
Abstract
Background
Several of the intended Plasmodium falciparum vaccine candidate antigens are highly polymorphic and could render a vaccine ineffective if their antigenic sites were not represented in the vaccine. In this study, characterization of genetic variability was performed in major B and T-cell epitopes within vaccine candidate antigens in isolates of P. falciparum from Peru.
Methods
DNA sequencing analysis was completed on 139 isolates of P. falciparum collected from endemic areas of the Amazon basin in Loreto, Peru from years 1998 to 2006. Genetic diversity was determined in immunological important regions in circumsporozoite protein (CSP), merozoite surface protein-1 (MSP-1), apical membrane antigen-1 (AMA-1), liver stage antigen-1 (LSA-1) and thrombospondin-related anonymous protein (TRAP). Alleles identified by DNA sequencing were aligned with the vaccine strain 3D7 and DNA polymorphism analysis and FST study-year pairwise comparisons were done using the DnaSP software. Multilocus analysis (MLA) was performed and average of expected heterozygosity was calculated for each loci and haplotype over time.
Results
Three different alleles for CSP, seven for MSP-1 Block 2, one for MSP-1 Block 17, three for AMA-1 and for LSA-1 each and one for TRAP were identified. There were 24 different haplotypes in 125 infections with complete locus typing for each gene.
Conclusion
Characterization of the genetic diversity in Plasmodium isolates from the Amazon Region of Peru showed that P. falciparum T and B cell epitopes in these antigens have polymorphisms more similar to India than to Africa. These findings are helpful in the formulation of a vaccine considering restricted repertoire populations.
Similar content being viewed by others

Background
Vaccine design for Plasmodium falciparum is hindered by polymorphisms in certain vaccine candidate loci [1, 2]. Highly polymorphic regions have been observed in P. falciparum antigenic surface proteins, such as the circumsporozoite protein (CSP), the merozoite surface protein 1 (MSP-1), the apical membrane antigen 1 (AMA-1), the liver stage antigen (LSA-1) and the thrombospondin-related anonymous protein (TRAP) [3].
One of the best characterized and widely accepted by many as a potential vaccine candidate for P. falciparum is CSP [4, 5]. CSP is a 58-kDa protein and is the major antigen on the surface of malaria sporozoites [6, 7]. The CSP protein can be subdivided into two non-repetitive regions (N- and C-termini) and a variable central region consisting of several repeats of four-residue long motifs; both regions exhibit polymorphisms [8–10]. Several T-cell epitopes have been found in the non-repeat regions while immunodominant B-cell epitopes have been identified in the central repeat region [8, 11]. RTS, S, AS02, a P. falciparum vaccine that consists of the repeat and C-terminal regions of CSP, has successfully completed Phase IIb trials in Mozambique [12, 13].
Another antigen that is considered as a vaccine candidate for P. falciparum is MSP-1. MSP-1 is a 195-kDa protein that is cleaved into an 83-kDa N-terminal fragment, two central fragments of 30- and 38-kDa and a 42-kDa C-terminal fragment [14, 15]. Just before invasion, the 42-kDa is further cleaved into 33- and 19-kDa fragments (MSP-133 and MSP-119). The MSP-119 protein fragment remains anchored to the merozoite surface at the time of erythrocyte invasion and because of its location is a major target of naturally-acquired antimalarial immunity [16]. Within the coding region of the 83-kDa fragment is Block 2, which is a principal target of antibodies associated with clinical immunity in African children [17, 18]. In contrast to Block 2, the Block 17 portion of Pf msp-1, which encodes the MSP-119 fragment, is conserved with only a few polymorphic sites that produce non-synonymous amino acid changes [16, 19].
AMA-1 has also been evaluated for inclusion in a multi-subunit vaccine for both P. falciparum and Plasmodium vivax. Recombinant AMA-1 induces protective immune responses in mouse and monkey models of malaria [20, 21] and both monoclonal and polyclonal antibodies to AMA-1 inhibit merozoite invasion of erythrocytes [22]. As with the other P. falciparum vaccine candidate sequences, Pf ama-1 is highly polymorphic [23–25] with most of polymorphisms occurring in domain I [22, 23, 26] making a broadly effective vaccine difficult to create.
The liver stage-specific antigen, LSA-1 is well conserved among P. falciparum isolates and is also considered a vaccine candidate. Cytokines, such as interferon gamma, have been implicated in the control of Plasmodium growth and with protection from reinfections with P. falciparum [27]. Studies have shown that the N-terminal and Pf LSA-1 protein junction (Pf LSA-J) regions of Pf LSA-1 protein, could induce INF-γ by CD8+ T-cells in adults [28].
Yet another candidate for inclusion in a vaccine for P. falciparum is TRAP [29, 30]. As with the many vaccine targets discussed above, TRAP protein is highly polymorphic. Studies designed to identify HABPs in TRAP successfully identified 21 loci, three of which contain B epitopes [31], while other studies using INF-gamma ELISPOT identified two CD8+ lymphocyte epitopes [32].
Knowledge of the distribution of polymorphic sites on malaria antigens is necessary to obtain a detailed understanding of their significance for vaccine development. This is the first report of the variants found in this part of the Amazon basin; moreover, this study includes infections occurring early in the Peruvian P. falciparum emergence (1998–1999) [33] as well as more recently occurring infections (2003–2006).
Methods
Malaria samples
Plasmodium falciparum isolates were collected from endemic areas in the Peruvian Amazon Department of Loreto during years 1998 to 2006 using human use approved protocols. Loreto is located in the northeast part of Peru and encompasses 30% of the Peruvian territory. The climate is warm and humid, with the rainy season (December – March) having temperatures reaching 36°C and the driest season (June – July) having temperatures as low as 17°C – 20°C.
