
Abstract
Background
In order to harmonize results for the detection and quantification of Plasmodium falciparum DNA by nucleic acid amplification technique (NAT)-based assays, a World Health Organization (WHO) collaborative study was performed, evaluating a series of candidate standard preparations.
Methods
Fourteen laboratories from 10 different countries participated in the collaborative study. Four candidate preparations based upon blood samples parasitaemic for P. falciparum were evaluated in the study. Sample AA was lyophilized, whilst samples BB, CC and DD were liquid/frozen preparations. The candidate standards were tested by each laboratory at a range of dilutions in four independent assays, using both qualitative and quantitative NAT-based assays. The results were collated and analysed statistically.
Results
Twenty sets of data were returned from the participating laboratories and used to determine the mean P. falciparum DNA content for each sample. The mean log10 "equivalents"/ml were 8.51 for sample AA, 8.45 for sample BB, 8.35 for sample CC, and 5.51 for sample DD. The freeze-dried preparation AA, was examined by accelerated thermal degradation studies and found to be highly stable.
Conclusion
On the basis of the collaborative study, the freeze-dried material, AA (NIBSC code No. 04/176) was established as the 1st WHO International Standard for P. falciparum DNA NAT-based assays and has been assigned a potency of 109 International Units (IU) per ml. Each vial contains 5 × 108 IU, equivalent to 0.5 ml of material after reconstitution.
Similar content being viewed by others

Background
Each year there are an estimated 300 million acute cases of malaria worldwide, accounting for more than one million deaths annually [1]. In humans, malaria is caused by infection with five species of Plasmodium (Plasmodium falciparum, Plasmodium malariae, Plasmodium ovale, Plasmodium vivax and Plasmodium knowlesi). Plasmodium falciparum is associated with the greatest morbidity and mortality, accounting for approximately 95% of deaths due to malaria [2]. Although most of these cases are found in endemic tropical regions of Africa, Asia, Central and South America, cases of imported malaria are reported in non-endemic areas. In Europe it has been estimated that there are in excess of 10,000 cases of imported malaria per year, with around 2,000 cases being recorded annually in the UK alone [3, 4]. Estimates by the Centers for Disease Control and Prevention (United States) have indicated that there were approximately 1,528 cases, including seven fatal cases, of imported malaria in the United States in 2005, largely due to travellers, immigrants and servicemen returning from malaria endemic regions [5]. Transfusion transmitted cases of malaria can also occur, by the use of contaminated blood, and may result in significant disease or even death in the recipients, particularly when P. falciparum is the causative agent [6].
Malaria diagnosis has relied upon microscopic analysis of Wright's or Wright-Giemsa stained blood smears to detect infection and determine parasite loads [7]. Slide interpretation requires considerable expertise and is difficult to standardize, particularly at low levels of parasitaemia or where mixed species infections occur [8, 9]. Nucleic acid amplification technique (NAT)-based assays, such as PCR are becoming increasingly employed in the diagnosis of malaria [10, 11]. Studies have shown that PCR can improve sensitivity and species discrimination, when compared to either microscopy or serological methods [12]. Although numerous PCR methods have been developed for the laboratory diagnosis and clinical management of malaria, the reported sensitivities of these assays vary widely. These differences may be a result of intrinsic variability in assay sensitivity or a consequence of calibration using different reference reagents, which are poorly standardized. In terms of quality assurance, it is of concern that variability in the implementation of assays between laboratories could give different results for the same patient sample.
