0040088.
Stewart JB, Beckenbach AT: Insect mitochondrial genomics: the complete mitochondrial genome sequence of the meadow spittlebug Philaenus spumarius (Hemiptera: Auchenorrhyncha: Cercopoidae). Genome. 2005, 48: 46-54. 10.1139/g04-090.
Saccone C, De GC, Gissi C, Pesole G, Reyes A: Evolutionary genomics in Metazoa: the mitochondrial DNA as a model system. Gene. 1999, 238: 195-209. 10.1016/S0378-1119(99)00270-X.
Junqueira AC, Lessinger AC, Torres TT, da Silva FR, Vettore AL, Arruda P, Zeredo Espin AM: The mitochondrial genome of the blowfly Chrysomya chloropyga (Diptera: Calliphoridae). Gene. 2004, 339: 7-15. 10.1016/j.gene.2004.06.031.
Ojala D, Montoya J, Attardi G: tRNA punctuation model of RNA processing in human mitochondria. Nature. 1981, 290: 470-474. 10.1038/290470a0.
Oliveira MT, Azeredo-Espin AML, Lessinger AC: The mitochondrial DNA control region of Muscidae flies: evolution and structural conservation in a Dipteran context. J Mol Evol. 2007, 64: 519-527. 10.1007/s00239-006-0099-6.
Saito S, Tamura K, Aotsuka T: Replication origin of mitochondrial DNA in insects. Genetics. 2005, 171: 1695-705. 10.1534/genetics.105.046243.
Cook CE: The complete mitochondrial genome of the stomatopod crustacean Squilla mantis. BMC Genomics. 2005, 6: 105-10.1186/1471-2164-6-105.
Caccone A, Garcia BA, Powell JR: Evolution of the mitochondrial DNA control region in the Anopheles gambiae complex. Insect Mol Biol. 1996, 5: 51-59. 10.1111/j.1365-2583.1996.tb00040.x.
Duarte GT, De Azeredo-Espin AM, Junqueira AC: The mitochondrial control region of blowflies (Diptera: Calliphoridae): a hot spot for mitochondrial genome rearrangements. J Med Entomol. 2008, 45: 667-76. 10.1603/0022-2585(2008)45[667:TMCROB]2.0.CO;2.
Schultheis AS, Weigt LA, Hendricks AC: Arrangement and structural conservation of the mitochondrial control region of two species of Plecoptera: utility of tandem repeat-containing regions in studies of population genetics and evolutionary history. Insect Mol Biol. 2002, 11: 605-610. 10.1046/j.1365-2583.2002.00371.x.
Anisimova M, Bielawski JP, Yang Z: Accuracy and power of Bayes prediction of amino acid sites under positive selection. Mol Biol Evol. 2002, 19: 950-958.
Anisimova M, Bielawski J, Dunn K, Yang Z: Phylogenomic analysis of natural selection pressure in Streptococcus genomes. BMC Evol Biol. 2007, 7: 154-10.1186/1471-2148-7-154.
Lehmann T, Hume JC, Licht M, Burns CS, Wollenberg K, Simard F, Ribeiro JM: Molecular evolution of immune genes in the malaria mosquito Anopheles gambiae. PLoS One. 2009, 4: e4549-10.1371/journal.pone.0004549.
Edwards FW: Diptera fam. Culicidae. Edited by: Wytsman P. 1932, Genera Insectorum. Desmet- Verteneuil, Brussels
Henning W: Insect phylogeny. 1981, Wiley, Chichester, UK
Reidenbach KR, Cook S, Bertone MA, Harbach RE, Wiegmann BM, Besansky NJ: Phylogenetic analysis and temporal diversification of mosquitoes (Diptera: Culicidae) based on nuclear genes and morphology. BMC Evol Biol. 2009, 9: 298-10.1186/1471-2148-9-298.
Rai KS, Black WC: Mosquito genomes: structure, organization and evolution. Adv Genet. 1999, 41: 1-33. full_text.
Müller J, Reisz RR: Four well-constrained calibration points from the vertebrate fossil record for molecular clock estimates. Bioessays. 2005, 27: 1069-1075. 10.1002/bies.20286.
Coyne JA, Orr HA: The evolutionary genetics of speciation. Philos Trans R Soc Lond B Biol Sci. 1998, 353: 287-305. 10.1098/rstb.1998.0210.
Wu CI, Ting CT: Genes and speciation. Nat Rev Genet. 2004, 5: 114-122. 10.1038/nrg1269.
Lehmann T, Diabate A: The molecular forms of Anopheles gambiae: a phenotypic perspective. Infect Genet Evol. 2008, 8: 737-746. 10.1016/j.meegid.2008.06.003.
Turner TL, Hahn MW, Nuzhdin SV: Genomic islands of speciation in Anopheles gambiae. PLoS Biol. 2005, 3 (9): e285-10.1371/journal.pbio.0030285.
Turner TL, Hahn MW: Locus- and population-specific selection and differentiation between incipient species of Anopheles gambiae. Mol Biol Evol. 2007, 24: 2132-2138. 10.1093/molbev/msm143.
Rich SM, Ayala FJ: Population structure and recent evolution of Plasmodium falciparum. PNAS. 2000, 97: 6994-7001. 10.1073/pnas.97.13.6994.
Carter R: Speculations on the origins of Plasmodium vivax malaria. Trends Parasitol. 2003, 19: 214-9. 10.1016/S1471-4922(03)00070-9.
Hume JC, Lyons EJ, Day KP: Human migration, mosquitoes and the evolution of Plasmodium falciparum. Trends Parasitol. 2003, 19: 144-9. 10.1016/S1471-4922(03)00008-4.
Acknowledgements
We thank Sara Bickersmith and Sasha McKeon (Wadsworth Center, NYSDOH) for assistance with analysis and experiments. Also thanks to Jose R. Loaiza (Smithsonian Tropical Institute, Panama) for valuable comments on this manuscript. This work was supported by NIH grant AI 2ROI 54139 to JEC.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
MM and OM participated in the design of the study, performed molecular and genetic analysis and wrote the manuscript. JK and AAJ advised on the analysis and assisted in preparing the manuscript. WPT and NLA provided samples used in this study and helped to draft the manuscript. JEC conceived and supervised the study, and assisted in the writing of the manuscript. All authors read and approved the final manuscript.
Electronic supplementary material
12936_2010_1208_MOESM1_ESM.DOC
Additional file 1: Sequence and position of the primers used for the amplification of the mtDNA of Anopheles darlingi. (DOC 40 KB)
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Moreno, M., Marinotti, O., Krzywinski, J. et al. Complete mtDNA genomes of Anopheles darlingi and an approach to anopheline divergence time. Malar J 9, 127 (2010). https://doi.org/10.1186/1475-2875-9-127
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-9-127