There were 139 P. falciparum isolates available for this study, all from P. falciparum-infected individuals living in or near the city of Iquitos. Twenty-seven of the isolates were collected in years 1998–1999 from patients diagnosed with severe and complicated malaria (courtesy of Dr. Richard Witzig; IRB NMRCD.2005.0004). Twenty-four samples were from patients enrolled in a sulphadoxine-pyrimethamine in vivo study conducted in 1999 (WRAIR #719). Thirty-two samples were collected between years 2003 to 2005 (11 from 2003 and 2005 each and 10 from 2004) from a community in the San Juan district, approximately five kilometers south of Iquitos, called Zungarococha from individuals participating in an active malaria detection study, including symptomatic and asymptomatic individuals (IRB PJT.NMRCD.015; IRB UAB) [33]. Also, fifty-six samples from year 2006 came from individuals presenting with malaria-like illness at clinics located in different communities near Iquitos (IRB NMRC.2000.0006). All samples obtained were blood samples confirmed to be positive for P. falciparum by microscopy.
Reference strain
Strain 3D7, which is the current vaccine strain used by the US Department of Defense malaria research programmes [34], was used for comparative genetic analysis with Peruvian sequences.
Genetic analysis of B- and T-cell epitopes
The genetic diversity of 11 DNA regions encoding mainly T and B cells epitopes in CSP (Pfcsp), MSP-1 (Pfmsp-1 Block 2 and Pfmsp-1 Block 17), AMA-1 (Pfama-1), LSA-1 (Pflsa-1) and TRAP (Pfssp-2) was determined in the isolates. DNA was purified from 200 μL of whole blood using the QIAamp DNA Blood mini kit. PCR fragments were generated using primers and conditions listed in Table 1 in order to amplify DNA regions encoding the selected T or B cell epitopes. PCR amplification was performed in a 30 μL reaction mixture containing 0.2 μM each of forward and reverse primers (for Pfmsp-1 0.8 μM of each primer was used), 200 μM each dNTP, 0.6 units of DNA polymerase recombinant, 3 μL of 10× PCR buffer, 1.5 mM of MgCl2 and 5 μL of the template extracted from blood.
The following PCR conditions were used:
Pfcsp genetic variability: 3 min at 94°C, 2 min at 58°C, 2 min at 72°C; 1 min at 94°C, 2 min at 58°C, 1 min at 72°C for 32 cycles and a final extension of 72°C for 10 min; obtaining a PCR product of 318 bp.
Pfmsp-1 genetic variability (Blocks 2 and 17): 5 min at 94°C; 30 sec at 91°C, 40 sec at 50°C, 40 sec at 70°C for 40 cycles and a final extension of 70°C for 5 min; obtaining a PCR product of approximately 400 bp.
Pfama-1 genetic variability: 2 min at 95°C; 1 min at 95°C, 1 min at 55°C, 2 min at 72°C for 30 cycles and a final extension of 72°C for 2 min; obtaining a PCR product of 803 bp.
Pflsa-1 genetic variability: 5 min at 94°C; 30 sec at 91°C, 40 sec at 45°C, 40 sec at 70°C for 40 cycles and a final extension of 70°C for 5 min; obtaining a PCR product of 336 bp.
Pfssp-2 genetic variability (Regions II and IV): 5 min at 94°C; 30 sec at 91°C, 40 sec at 50°C, 40 sec at 70°C for 35 cycles; and a final extension of 70°C for 5 min; obtaining PCR products of 1000 bp and 650 bp, respectively.
PCR products were purified using Qiagen PCR spin columns and sequenced using BigDye terminator v3.1 cycle (Applied Biosystem, Foster City, CA) sequencing kit, the primers listed in Table 1 and an ABI 3100 automated sequencer.
Sequence analysis
Sequences were analysed using the Sequencher version 4.7 software (Gene Codes Corporation, Michigan) and comparative alignments to 3D7 were performed using the MEGA version 3.1 software [35]. Additionally, strains 7G8, D6 and W2 were sequenced and used as regional reference strains. Nucleotide diversity, Pi (π) [36, 37], which is the average number of substitutions between any two sequences, and haplotype (gene) diversity values [36] were estimated with the DnaSP 4.0 software [38]. The standard deviation (or standard error) was calculated for both measures. Statistical Analysis Software (SAS Version 9.3, Cary, NC) was used to test multilocus linkage association (MLA) by Fisher Exact Test (FET); two-tailed with alpha = 0.05. The p-value was multiplied by four to correct for conducting pairwise comparisons of four antigens. The antigen diversity was calculated by determining the heterozygosity of alleles detected in each antigen, where heterozygosity (H) = (1 - Sp2), with p2 being the squared frequency of each allele variant. Wright's F-statistic (Fst) to evaluate gene flow among the different study years was determined using DnaSP 4.0 software.
Results
Samples were analysed in three major groups, called study-years: 1998–99, 2003–05 and 2006. Amplified products from 139 isolates were obtained for Pf csp; 135 for Pf msp-1, 128 for Pf ama-1, 137 for Pf lsa-1 and 131 for Pfss p-2. In general, the observed polymorphisms in Peruvian isolates have been previously reported in other populations from South America [9, 23]. Nonsynonymous substitutions were found in all regions encoding T and B cell epitopes, except in Pfss p-2. From all study years, diversity was lowest in the 1998–99 isolates.
Pfcsp
DNA sequences of the 3' region spanning the Th2R and Th3R epitopes at the C-terminal portion of CSP were obtained from the 139 isolates. Results from all samples showed four different alleles (Table 2), none of them similar to the 3D7 strain. In comparison to the 3D7 sequence, mutation points were observed at residues 329 (Lys→Gln), 332 (Lys→Glu), 333 (Glu→Gln), 336 (Asn→Lys/Gln), 339 (Gln→Lys/Arg), 341 (Ser→Ala), 342 (Leu→Ile), 367 (Asn→Gly), 369 (Pro→Ser), 372(Glu→Gln), 374 (Asp→Asn) and 376 (Ala→Glu), all located in Th2R and Th3R regions. From the four different alleles found in Peruvian isolates, allele 1 was the prevalent allele in 1998–99 while in 2003–05 and 2006 both alleles 1 and 2 were commonly found (Table 3). The Peruvian Pf csp sequences were more closely related to 7G8, HB3 and Ven 765 alleles, which was expected due to the close geographical origin of the isolates [39]. All polymorphisms were non-synonymous, and most mutations were at the first or second position of the codons, corroborating earlier studies [7]. In comparison with 7G8, W2 and HB3 strains, alleles 1 and 2 (Table 2) were exactly the same to 7G8 and HB3 sequences, respectively. It is also interesting to notice that all the isolates in the present study that share identical Th2R sequence also share identical Th3R sequence.