In the last ten years, a number of International Standards have been developed by the World Health Organization (WHO) for NAT-based assays. The first was established in 1997 for hepatitis C virus (HCV) RNA [13]. This standard was pivotal in the introduction of HCV RNA NAT screening of blood and plasma donations, used in transfusion and the manufacture of plasma derived medicinal products, where previously there had been transmissions of HCV. Further standards have been established for hepatitis B virus (HBV) DNA, HIV-1 RNA, parvovirus B19 DNA, hepatitis A virus (HAV) RNA [14–17]. The standards have allowed the development of regulatory requirements for the detection of these blood borne viruses in plasma, by setting commonly recognized thresholds and limits of detection. The standards have been used in the calibration of secondary standards and working reagents, and have been used in the validation of assays for both the qualitative and quantitative NAT-based techniques. These blood-borne virus standards have also found use in the clinical setting, both for diagnosis and for monitoring viral loads in response to antiviral therapy. Moreover, results in a number of commercially available assays for blood borne viruses are expressed in terms of WHO International Units associated with the respective standards.
This paper describes a collaborative study to evaluate four candidate standards for their suitability for use as a WHO International Standard for P. falciparum DNA for use in NAT-based assays. The value of such a standard will be to ensure consistent assay implementation between laboratories; allow inter-laboratory comparisons; provide materials for assay validation; and in particular to generate secondary standards to monitor routine assay use. The need for such a material was recognized by the WHO Expert Committee on Biological Standardization (ECBS) in 2004.
Methods
Candidate standards
Four candidate standards were included in this study. Sample AA was a freeze-dried preparation of blood, obtained from a patient infected with P. falciparum following an exchange transfusion. Local Research Ethical Committee approval was obtained at the London Hospital for Tropical Diseases for the collection and subsequent use of this sample. The blood used to prepare sample AA, was determined to have an overall parasitaemia of approximately 9.8% by light microscopy. Sample AA was stored at -70°C until lyophilization. The conditions for lyophilization were as previously described [13, 14]. Lyophilized vials of AA have been stored at -20°C with constant temperature monitoring, at the National Institute for Biological Standards and Control (NIBSC). Sample BB was a liquid preparation, of in vitro cultured P. falciparum (3D7 strain) prepared in leucodepleted blood, with a parasitaemia of approximately 10%. Sample CC was a liquid preparation of approximately 6.9% parasitaemia of P. falciparum infected blood, obtained from the same patient as sample AA. Sample DD was a 1:1000 dilution of sample CC prepared in leucodepleted blood. Samples BB, CC and DD were dispensed in 0.5 ml volumes and stored as liquid/frozen preparations at -70°C. All samples tested negative by PCR for the following viral markers: HBV DNA, HCV RNA, HAV RNA, HIV-1 RNA and parvovirus B19 DNA.
Design of the study
Fourteen laboratories participated in the collaborative study. Laboratories were sent four vials of each of the candidate standards. The participants were requested to store samples at or below -70°C until analysis. The lyophilized preparation (AA) was reconstituted using 0.5 ml of nuclease-free deionized water immediately prior to analysis, with gentle agitation for 20 minutes to fully dissolve the contents. The liquid preparations (BB, CC and DD) were to be thawed quickly before use. Participants were requested to test the panel of candidate standards in four independent assays for P. falciparum DNA, using fresh vials of the four candidates for each assay run. In the case of qualitative assays, serial dilutions of the samples were analysed in four independent assays. In the first qualitative assay, ten-fold dilutions were performed to determine the end point for the detection of P. falciparum DNA. In each of the subsequent three assays, a minimum of two half-log10 (i.e. 1:3.2) dilutions either side of the pre-determined end-point were assayed, and results reported as positive or negative. Participants were requested to prepare dilutions in the sample diluent normally used in their assay system. In the case of quantitative assays for P. falciparum DNA, results were used directly as returned by laboratories, after correction for any dilutions made.
Statistical methods
The qualitative end-point assays were analyzed using the Poisson model, as previously described for collaborative studies for other NAT standards [13, 15]. All estimates were expressed as log10 PCR detectable units/ml. Overall means were calculated as arithmetic means of the log10 estimates.
