
Abstract
Background
As well as being inducible by haem, haemoxygenase -1 (HO-1) is also induced by interleukin-10 and an anti-inflammatory prostaglandin, 15d PGJ2, the carbon monoxide thus produced mediating the anti-inflammatory effects of these molecules. The cellular distribution of HO-1, by immunohistochemistry, in brain, lung and liver in fatal falciparum malaria, and in sepsis, is reported.
Methods
Wax sections were stained, at a 1:1000 dilution of primary antibody, for HO-1 in tissues collected during paediatric autopsies in Blantyre, Malawi. These comprised 37 acutely ill comatose patients, 32 of whom were diagnosed clinically as cerebral malaria and the other 5 as bacterial diseases with coma. Another 3 died unexpectedly from an alert state. Other control tissues were from Australian adults.
Results
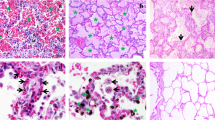
Apart from its presence in splenic red pulp macrophages and microhaemorrhages, staining for HO-1 was confined to intravascular monocytes and certain tissue macrophages. Of the 32 clinically diagnosed cerebral malaria cases, 11 (category A) cases had negligible histological change in the brain and absence of or scanty intravascular sequestration of parasitized erythrocytes. Of these 11 cases, eight proved at autopsy to have other pathological changes as well, and none of these eight showed HO-1 staining within the brain apart from isolated moderate staining in one case. Two of the three without another pathological diagnosis showed moderate staining of scattered monocytes in brain vessels. Six of these 11 (category A) cases exhibited strong lung staining, and the Kupffer cells of nine of them were intensely stained. Of the seven (category B) cases with no histological changes in the brain, but appreciable sequestered parasitised erythrocytes present, one was without staining, and the other six showed strongly staining, rare or scattered monocytes in cerebral vessels. All six lung sections not obscured by neutrophils showed strong staining of monocytes and alveolar macrophages, and all six available liver sections showed moderate or strong staining of Kupffer cells. Of the 14 (category C) cases, in which brains showed micro-haemorrhages and intravascular mononuclear cell accumulations, plus sequestered parasitised erythrocytes, all exhibited strong monocyte HO-1 staining in cells forming accumulations and scattered singly within cerebral blood vessels. Eleven of the available and readable 13 lung sections showed strongly staining monocytes and alveolar macrophages, and one stained moderately. All of the 14 livers had strongly stained Kupffer cells. Of five cases of comatose culture-defined bacterial infection, three showed a scattering of stained monocytes in vessels within the brain parenchyma, three had stained cells in lung sections, and all five demonstrated moderately or strongly staining Kupffer cells. Brain sections from all three African controls, lung sections from two of them, and liver from one, showed no staining for HO-1, and other control lung and liver sections showed few, palely stained cells only. Australian-origin adult brains exhibited no staining, whether the patients had died from coronary artery disease or from non-infectious, non-cerebral conditions
Conclusions
Clinically diagnosed 'cerebral malaria' in children includes some cases in whom malaria is not the only diagnosis with the hindsight afforded by autopsy. In these patients there is widespread systemic inflammation, judged by HO-1 induction, at the time of death, but minimal intracerebral inflammation. In other cases with no pathological diagnosis except malaria, there is evidence of widespread inflammatory responses both in the brain and in other major organs. The relative contributions of intracerebral and systemic host inflammatory responses in the pathogenesis of coma and death in malaria deserve further investigation.
Similar content being viewed by others

Introduction
Falciparum malaria is a complex multi-organ disease. There is no simple or accepted explanation for how small numbers of parasites can cause such severe illness, or how this infection can cause such wide-spread pathology, since only hepatocytes and erythrocytes are invaded by the pathogen. Undoubtedly parasitized red cells sequester in capillaries and venules, but in recent times the traditional idea that this is the primary cause of organ failure and death through obstructing blood flow has needed modifying. In particular, it has had to accommodate the evident involvement of excessive systemic release of pro-inflammatory cytokines, triggered by malarial toxins. For the last decade, many researchers have focussed their efforts on the pathophysiological implications of the ability of these mediators to generate inducible nitric oxide synthase (iNOS), and thus produce a continuous, potentially large, supply of nitric oxide in tissues that normally experience only low, tightly controlled, levels of this ubiquitous cellular messenger. Despite the harmful effects of iNOS-induced nitric oxide (NO) when produced in unusually large amounts [1–3], more commonly it provides negative feedback that suppresses production of the inflammatory cytokines that generate it, and a range of other downstream harmful mediators, through inhibiting NF kappa B, a major activator of protein transcription [4].
Carbon monoxide (CO), another endogenous gas with a similar structure, also inhibits TNF generation [5] again through inhibiting NF kappa B [6]. Both molecules are generated by enzymes that have at least one constitutive form, and another, iNOS and haemoxygenase-1 (HO-1) respectively, induced by inflammatory cytokines. NO and CO act interactively as second messengers in ways that are still being elucidated [7]. For instance, both NO and CO can activate soluble guanylate cyclase to generate cyclic GMP [8], and thus dilate blood vessel walls, as well as perform their immunosuppressive roles.