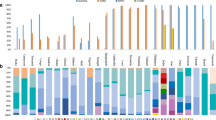
According to DNA polymorphism analysis per study-years, haplotype diversity values have increased since 1998–99 (0.21882) until 2006 (0.53766); also, the nucleotide diversity value for 2006 samples (0.01252) showed an increment of almost 2.3 times more comparing to the value obtained for samples from years 1998–99 (0.00539).
Pfmsp-1
The genetic variability in Blocks 2 and 17 of Pfmsp-1 was determined and compared to the vaccine strain 3D7. From the 135 samples analysed, 85 samples had the Pfmsp-1 Block 2 (Pfmsp-1-B2) K1 type and 50 had the Pfmsp-1-B2 MAD-20 type. Seven different Pfmsp-1-B2 alleles were found, five from the K1 type and 2 from the MAD-20 type (Table 2). Pfmsp-1-B2 allele frequencies differed by study-year. Considering protein (MSP-1B2) allele frequencies, study-year 1998–99 only had two MSP1B2 alleles at a frequency of >10%, while study-year 2003–05 had three MSP1B2 alleles at a frequency of >10% (Table 3).
Allele 1 (a K1 type) and Allele 2 (a MAD-20 type) were the most frequently found in all Peruvian samples. K1 type alleles commenced with the hexapeptide SAQSGT or SAQSGA and ended with SGPSGT. Most diversity was due to duplications or deletions of the repeat motifs SAQ, SGT and SGP. The MAD-20 type allele started with SGGSVT and ended with SVASGG and diversity was due to repetitions of SGG and SVA. Synonymous substitutions were observed for Alanine (GCA/GCT) in K1 isolates and for Glycine (GGC/GGT) in MAD-20 isolates. Also, Block 2 PCR products of the same size did not necessarily correspond to the same allele family since there were two K1 alleles that were the same base-pair length, but had different sequence (Table 3).
Nucleotide sequences for the C-terminal 19-kDa region of MSP-1 (Block 17) were also determined. Substitutions at amino acid positions 1644 (Glu→Gln), 1691 (Thr→Lys), 1700 (Ser→Asn), 1701(Arg→Gly) and 1716 (Leu→Phe) have been previously reported [16, 40] but all isolates from Peru only contained amino acids Gln, Lys, Asn, Gly and Phe in these positions.
Pfama-1
The alignment of Pf AMA-1 domain I included 128 isolates. Regions 259–271 (R1) and 279–287 (R2) are naturally immunogenic T and B cell epitopes and non-synonymous substitutions were found within these resgions in positions 267 (Glu→Gln) and 282 (Ile→Lys) (Table 2). Overall Pfama-1 Domain I Pervian nucleotide sequences, 24 variable sites were found, 23 of them corresponding to nonsynonymous substitutions and only one corresponding to a synonymous substitution. Three different alleles were found in Peru; from which allele 1 had identical amino acid sequence to the vaccine strain 3D7 with only one nucleotide different (AAA/AAG) in codon 292 (Lys). This was the allele most commonly found, followed by allele 2, and then followed by allele 3, which was only detected in study-year 2006 (Table 3).
Pflsa-1
Sequences of the N-terminal region of Pflsa-1, designated T1, were aligned from 137 isolates with the 3D7 sequence. The N-terminal region of LSA-1 has been shown to induce interferon gamma production in peripheral blood mononuclear cells (PBMC). The alignment showed two non-synonymous substitutions in aa 85 (Thr→Ser) and aa 96 (Asn→Tyr) and that the most abundant allele for T1 was allele 1. Alleles 2 and 3 were less frequent (Table 3), the latter having the same nucleotide sequence as 3D7 (Table 2).
Pfssp-2
The antigenic variability of TRAP, encoded by Pfssp-2, was assessed at previous discovered CD8+ T-lymphocyte TRAP epitopes located at amino acids 78–88 (P1) and 504–513 (P5) as well as at B cell epitopes located at amino acids 201–215 (P2), 421–435 (P3) and 461–475 (P4) [31] (Table 2). The alignment of 131 sequences revealed that epitopes corresponding to P1, P2, P3, P4 and P5 are highly conserved in Peruvian isolates. In comparison to the 3D7 sequence, two substitutions were found at positions 82 (Asp→Glu) and 434 (Ser→Pro), and all Peruvian isolates had the same sequence.
Multilocus analysis
For each sample, the combination of alleles detected in the MSP-1B2, CSP, AMA-1, and LSA-1 (multilocus haplotypes) were determined. The order, listed above, was based upon pairwise multilocus association (MLA) analysis demonstrating the most consistent linkage between MSP-1B2 and the other antigens. The result is a four digit code where each digit is the allele detected for the respective loci: twenty-four different multilocus haplotypes were detected (Table 4). Considering all study-years (1998–99, 2003–05, and 2006 samples all together), MSP-1B2 alleles were associated with each of the other loci (p ≤ 0.0001, Fisher Exact Test [FET]) and CSP alleles were associated with MSP-1B2 and AMA-1 (p ≤ 0.0001, FET). CSP was also associated with LSA-1 (p = 0.0003, FET); however, the linkage was less specific: 47.5% were allele 1 and 1, but 19.0% were allele 1 with 2 and 25.6 were allele 2 with 1. AMA-1 was only associated with LSA-1, and this was marginal (p = 0.0401). The only linkage that was observed in each of the year-study samples was MSP-1B2 with AMA-1 (p < 0.0142, FET), with the allele 1 and allele 1 (and NOT allele 1 and NOT allele 1) linkage being observed each year-study. The linkage between CSP and AMA-1 and the linkage between CSP and MSP-1B2 was observed in the 1998–99 study-year and the 2006 study-year. The MSP-1B2 and LSA-1 allele linkage (allele 1 with 1 and allele 2 with 2) was only observed in 1998–99. The only MLA observed in the 2003–05 samples (active case detection) was between MSP-1B2 and AMA-1 (p = 0.0005, FET), with MSP-1B2 allele 3 and AMA-1 allele 2, being detected together 14.3% of the time.