Stability studies
The lyophilized candidate standard (AA) was analysed for its stability over time in accelerated thermal degradation studies. Samples were incubated in temperature controlled environments, withdrawn at specified times and analysed for P. falciparum DNA content as described below. Following reconstitution, 200 μl volumes of sample for AA were extracted using the MagNA Pure LC instrument with software version 4.0 (Roche Applied Science, Mannheim, Germany). Samples were extracted using the DNA Isolation Kit I (Roche Applied Science, Mannheim, Germany) according to the manufacturer's instructions. Elution was performed with 100 μl of elution buffer. Real-time PCRs were performed on the LightCycler 2.0 instrument (Roche Applied Science, Mannheim, Germany). An in-house TaqMan assay was performed using primers selected from the most conserved regions of the 18S ribosomal RNA (rRNA) gene. The primer sequences were as follows: forward primer, 5' CAG ATG TCA GAG GTC AAA TTC TAA GAT T 3'; reverse primer, 5' TCC CTT AAC TTT CGT TCT TGA TTA ATG 3'. The sequence of the fluorogenic hydrolysis probe was as follows: 5' (FAM) CTG GAG ACG GAC TAC TGC GAA AGC ATT TG (TAMRA) 3'. Amplification reactions were performed using the LightCycler FastStart DNA Master Hybprobe kit (Roche Applied Science, Mannheim, Germany). The amplification conditions were as follows: 95°C for 10 min, then 45 cycles of the following sequential steps: 95 °C for 15s, 60°C for 1 min. Fluorescence data was collected during the combined annealing/extension step and detected at 530 nm. A standard curve was generated using serial ten-fold dilutions of a sample with a concentration of 180,000 parasites per μl, as determined by Giemsa-stained thin film microscopy by a proficient operator, with independent confirmation.
Results
Data returned and data analysis
Fourteen laboratories from 10 countries, returned results from four assays for each of the four separate preparations. The participants included parasitology laboratories from tropical medicine institutes, universities, hospitals, quality control laboratories, manufacturers of diagnostic kits and other laboratories. The majority of assays used by the participating laboratories were ones developed in-house, although participants returned results from commercial assays. Throughout this study, a code number has been allocated at random for each laboratory and does not necessarily represent the order of the participants described in Table 1. The types of assays used by participants are listed in Table 2.
Participating laboratories were requested to test the samples in qualitative or quantitative format in order to determine the P. falciparum DNA content for each of the four candidate standards. The majority of laboratories provided data from qualitative end-point dilution assays. These laboratories reported the number of samples positive out of the number tested at various dilutions. These were treated as a dilution series and used to provide a single estimate of "PCR-detectable units/ml" in the undiluted sample using the method of maximum likelihood for "dilution assays" as described for the analysis of other NAT standards [13, 15]. The model assumes that the probability of a positive result at a given dilution follows a Poisson distribution (with the mean given by the expected number of "copies" in the sample tested, and that a single copy will lead to a positive result). Calculations were carried out using the statistical package GLIM [13, 15].
Where a laboratory marked a result as uncertain or +/-, it was treated as negative. Where a laboratory performed more than one assay method, the results were analyzed separately, treated as if from separate laboratories, and coded as for example as laboratories 1A and 1B. Laboratory 1B did not continue the dilution series for sample AA until negative responses were observed. It was therefore not possible to calculate an estimate of units/ml for this sample (see below). For assays 2–4, Laboratory 9 only tested single log dilutions to the endpoint rather than half-log dilutions at the supposed end point. It was still possible to determine estimates of detectable units/ml, but they may be less precise than for other laboratories. Laboratory 10 (10A & 10B) had some positives at very high dilutions. Because the Poisson model could not be fitted with these results, they were excluded as potential false positives. Laboratory 11 (11A & 11B) reported "dilutions not well executed" for assay 2, which was therefore excluded. It was not possible to obtain an estimate of detectable units/ml for sample CC for 11A, as all results were positive. Some of the remaining results from 11B were also inconsistent. Laboratory 14B had some inconsistent negatives at 10-1 and 10-2 for sample DD in assay 2, which were excluded.