Multilocus nucleotide genetic diversity was determined and compared for each study-year sampling (1998–99, 2003–05, and 2006). Combining all loci together, the nucleotide diversity was similar in all study-years (ranging from 0.2725 to 0.2794); however, the samples collected in study-year 2003–05 (samples collected in active case detection) had higher amino acid haplotype diversity (0.8466 +/-SD 0.033) in comparison to the 1998–99 and 2006 study-years (0.6910 +/- SD 0.055 and 0.7543 +/- SD 0.045, respectively). Likewise, comparing the average allelic heterozygosity for each antigen, the average across all antigens was highest in the 2003–05 samples (H = 0.4474) versus 1998–99 (H = 0.4366) and 2006 (H = 0.3975).
The genetic distance, or gene flow, among the 1998–1999, 2003–2005 and 2006 study-years was determined using Wright's F-statistic, Fst. The Fst for each pairwise comparison was low, ranging from -0.0168 to -0.0050. Therefore, there was no evidence of genetic isolation by year-study. The 2003–2005 and 2006 study-years were the most genetically similar (Fst = -0.0168).
Discussion
Investigating the extent of genetic variation can assist in the laborious process required in selecting antigens for further vaccine development. Several P. falciparum and P. vivax vaccine candidate antigens are highly polymorphic which could pose a serious problem in the formulation of vaccines from a single, well-characterized strain. Previous studies have addressed the diversity and maintenance of several malarial vaccine candidates in Africa [6]. An understanding of genetic variability in T and B cell epitopes of vaccine candidate antigens in P. falciparum Peruvian isolates from the Amazon basin is presented in this study.
According to the Pf csp nucleotide and haplotype diversity values, Th2R and Th3R regions have higher values than those obtained for other antigens in this Peru study; nevertheless, Pf csp nucleotide and haplotype diversity values in Peru for Th2R and Th3R regions are much lower than those obtained in Kenya (π = 0.09772 and 0.07442), Gambia (π = 0.08789 and 0.06454) and Venezuela (π = 0.08466 and 0.08025). However, Peruvian isolates exhibit similar Th2R nucleotide diversity values to samples from Brazil, Vietnam and India [7, 9]. Allele 1 has been previously reported in Iran [41], Brazil [42] and Gambia [43]; allele 2 in Thailand [44], Iran [41] and Myanmar (de Stricker et al., unpublished); and allele 3 in Venezuela [9].
In addition, haplotype diversity for the entire 3' end of CSP was influenced by both Th2R and Th3R regions exclusively, since all 7 variable sites were contained in these regions and all of the polymorphims produced nonsynonymous substitutions. Linkage and recombination events between the Th2R and Th3R regions have been described in previous studies [9]. In the present study, haplotype linkage between Pf csp Th2R and Th3R polymorphisms was also observed. Nucleotide and haplotype diversity values for the entire 3' end of CSP of all samples were 0.01102 and 0.46387, respectively. These values show that Peruvian isolates are less diverse than African and more similar to Indian isolates [9].
CSP is one of the most widely characterized malaria vaccine candidate antigens, and the only one whose components have gone so far as completion of a successful phase II b clinical trial [12, 45], the generation of relevant genetic, epidemiologic and immunologic data for the CSP gene is important, particularly for regions of low malaria endemicity. It is apparent that diversity in Pf csp is regionally restricted, and that Peru has low genetic polymorphism with one predominant allele and variants in a small number, as in India [10], Vietnam [7] and Thailand [5]. This suggests that since polymorphisms are restricted and can be grouped, allelic variants can be included in a polyvalent vaccine that could be widely effective.
On the other hand, diversity in Pfmsp-1 Block 2 was lower than reported in P. falciparum isolates studied in other geographic regions [16]. Neither RO33 nor MR Block 2 alleles were found; although these Block 2 alleles have been reported in other studies, including Venezuela [15, 46]. The number of alleles detected using this direct sequencing method is lower than those detected using PCR-based genotyping methods of samples from other populations such as Kenya [15] with 20 K1 and 15 MAD-20 alleles detected by nested PCR from 362 samples. However, similar results to the Peruvian samples were found in 104 isolates from Brazil with eight alleles verified by Southern blot and SSCP-PCR [47]. It is important to remark that the direct sequencing used in this Peruvian study only detected one Pfmsp-1 Block 2 per infection (sample) and, therefore, could underestimate the genetic diversity by only detecting the more high-density/dominant allele in each infection. Results indicated that 63% of the isolates in this study where K1 alleles. Although the 3D7 vaccine strain is in the K1 allele family, none of the Peruvian isolates had the same K1 allele (sequence) as the vaccine strain 3D7.
Strikingly, nucleotide sequences for the C-terminal 19-kDa region of MSP-1 (Block 17) showed no diversity at positions 1644 (Gln), 1691 (Lys), 1700 (Asn), 1701 (Gly) and 1716 (Phe), while polymorphisms in these positions have been reported in Brazil, Vietnam and Kenya [48, 49]. The 3D7 vaccine strain has the MSP-1 19 kDa haplotype ETSRL, suggesting that if immunity conferred by monovalent vaccines is allele specific, it would have low overall efficacy in populations where the target allele is in the minority [50].
Results also showed two non-synonymous substitutions in R1 and R2 regions of Pfama-1. The extensive number of non-synonymous polymorphisms outside R1 and R2 described in this study revealed that this gene could undergo intense selective pressure in these other regions of AMA-1 upon administration of a full-length peptide vaccine having a single allele type. The overall genetic diversity in Pfama-1 domain I (π = 0.00697) is lower than in the Kenyan and Southeast Asian isolates (π values of 0.01361 and 0.01196, respectively) [23]. From the three different Pfama-1 alleles found, allele 3 was only present in samples from 2006 and could have arisen by a new introduction of this R1-R2 haplotype from another geographic region or by recombination between alleles 1 and 2 which existed in prior study-years.
Two non-synonymous and no synonymous substitutions were found in LSA-1 T1 in Peruvian isolates. In comparison, within Brazilian, Papua New Guinean and Kenyan isolates two synonymous substitutions (positions 87 and 104) and three different non-synonymous substitutions (positions 92, 95 and 104) have been reported [51]. These data suggest that T cell epitopes of P. falciparum LSA-1 are highly conserved in field isolates from geographically diverse regions with varying transmission patterns, since non much allelic diversity is observed. However, even in this low and recent transmission region of Peru, we detected two non-synonymous substitutions.
Pf ssp-2 HABPs sequences were highly conserved among these Peruvian isolates. Part of the peptide 2 sequence has been reported to be a B cell epitope, recognized by sera from malaria immune humans living in endemic areas [52]. Peptides 3 and 4 are located within a region characterized by the presence of a great number of Asn, Lys and Pro residues. This long stretch of amino acids suggests configuration binding sites, which could be altered with just minimal changes in amino acid sequence [31]. For instance, results showed Pro in site 434 instead of Ser as in the 3D7 sequence; also, P3 showed a characteristic sequence in which amino acids Asp and Pro are present each forth residue and that may allow binding in different registers [28]. P1 and P5 have been identified as CD8+ T-lymphocyte epitopes and also showed to be highly conserved, with just one variation in aa 82 (Glu/Asp) compared to 3D7 strain.
Most of the genetic diversity detected in Peruvian isolates was attributable to non-synonomous, amino acid changes. Over time, comparing the allele frequencies in the 1998–1999, 2003–2005 and 2006 study-years, there were changes in allele frequencies with new alleles being detected. The 2006 samples had the highest allele counts. Although multilocus associations were detected, these associations were most evident in the year 1998–1999 samples. Interestingly, the 2003–2005 study-year samples had the least multilocus associations (only MSP-1B2 and AMA-1 polymorphisms were determined to be associated) and had the greatest overall allelic diversity (H = 0.8466). The diversification observed over time might be attributed to natural selection from a specific immune response. In particularly, regarding the 2003–2005 samples, these samples were collected in active case detection in a population where the frequency of asymptomatic infections suggests immune pressure [37].
Several studies have demonstrated the importance of T and B cell mediated immunity to malaria and how nonsynonymous substitutions may affect the conformation of epitopes and in consequence the immunological response [53]. Nevertheless, more studies need to be done in order to understand the immunological implications of amino acid changes in these malaria vaccine candidate antigens. Studying different genes and their alleles help us to understand if they interact to influence in malaria infection and if minimal changes in their sequence could render a vaccine ineffective. An alternative to improve the effectiveness of a vaccine would be to create a construct using the most common regional specific alleles considering the genetic diversity found in the area. By providing information about the prevalence and dynamics of vaccine candidate antigens polymorphisms, an accurate construct could be built.
Conclusion
Peruvian isolates are less polymorphic than African and more like Indian populations. Although, conserved epitopes were found in Peru, the observation of uneven geographic distribution of polymorphisms and the high number of alleles distributed worldwide, especially for CSP and MSP-1 Block 2 may have an adverse impact on the effectiveness of vaccines. The number of allelic variants increased over time in this study, suggesting that even in geographic regions with low transmission, vaccine strategy development should include conduction of diversity studies. The uneven geographic distribution of alleles may jeopardize the formulation and use of vaccines directed on a specific variable loci since local variants may not be considered in the vaccine design.
Glossary
Interferon-gamma (IFN-γ): a dimerized soluble cytokine that is the only member of the type II class of interferons.
T cell epitopes: antigenic determinants recognized and bound by the T-cell receptor.
B-cell epitopes: antigenic determinants recognized and bound by the B-cell receptor.
Abbreviations
- CSP:
-
Plasmodium falciparum circumsporozoite protein
- MSP-1:
-
Plasmodium falciparum merozoite surface protein-1
- AMA-1:
-
Plasmodium falciparum apical membrane antigen-1
- TRAP:
-
Plasmodium falciparum thrombospondin related anonymous protein
- LSA-1:
-
Plasmodium falciparum liver stage antigen-1
- HABP:
-
high affinity binding peptide
- Pfcsp :
-
gene encoding CSP
- Pfmsp-1 :
-
gene encoding MSP-1
- Pfama-1 :
-
gene encoding AMA-1
- Pfspp-2 :
-
gene encoding TRAP
- FET:
-
Fisher Exact Test
- SD:
-
standard deviation
References
Hughes AL, Verra F: Extensive polymorphism and ancient origin of Plasmodium falciparum. Trends Parasitol. 2002, 18: 348-351. 10.1016/S1471-4922(02)02290-0.
Tongren JE, Zavala F, Roos DS, Riley EM: Malaria vaccines: if at first you don't succeed. Trends Parasitol. 2004, 20: 604-610. 10.1016/j.pt.2004.09.005.
Escalante AA, Lal AA, Ayala FJ: Genetic polymorphism and natural selection in the malaria parasite Plasmodium falciparum. Genetics. 1998, 149: 189-202.
Nussenzweig RS, Nussenzweig V: Antisporozoite vaccine for malaria: experimental basis and current status. Rev Infect Dis. 1989, 11 (Suppl 3): S579-585.