Two laboratories provided quantitative results. Laboratory 3B provided quantitative results using a commercial kit (Artus RealArt Malaria LC PCR kit, Qiagen, Hamburg, Germany) for a range of dilutions for each sample. The results were quoted as copies/μl. Laboratory 5, provided results from an in-house quantitative assay, giving results as a mean log10 Par/ml based on two sets of triplicate determinations for each sample.
Estimates of PCR-detectable units/ml
The estimated log10 PCR-detectable units/ml for each of the four candidate standards from the qualitative end-point assays are shown in histogram form in Figure 1 and in Table 3. Each box represents the estimate from one laboratory, and is labeled with the laboratory code number. The overall means, range and between laboratory standard deviations are shown in Table 4. The agreement between laboratories is similar to that observed in the collaborative study to establish the 1st International Standard for HCV RNA for NAT-based assays [13]. The quantitative results from laboratories 3B and 5 are shown in Table 5. For laboratory 3B, the results are based on the neat and 1 in 10 dilutions only, as the results did not appear linear for dilutions beyond this. Results were expressed as "copies/ul". For laboratory 5, results were expressed as "Par/ml". The estimates from laboratory 3B are much higher than the results from the qualitative assays, particularly for samples AA, BB and CC. For laboratory 5, the estimates for samples AA, CC and DD are much lower than those from the qualitative assays. The estimate for sample BB is of the same order as the qualitative assays, but 4 log10 higher than the equivalent estimates of AA and CC. This may be due to the fact that this particular assay is amplifying RNA and not DNA, compared to the other assays. Samples AA, CC and DD were subject to multiple rounds of freeze-thawing during their preparation, and due to cell lysis, the labile nature of RNA and its susceptibility to degradation by nucleases have resulted in loss of potency of these preparations when RNA levels are examined. Sample BB only underwent a single freeze-thaw cycle before dispatch to study participants and hence has a higher potency in this particular assay for Laboratory 5. These results emphasize that this candidate standard is not suitable for RNA assays.

Histograms of laboratory estimates of polymerase chain reaction (PCR)-detectable units/ml (log 10 ) for samples AA, BB, CC and DD (panels A, B, C and D respectively). The laboratory code numbers are inicated in each box in the respective histograms.
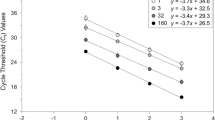
Figure 2 shows the performance of the four candidate standard preparations in a real-time TaqMan PCR assay (as described in Stability Studies section of the Materials and Methods), which further demonstrates mean estimated PCR detectable units/ml values shown in Table 4. Near identical crossing cycle (Ct) values are found for samples AA, BB and CC, with sample DD generating a much later Ct values reflecting the dilution of this material when it was originally prepared. It should be noted that sample AA was derived from blood from an exchange transfusion that was determined to have an overall parasitaemia of approximately 9.8% which had been determined by light microscopy. Sample BB was a liquid preparation of in vitro cultured P. falciparum (3D7 strain) with a parasitaemia of approximately 10% was also evaluated in this study. The mean estimates of these two preparations AA and BB are 8.51 and 8.45 log10 PCR detectable units/ml respectively based upon the results of the end point dilution assays, demonstrating that the patient derived sample AA gives results that are commutable to the in vitro propagated sample derived from the 3D7 laboratory strain of P. falciparum.

Amplification plot of samples AA, BB, CC and DD using a TaqMan assay targeting the 18S rRNA gene of P. falciparum , as described in Stability Section of the Materials and Methods. The different samples are indicated on the figure. No template control (NTC).