Kumkhaek C, Phra-ek K, Renia L, Singhasivanon P, Looareesuwan S, Hirunpetcharat C, White NJ, Brockman A, Gruner AC, Lebrun N: Are Extensive T Cell Epitope Polymorphisms in the Plasmodium falciparum Circumsporozoite Antigen, a Leading Sporozoite Vaccine Candidate, Selected by Immune Pressure?. J Immunol. 2005, 175: 3935-3939.
Alloueche A, Milligan P, Conway DJ, Pinder M, Bojang K, Doherty T, Tornieporth N, Cohen J, Greenwood BM: Protective efficacy of the RTS,S/AS02 Plasmodium falciparum malaria vaccine is not strain specific. Am J Trop Med Hyg. 2003, 68: 97-101.
Jalloh A, van Thien H, Ferreira MU, Ohashi J, Matsuoka H, Kanbe T, Kikuchi A, Kawamoto F: Sequence variation in the T-cell epitopes of the Plasmodium falciparum circumsporozoite protein among field isolates is temporally stable: a 5-year longitudinal study in southern Vietnam. J Clin Microbiol. 2006, 44: 1229-1235. 10.1128/JCM.44.4.1229-1235.2006.
Alloueche A, Silveira H, Conway DJ, Bojang K, Doherty T, Cohen J, Pinder M, Greenwood BM: High-throughput sequence typing of T-cell epitope polymorphisms in Plasmodium falciparum circumsporozoite protein. Mol Biochem Parasitol. 2000, 106: 273-282. 10.1016/S0166-6851(99)00221-2.
Escalante AA, Grebert HM, Isea R, Goldman IF, Basco L, Magris M, Biswas S, Kariuki S, Lal AA: A study of genetic diversity in the gene encoding the circumsporozoite protein (CSP) of Plasmodium falciparum from different transmission areas–XVI. Asembo Bay Cohort Project. Mol Biochem Parasitol. 2002, 125: 83-90. 10.1016/S0166-6851(02)00216-5.
Bhattacharya PR, Bhatia V, Pillai CR: Genetic diversity of T-helper cell epitopic regions of circumsporozoite protein of Plasmodium falciparum isolates from India. Trans R Soc Trop Med Hyg. 2006, 100: 395-400. 10.1016/j.trstmh.2005.07.012.
Tsuji M, Zavala F: Peptide-based subunit vaccines against pre-erythrocytic stages of malaria parasites. Mol Immunol. 2001, 38: 433-442. 10.1016/S0161-5890(01)00079-7.
Alonso PL, Sacarlal J, Aponte JJ, Leach A, Macete E, Milman J, Mandomando I, Spiessens B, Guinovart C, Espasa M: Efficacy of the RTS,S/AS02A vaccine against Plasmodium falciparum infection and disease in young African children: randomised controlled trial. Lancet. 2004, 364: 1411-1420. 10.1016/S0140-6736(04)17223-1.
Alonso PL, Sacarlal J, Aponte JJ, Leach A, Macete E, Aide P, Sigauque B, Milman J, Mandomando I, Bassat Q: Duration of protection with RTS,S/AS02A malaria vaccine in prevention of Plasmodium falciparum disease in Mozambican children: single-blind extended follow-up of a randomised controlled trial. Lancet. 2005, 366: 2012-2018. 10.1016/S0140-6736(05)67669-6.
Espejo F, Bermudez A, Torres E, Urquiza M, Rodriguez R, Lopez Y, Patarroyo ME: Shortening and modifying the 1513 MSP-1 peptide's alpha-helical region induces protection against malaria. Biochem Biophys Res Commun. 2004, 315: 418-427. 10.1016/j.bbrc.2004.01.072.
Takala SL, Escalante AA, Branch OH, Kariuki S, Biswas S, Chaiyaroj SC, Lal AA: Genetic diversity in the Block 2 region of the merozoite surface protein 1 (MSP-1) of Plasmodium falciparum: additional complexity and selection and convergence in fragment size polymorphism. Infect Genet Evol. 2006, 6: 417-424. 10.1016/j.meegid.2006.01.009.
Ferreira MU, Ribeiro WL, Tonon AP, Kawamoto F, Rich SM: Sequence diversity and evolution of the malaria vaccine candidate merozoite surface protein-1 (MSP-1) of Plasmodium falciparum. Gene. 2003, 304: 65-75. 10.1016/S0378-1119(02)01180-0.
Conway DJ, Greenwood BM, McBride JS: Longitudinal study of Plasmodium falciparum polymorphic antigens in a malaria-endemic population. Infect Immun. 1992, 60: 1122-1127.
Cavanagh DR, Dodoo D, Hviid L, Kurtzhals JA, Theander TG, Akanmori BD, Polley S, Conway DJ, Koram K, McBride JS: Antibodies to the N-terminal block 2 of Plasmodium falciparum merozoite surface protein 1 are associated with protection against clinical malaria. Infect Immun. 2004, 72: 6492-6502. 10.1128/IAI.72.11.6492-6502.2004.
Ballou WR, Arevalo-Herrera M, Carucci D, Richie TL, Corradin G, Diggs C, Druilhe P, Giersing BK, Saul A, Heppner DG: Update on the clinical development of candidate malaria vaccines. Am J Trop Med Hyg. 2004, 71: 239-247.
Anders RF, Crewther PE, Edwards S, Margetts M, Matthew ML, Pollock B, Pye D: Immunisation with recombinant AMA-1 protects mice against infection with Plasmodium chabaudi. Vaccine. 1998, 16: 240-247. 10.1016/S0264-410X(97)88331-4.
Crewther PE, Matthew ML, Flegg RH, Anders RF: Protective immune responses to apical membrane antigen 1 of Plasmodium chabaudi involve recognition of strain-specific epitopes. Infect Immun. 1996, 64: 3310-3317.