Stability studies on candidate sample AA
The stability of sample AA was examined over time in accelerated degradation studies. Samples were incubated in temperature controlled environments, withdrawn at specified times and tested for P. falciparum DNA content. A baseline titre was determined by comparison to vials of sample AA stored at -20°C (i.e. the long term storage temperature for lyophilized standards). The overall geometric means (log10) for the estimated parasites/ul were determined for the sample AA at each of the different incubation temperatures. This is displayed as the log10 drop (Table 6). At the higher temperatures there is clearly some degradation, with a drop of around 0.3 log10 after a year at +20°C, and around 0.6 log10 at higher temperatures. There was little observed loss at +4°C however, with an apparent increase at 8 months, and a loss of 0.06 log10 at 12 months. This data is based on a limited number of vials however, and these differences are within the expected assay variability. If the Arrhenius model for accelerated degradation [24, 25] is applied to this data, a predicted loss per year at -20°C of around 20%, or 0.1 log10 is obtained. However this is not a precise estimate. This prediction does not appear to be consistent with the observed data for the sample stored at +4°C, which has not shown degradation of the order of that predicted for the -20°C sample. It is not clear whether the Arrhenius model, which is based on a simple first order degradation process, is appropriate for material of this type. Although there is degradation at higher temperatures, there is little observed degradation after 12 months at +4°C and this observed stability is in line with that observed for the WHO International Standards for the blood borne viruses [13, 15, 17]. Studies of real-time stability are of sample AA are ongoing.
Discussion
The use of NAT-based assays for the detection of P. falciparum DNA in blood is now being increasingly used in the management and diagnosis of malaria, particularly in countries where travellers are returning from malaria endemic regions. Such NAT-based assays are also being used, on occasion in the primary screening or confirmatory testing of blood used for transfusion [Dr. S. Salueda, personal communication]. In some centres performing vaccine trials, there are reports of using DNA-based assays to monitor parasitaemia using real-time PCR [26]. However there is widespread variation in the sensitivity and specificity of these assays and the aim of this study was to evaluate candidate reference preparations for P. falciparum DNA that will be useful for standardization purposes.
The candidate standards were evaluated by a wide range of assays from a large number of laboratories worldwide, as is the requirement of such WHO international collaborative studies [27]. During this study, the participating laboratories used a variety of different extraction and amplification methods for the detection of P. falciparum DNA (Table 2). The majority of these assays were developed in-house and therefore it was not unsurprising to see such a spread of results reported. The participating laboratories mainly returned qualitative results for the NAT-based assays, with very few data returned for quantitative assays. As a consequence, some variability was expected in the range of log10 PCR-detectable units/ml, and indeed was found to be comparable to that seen in previous studies carried out to establish other International Standard for NAT-based assays for the blood borne viruses such as HCV [13–17]. With the exclusion of outlier results, the ranges seen in the other studies are typically between 1.5 and 2.5 log10 PCR-detectable units/ml. Whilst many of assays targeted the 18S rRNA gene, other regions of the P. falciparum genome were used for analysis including the small subunit rRNA gene, the β-tubulin gene and the conserved ATS domain of var genes (Table 2). Despite the differences in the genes targeted by the different assays, there is still a wide range of results, as described above, even when the same region of the P. falciparum genome is targeted, such as the multicopy 18S rRNA gene. In the case of Laboratory 3A, results were reported for a PCR assay directed against the 18S rRNA gene, which gave comparable results to laboratory 7 targeting the single copy β-tubulin gene. In a recent study [28], comparing PCR-based detection methods for P. falciparum on a panel of field samples from Nigeria, a range of assays based upon single or multicopy genes gave a range, the sensitivities of which broadly correlated with copy number, however, this is based upon the work of a single group and not a multicentre study. It would appear that even when the same genes are targeted the difference in response observed by the laboratories will be dependent upon the way the assays are designed (e.g. primer and probe sequences) and implemented (reaction mixes, buffer components, performance characteristics and calibration status of thermal cyclers etc.). The availability of an International Standard will help to reconcile differences such as those mentioned above.