Coley AM, Parisi K, Masciantonio R, Hoeck J, Casey JL, Murphy VJ, Harris KS, Batchelor AH, Anders RF, Foley M: The most polymorphic residue on Plasmodium falciparum apical membrane antigen 1 determines binding of an invasion-inhibitory antibody. Infect Immun. 2006, 74: 2628-2636. 10.1128/IAI.74.5.2628-2636.2006.
Escalante AA, Grebert HM, Chaiyaroj SC, Magris M, Biswas S, Nahlen BL, Lal AA: Polymorphism in the gene encoding the apical membrane antigen-1 (AMA-1) of Plasmodium falciparum. X. Asembo Bay Cohort Project. Mol Biochem Parasitol. 2001, 113: 279-287. 10.1016/S0166-6851(01)00229-8.
Cortes A, Mellombo M, Mueller I, Benet A, Reeder JC, Anders RF: Geographical structure of diversity and differences between symptomatic and asymptomatic infections for Plasmodium falciparum vaccine candidate AMA1. Infect Immun. 2003, 71: 1416-1426. 10.1128/IAI.71.3.1416-1426.2003.
Marshall VM, Zhang L, Anders RF, Coppel RL: Diversity of the vaccine candidate AMA-1 of Plasmodium falciparum. Mol Biochem Parasitol. 1996, 77: 109-113. 10.1016/0166-6851(96)02583-2.
Kocken CH, Narum DL, Massougbodji A, Ayivi B, Dubbeld MA, Wel van der A, Conway DJ, Sanni A, Thomas AW: Molecular characterisation of Plasmodium reichenowi apical membrane antigen-1 (AMA-1), comparison with P. falciparum AMA-1, and antibody-mediated inhibition of red cell invasion. Mol Biochem Parasitol. 2000, 109: 147-156. 10.1016/S0166-6851(00)00250-4.
Connelly M, King CL, Bucci K, Walters S, Genton B, Alpers MP, Hollingdale M, Kazura JW: T-cell immunity to peptide epitopes of liver-stage antigen 1 in an area of Papua New Guinea in which malaria is holoendemic. Infect Immun. 1997, 65: 5082-5087.
Bongartz M, Rezbach P, Borrmann S, Hollingdale MR, Kremsner PG, Luty AJ: Age-dependent enhancement of IFN-gamma responses to Plasmodium falciparum liver stage antigen-1 T cell epitopes. Parasitol Res. 2002, 2002 Aug 1021.,
Lopez-Estrano C, Bhattacharjee S, Harrison T, Haldar K: Cooperative domains define a unique host cell-targeting signal in Plasmodium falciparum-infected erythrocytes. Proc Natl Acad Sci USA. 2003, 12003 Sep 12426.,
Rogers WO, Malik A, Mellouk S, Nakamura K, Rogers MD, Szarfman A, Gordon DM, Nussler AK, Aikawa M, Hoffman SL: Characterization of Plasmodium falciparum sporozoite surface protein 2. Proc Natl Acad Sci USA. 1992, 89: 9176-9180. 10.1073/pnas.89.19.9176.
Lopez R, Curtidor H, Urquiza M, Garcia J, Puentes A, Suarez J, Ocampo M, Vera R, Rodriguez LE, Castillo F: Plasmodium falciparum: binding studies of peptide derived from the sporozoite surface protein 2 to Hep G2 cells. J Pept Res. 2001, 58: 285-292. 10.1034/j.1399-3011.2001.00902.x.
Gonzalez JM, Peter K, Esposito F, Nebie I, Tiercy JM, Bonelo A, Arevalo-Herrera M, Valmori D, Romero P, Herrera S: HLA-A*0201 restricted CD8+ T-lymphocyte responses to malaria: identification of new Plasmodium falciparum epitopes by IFN-gamma ELISPOT. Parasite Immunol. 2000, 22: 501-514. 10.1046/j.1365-3024.2000.00331.x.
Branch O, Casapia WM, Gamboa DV, Hernandez JN, Alava FF, Roncal N, Alvarez E, Perez EJ, Gotuzzo E: Clustered local transmission and asymptomatic Plasmodium falciparum and Plasmodium vivax malaria infections in a recently emerged, hypoendemic Peruvian Amazon community. Malar J. 2005, 4: 27-10.1186/1475-2875-4-27.
Garcon N, Heppner DG, Cohen J: Development of RTS,S/AS02: a purified subunit-based malaria vaccine candidate formulated with a novel adjuvant. Expert Rev Vaccines. 2003, 2: 231-238. 10.1586/14760584.2.2.231.
Kumar S, Tamura K, Jakobsen IB, Nei M: MEGA2: molecular evolutionary genetics analysis software. Bioinformatics. 2001, 17: 1244-1245. 10.1093/bioinformatics/17.12.1244.
Nei M: Molecular Evoultionary Genetics. 1987, Columbia Univ Press
Nei MaJCM: A simple method for estimating average number of nucleotide substitutions within and between populations from restriction data. Genetics. 1990, 125: 873-879.
Rozas J, Sanchez-DelBarrio JC, Messeguer X, Rozas R: DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics. 2003, 19: 2496-2497. 10.1093/bioinformatics/btg359.
Cortese JF, Caraballo A, Contreras CE, Plowe CV: Origin and dissemination of Plasmodium falciparum drug-resistance mutations in South America. J Infect Dis. 2002, 2002 Sep 1009.,
Hoffmann EH, Ribolla PE, Ferreira MU: Genetic relatedness of Plasmodium falciparum isolates and the origin of allelic diversity at the merozoite surface protein-1 (MSP-1) locus in Brazil and Vietnam. Malar J. 2003, 2: 24-10.1186/1475-2875-2-24.
Zakeri S, Avazalipoor M, Mehrizi AA, Djadid ND, Snounou G: Restricted T-cell epitope diversity in the circumsporozoite protein from Plasmodium falciparum populations prevalent in Iran. Am J Trop Med Hyg. 2007, 76: 1046-1051.