To serve as an International Standard, ideally the preparation should be stable for many years and this is the reason that lyophilized preparations are preferred, where residual moisture and oxygen are minimized [27]. From the Arrhenius model for accelerated degradation [24, 25] the estimates of the potency for the samples stored at elevated temperatures can be used to predict the long term stability at various temperatures. As a consequence accelerated thermal degradation studies were carried out. In the initial stability studies performed on the lyophilized candidate AA, it was observed that there was very little degradation at lower temperatures (i.e. +4°C and +20°C). There was some observed degradation to the samples stored at +37°C and +45°C. This may be due to natural degradation expected at these temperatures, but may also be the result of increasing difficulty in resuspending samples stored at elevated temperatures over time. Whilst the results for the stability of candidate AA are not such a good fit with the model, they nevertheless suggest that this preparation is of adequate stability and support the long term use of the material. Moreover, the results of the stability studies show that a short time at elevated temperatures during shipment of the standard at ambient temperatures should not cause unacceptable loss of potency. Studies are continuing in order to monitor the real-time stability of this material.
The discussions outlined above suggest that candidate AA would be the most suitable to serve as the International Standard. It also has the advantage of being based on blood from a naturally occurring P. falciparum infection and would therefore most closely simulate the field situation. However it should still be noted that the results for candidate BB, based upon the P. falciparum 3D7 clone cultured in leucodepleted blood, demonstrated that the potencies of the two preparations were near identical despite their different formulation and that results are commutable for these two preparations. Candidate preparation AA was therefore selected as the International Standard as it demonstrated that the DNA concentration was in a suitable range that would be acceptable for use, it was suitable for use in a wide variety of assays utilized by the study participants and it demonstrated good stability. The preparation was assigned an arbitrary unitage in International Units (IU), unrelated to the level of parasitaemia or DNA copy number. These arbitrary units can be used to compare assays or laboratory performance by the response given for the International Standard.
Since the introduction of the 1st WHO International Standard HCV RNA International Standard ten years ago, the number of quantitative assays available has greatly increased. In a recent study to replace the 2nd WHO International Standard for HCV RNA for NAT-based assays [29] there was a marked increase in the number of quantitative assays and very good agreement with the determinations of the potency of the current standard and the candidate replacements. Such standards can be used for quality control and in the determination of the analytical sensitivity of different testing procedures. They also provide a source of material for assay validation and the production of secondary standards to be used as run controls or working reagents. Indeed the data generated in this present study, produced from a large number of laboratories, using a variety of different assays for P. falciparum DNA detection, demonstrate the wide range of responses to the panel of candidate standards and reinforce the need for a common standard to allow inter-laboratory comparisons of both in-house and commercial assay sensitivities by traceability to a "gold standard" with a physical existence.
Conclusion
In October 2006 the ECBS established this preparation as the 1st WHO International Standard for Plasmodium falciparum DNA NAT-based assays. This standard has been assigned a unitage of 109 IU/ml. (5 × 108 IU per vial, to be reconstituted in 0.5 ml). The 1st WHO International Standard for P.falciparum DNA NAT-based assays is stored at the National Institute for Biological Standards and Control (NIBSC code number 04/176). Details on requesting the standard are available on the NIBSC website.
References
World Health Organization: Severe falciparum malaria. Trans R Soc Trop Med Hyg. 2000, 94: 1-90. 10.1016/S0035-9203(00)90300-6.
Stoppacher R, Adams SP: Malaria deaths in the United States: case report and review of deaths, 1979–1998. J Forensic Sci. 2003, 48: 404-408.
Muentener P, Schlagenhauf P, Steffen R: Imported malaria (1985–95): trends and perspectives. Bull World Health Organ. 1999, 77: 560-566.
Jerrard DA, Broder JS, Hanna JR, Colletti JE, Grundmann KA, Geroff AJ, Mattu A: Malaria: a rising incidence in the United States. J Emerg Med. 2002, 23: 23-33. 10.1016/S0736-4679(02)00457-2.
Centers for Disease Control and Prevention: Malaria surveillance – United States, 2005. Morbid Mortal Wkly Rep. 2007, 56: 23-38.
Kitchen AD, Chiodini PL: Malaria and blood transfusion. Vox Sang. 2006, 90: 77-84. 10.1111/j.1423-0410.2006.00733.x.
Kain KC, Harrington MA, Tennyson S, Keystone JS: Imported malaria: prospective analysis of problems in diagnosis and management. Clin Infect Dis. 1998, 27: 142-149. 10.1086/514616.
Payne D: Use and limitations of light microscopy for diagnosing malaria at the primary health care level. Bull World Health Organ. 1988, 66: 621-626.
O'Meara WP, Barcus M, Wongsrichanalai C, Muth S, Maguire JD, Jordan RG, Prescott WR, McKenzie FE: Reader technique as a source of variability in determining malaria parasite density by microscopy. Malar J. 2006, 5: 118-10.1186/1475-2875-5-118.
Padley D, Moody AH, Chiodini PL, Saldanha J: Use of a rapid, single-round, multiplex PCR to detect malarial parasites and identify the species present. Ann Trop Med Parasitol. 2003, 97: 131-137. 10.1179/000349803125002977.
Johnston SP, Pieniazek NJ, Xayavong MV, Slemenda SB, Wilkins PP, da Silva AJ: PCR as a confirmatory technique for laboratory diagnosis of malaria. J Clin Microbiol. 2006, 44: 1087-1089. 10.1128/JCM.44.3.1087-1089.2006.
Perandin F, Manca N, Piccolo G, Calderaro A, Galati L, Ricci L, Medici MC, Arcangeletti C, Dettori G, Chezzi C: Identification of Plasmodium falciparum, P. vivax, P. ovale, and P. malariae and detection of mixed infection in patients with imported malaria in Italy. New Microbiol. 2003, 26: 91-100.
Saldanha J, Lelie N, Heath A: Establishment of the first international standard for nucleic acid amplification technology (NAT) assays for HCV RNA. WHO Collaborative Study Group. Vox Sang. 1999, 76: 149-158. 10.1159/000031040.
Saldanha J, Gerlich W, Lelie N, Dawson P, Heermann K, Heath A, WHO Collaborative Study Group: An international collaborative study to establish a World Health Organization international standard for hepatitis B virus DNA nucleic acid amplification techniques. Vox Sang. 1999, 80: 63-71. 10.1046/j.1423-0410.2001.00003.x.
Holmes H, Davis C, Heath A, Hewlett I, Lelie N: An international collaborative study to establish the 1st international standard for HIV-1 RNA for use in nucleic acid-based techniques. J Virol Methods. 2001, 92: 141-150. 10.1016/S0166-0934(00)00283-4.
Saldanha J, Lelie N, Yu MW, Heath A, the B19 Collaborative Study Group: Establishment of the first World Health Organization International Standard for human parvovirus B19 DNA nucleic acid amplification techniques. Vox Sang. 2002, 82: 24-31. 10.1046/j.1423-0410.2002.00132.x.
Saldanha J, Heath A, Lelie N, Pisani G, Yu MY, and the Collaborative Study Group: A World Health Organization International Standard for hepatitis A virus RNA nucleic acid amplification technology assays. Vox Sang. 2005, 89: 52-8. 10.1111/j.1423-0410.2005.00633.x.
Snounou G, Viriyakosol S, Jarra W, Thaithong S, Brown KN: Identification of the four human malaria parasite species in field samples by the polymerase chain reaction and detection of a high prevalence of mixed infections. Mol Biochem Parasitol. 1993, 58: 283-292. 10.1016/0166-6851(93)90050-8.
Hulier E, Pétour P, Snounou G, Nivez MP, Miltgen F, Mazier D, Rénia L: A method for the quantitative assessment of malaria parasite development in organs of the mammalian host. Mol Biochem Parasitol. 1996, 77: 127-135. 10.1016/0166-6851(96)02584-4.