Tanabe K, Sakihama N, Hattori T, Ranford-Cartwright L, Goldman I, Escalante AA, Lal AA: Genetic distance in housekeeping genes between Plasmodium falciparum and Plasmodium reichenowi and within P. falciparum. J Mol Evol. 2004, 59: 687-694. 10.1007/s00239-004-2662-3.
Weedall GD, Preston BM, Thomas AW, Sutherland CJ, Conway DJ: Differential evidence of natural selection on two leading sporozoite stage malaria vaccine candidate antigens. Int J Parasitol. 2007, 37: 77-85. 10.1016/j.ijpara.2006.09.001.
Jongwutiwes S, Tanabe K, Hughes MK, Kanbara H, Hughes AL: Allelic variation in the circumsporozoite protein of Plasmodium falciparum from Thai field isolates. Am J Trop Med Hyg. 1994, 51: 659-668.
Bojang KA, Olodude F, Pinder M, Ofori-Anyinam O, Vigneron L, Fitzpatrick S, Njie F, Kassanga A, Leach A, Milman J: Safety and immunogenicty of RTS,S/AS02A candidate malaria vaccine in Gambian children. Vaccine. 2005, 23: 4148-4157. 10.1016/j.vaccine.2005.03.019.
Cavanagh DR, Elhassan IM, Roper C, Robinson VJ, Giha H, Holder AA, Hviid L, Theander TG, Arnot DE, McBride JS: A longitudinal study of type-specific antibody responses to Plasmodium falciparum merozoite surface protein-1 in an area of unstable malaria in Sudan. J Immunol. 1998, 161: 347-359.
Sallenave-Sales S, Ferreira-da-Cruz MF, Faria CP, Cerruti C, Daniel-Ribeiro CT, Zalis MG: Plasmodium falciparum: limited genetic diversity of MSP-2 in isolates circulating in Brazilian endemic areas. Exp Parasitol. 2003, 103: 127-135. 10.1016/S0014-4894(03)00091-2.
Kaneko O, Kimura M, Kawamoto F, Ferreira MU, Tanabe K: Plasmodium falciparum: allelic variation in the merozoite surface protein 1 gene in wild isolates from southern Vietnam. Exp Parasitol. 1997, 86: 45-57. 10.1006/expr.1997.4147.
Pacheco MA, Poe AC, Collins WE, Lal AA, Tanabe K, Kariuki SK, Udhayakumar V, Escalante AA: A comparative study of the genetic diversity of the 42 kDa fragment of the merozoite surface protein 1 in Plasmodium falciparum and P. vivax. Infect Genet Evol. 2007, 7: 180-187. 10.1016/j.meegid.2006.08.002.
Takala SL, Coulibaly D, Thera MA, Dicko A, Smith DL, Guindo AB, Kone AK, Traore K, Ouattara A, Djimde AA: Dynamics of polymorphism in a malaria vaccine antigen at a vaccine-testing site in mali. PLoS Med. 2007, 4: e93-10.1371/journal.pmed.0040093.
Ravichandran M, Doolan DL, Cox-Singh J, Hoffman SL, Singh B: Research note: HLA degenerate T-cell epitopes from Plasmodium falciparum liver stage-specific antigen 1 (LSA-1) are highly conserved in isolates from geographically distinct areas. Parasite Immunol. 2000, 22: 469-473. 10.1046/j.1365-3024.2000.00324.x.
Charoenvit Y, Fallarme V, Rogers WO, Sacci JB, Kaur M, Aguiar JC, Yuan LF, Corradin G, Andersen E, Wizel B: Development of two monoclonal antibodies against Plasmodium falciparum sporozoite surface protein 2 and mapping of B-cell epitopes. Infect Immun. 1997, 65: 3430-3437.
Kumar KA, Sano G, Boscardin S, Nussenzweig RS, Nussenzweig MC, Zavala F, Nussenzweig V: The circumsporozoite protein is an immunodominant protective antigen in irradiated sporozoites. Nature. 2006, 444: 937-940. 10.1038/nature05361.
Acknowledgements
The views expressed in this article are those of the authors and do not necessarily reflect the official policy of the Department of the Navy, Department of Defence, nor the US government. This work was supported by funds provided by the Department of military Infectious Disease Research Program under the unit number 6000.RAD1.F.A0309. The study protocols were approved by various Institutional Review Boards (NMRC, NMRCD and UAB) in compliance with all applicable Federal regulations governing the protection of human subjects. One of the authors, (DJB), is a military service member. This work was prepared as part of his official duties. Title 17 U.S.C. §105 provides that "Copyright protection under this title is not available for any work of the United Stated Government". Title 17 U.S.C §101 defines a U.S. Government work as a work prepared by a military service number or employee of the U.S. government as part of that person's official duties. We are grateful to personnel from the Parasitology staff working at the NMRCD-Iquitos Laboratory for collecting samples for the work and personnel from the Virology staff at NMRCD-Lima for their support. We are grateful to the staff in the Malaria Immunology and Genetics in the Amazon project for collecting the samples in Zungarococha (2003–2005) and extraction of these DNA samples. We also like to thank Mr. Pablo Tsukayama for his invaluable comments and suggestions.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
SMC conducted the molecular genetic studies, analysed the data and wrote the first draft of the manuscript, OB provided isolates, participated in the genetic diversity, linkage, and study-year comparisons and assisted in writing and editing the manuscript, AAE participated in the DNA polymorphisms analysis and assisted in writing and editing the manuscript, CML participated in the coordination of the laboratory component of the study and assisted in editing the manuscript, DJB designed the project, supervised and directed the research team and contributed to the writing and editing of the manuscript. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Chenet, S.M., Branch, O.H., Escalante, A.A. et al. Genetic diversity of vaccine candidate antigens in Plasmodium falciparum isolates from the Amazon basin of Peru. Malar J 7, 93 (2008). https://doi.org/10.1186/1475-2875-7-93
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-7-93