Rougemont M, Van Saanen M, Sahli R, Hinrikson HP, Bille J, Jaton K: Detection of four Plasmodium species in blood from humans by 18S rRNA gene subunit-based and species-specific real-time PCR assays. J Clin Microbiol. 2004, 42: 5636-5643. 10.1128/JCM.42.12.5636-5643.2004.
Boom R, Sol CJ, Salimans MM, Jansen CL, Wertheim-van Dillen PM, Noordaa van der J: Rapid and simple method for purification of nucleic acids. J Clin Microbiol. 1990, 28: 495-503.
Price RN, Uhlemann AC, Brockman A, McGready R, Ashley E, Phaipun L, Patel R, Laing K, Looareesuwan S, White NJ, Nosten F, Krishna S: Mefloquine resistance in Plasmodium falciparum and increased pfmdr1 gene copy number. Lancet. 2004, 364: 438-447. 10.1016/S0140-6736(04)16767-6.
Perandin F, Manca N, Calderaro A, Piccolo G, Galati L, Ricci L, Medici MC, Arcangeletti MC, Snounou G, Dettori G, Chezzi C: Development of a Real-Time PCR Assay for Detection of Plasmodium falciparum, Plasmodium vivax, and Plasmodium ovale for Routine Clinical Diagnosis. J Clin Microbiol. 2004, 42: 1214-1219. 10.1128/JCM.42.3.1214-1219.2004.
Kirkwood TBL: Predicting the stability of biological standards and products. Biometrics. 1977, 33: 736-742. 10.2307/2529472.
Tydeman MS, Kirkwood TBL: Design and analysis of accelerated thermal degradation tests for the stability of biological standards I. Properties of maximum likelihood estimators. J Biol Standardization. 1984, 12: 195-206. 10.1016/S0092-1157(84)80054-2.
Hermsen CC, De Vlas SJ, Van Gemert GJA, Telgt DS, Verhage DF, Sauerwein RW: Testing vaccines in human experimental malaria; statistical analysis of parasitemia measured by a quantitative real-time polymerase chain reaction. Am J Trop Med Hyg. 2004, 71: 196-201.
World Health Organization: Recommendations for the preparation, characterization and establishment of international and other biological reference materials. WHO/BS/04.119 (revised 2004)
Oyedeji SI, Awobode HO, Monday GC, Kendjo E, Kremsner PG, Kun JF: Comparison of PCR-based detection of Plasmodium falciparum infections based on single and multicopy genes. Malar J. 2007, 6: 112-10.1186/1475-2875-6-112.
Baylis SA, Heath A, and the collaborative study group: WHO collaborative study to establish a replacement WHO International Standard for hepatitis C virus RNA nucleic acid amplification technique (NAT)-based assays. WHO ECBS Report 2007: WHO/BS/07.2055
Acknowledgements
We thank Claire Swales at the London School of Hygiene and Tropical Medicine for the preparation of sample BB used in this study.
Author information
Authors and Affiliations
Consortia
Corresponding author
Additional information
Authors' contributions
DJP: Design and coordination of the study; molecular studies of the stability of 04/176. Drafting and preparation of the manuscript. ABH: Statistical analysis. PLC & CS: Facilitated the collection of materials for samples AA, CC and DD. Participated in the design of the study. SAB: Participation in the design and coordination of the study. Critically revising the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under license to BioMed Central Ltd. This is an Open Access article distributed under the terms of the Creative Commons Attribution License (http://creativecommons.org/licenses/by/2.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Padley, D.J., Heath, A.B., Sutherland, C. et al. Establishment of the 1st World Health Organization International Standard for Plasmodium falciparum DNA for nucleic acid amplification technique (NAT)-based assays. Malar J 7, 139 (2008). https://doi.org/10.1186/1475-2875-7-139
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-7-139