This shared activity of NO and CO duplicates that of interleukin-10 (IL-10), the prototype anti-inflammatory cytokine, which also suppresses generation of tumour necrosis factor (TNF) and interleukin-1β through inhibiting NF kappa B [9]. Thus the high circulating levels of IL-10 seen in human malaria [10, 11] and sepsis [12], have been proposed to suppress disease severity through inhibiting the systemic inflammatory effects of TNF [13]. These apparently disparate observations are now appreciated to be different parts of the same chain of events, with IL-10 producing its strong anti-TNF effect through inducing HO-1, and thus generating CO [14]. The most plausible explanation for the early observation that TNF induces HO-1 [15] is now therefore its ability to induce IL-10 [16]. Likewise, the anti-inflammatory effect of 15d prostaglandin J2 (15d PGJ2), which is present in tissues during inflammation [17], also operates through HO-1 induction and subsequent generation of CO [18]. As in sepsis, cyclooxygenase-2, which generates 15d PGJ2, is induced in severe malaria [19]. Thus, rather than HO-1 simply being a marker for haem degradation and a generator of anti-oxidant defences, it is now recognized to be an integral part of the network of inflammatory mediators. It therefore serves as a convenient and sensitive marker for such activity.
Accordingly, brain, lung and liver from 40 African children who had died of malaria, sepsis or unrelated conditions were stained for HO-1 in order to identify cellular sites where the CO-mediated anti-inflammatory activity of IL-10 might be located in these infectious diseases. These tissues had previously been stained for migration inhibitory factor (MIF) and inducible nitric oxide synthase (iNOS) [20]. Before the IL-10 or prostaglandin links of HO-1 were appreciated, others [21, 22] have immunostained brains, but no other tissues, from adult malaria cases, to detect this enzyme. The present study provides further evidence for the presence of multi-organ inflammatory changes in children fulfilling the clinical criteria of 'cerebral malaria', whether or not malaria was the principal or only pathological diagnosis.
Materials and Methods
Case Tissues
As described earlier [20], all 40 subjects (age range six months to 12 years; 22 females) were children who had been admitted to the Malaria Project wards in the Department of Paediatrics at the Queen Elizabeth Central Hospital in Blantyre, Malawi (Table). Evaluation, diagnoses, treatment, autopsy permissions were as previously described [20]. Autopsies were performed as quickly after death as possible, with post-mortem intervals ranging from two to 14.5 hrs. Tissue samples were placed into 10% neutral buffered formalin for fixation. The project was approved by the ethics and research boards of the College of Medicine (University of Malawi), the University of Liverpool and the Australian National University.
Control tissues
Tissues from three Malawian children who were enrolled in this study served as local non-comatose controls. No coma was present at any stage in two of these (patients 41 and 50; see Table). The former grew Salmonella typhimurium from cerebrospinal fluid and blood, and died, having been alert a short time before, after an acute gastrointestinal haemorrhage, and the other grew scanty Streptococcus pneumoniae from the cerebrospinal fluid. The third (patient 43) had been diagnosed as cerebral malaria but, after recovering to an alert state, died from a cardiopulmonary arrest. In addition, various adult controls from Australian sources were studied. These comprised sections of five blocks of tissue, trimmed from the periphery of tumour excisions from adult chest wall, and containing skeletal muscle, adipose tissue and small blood vessels. Midbrain sections from three adults who had died of coronary artery disease, and from another three who died of non-infectious, non-cerebral conditions (Brain Bank for Sydney Central Area Health Science Approval X980216), were also stained. A section of an inflamed pilonidal sinus was routinely included as a positive control.
Immunohistochemistry
Formalin-fixed tissue samples were embedded in paraffin, sectioned (4 microns) on to polylysine-coated slides, and stained with haematoxylin and eosin (H&E) for routine morphology. A monoclonal anti-HO-1 antibody was purchased from StressGen; (Cat. No. OSA-110). Other monoclonals were used as irrelevant primary control antibodies, and in other controls the primary antibody was omitted. As previously [20], antigen retrieval was performed by immersion in 0.01 M citrate buffer, pH 6.0, in a waterbath at 95°C for 20 min and then cooling to room temperature while still immersed in buffer. After quenching with 3% H2O2 and treating with primary antibody (dilution of the stock solution 1:500 to 1:2000) at room temperature for 1 hr, biotin-conjugated secondary antibody and streptavidin-conjugated horseradish peroxidase from an LSAB+ kit (DAKO) were applied to sections for 20 min at room temperature to amplify the antigen signal for subsequent 3,3'-diaminobenzidine (DAB) staining. Known positive controls were stained in each run, and runs were often duplicated on different days to confirm repeatability. Sections were counterstained with haematoxylin, and outcomes with a dilution of primary antibody of 1:1000 are shown to illustrate the observed changes. Anti-CD68 antibody (Clone PG-M1) was obtained from DAKO, and used, with antigen retrieval, at a primary antibody dilution of 1:500.
Histological examination
In a recent investigation of the distribution of MIF and iNOS [20], in which 32 cases that had been clinically diagnosed as cerebral malaria were studied, they were classified into three categories on the basis of the presence or absence of sequestered intracerebral parasites and brain pathology. Category A (n = 11) had no or scanty intracerebral parasites and negligible brain pathology detected, category B (n=seven) had sequestered parasites in brain vessels, again with negligible brain pathology, and category C (n = 14) had both sequestered parasites and inflammatory brain pathology in the form of intravascular monocyte aggregations, fibrin deposition and/or microhaemorrhages. Here this same terminology is retained, and the outcome of immunostaining to detect HO-1 in the brain, lung and liver of these same cases is reported. In most of the category A patients, autopsy revealed another likely cause of death: this was pneumonia in five cases, hepatic necrosis in one, severe anaemia with pulmonary oedema in one, and ruptured cerebral aneurysm in one. No alternative causes were identified in category B and C patients.
One hundred and forty sections from 49 brains were stained for HO-1 and examined. Samples were from frontal lobe, parietal lobe, temporal lobe, occipital calcarine fissure, hippocampus, caudate nucleus, basal ganglia, thalamus, midbrain, pons and medulla, with frontal lobe, occipital region, midbrain and pons most commonly included. Two sections were considered ample to record a result on sections where staining was readily detected, since up to seven were examined in some cases, with no difference in outcome from the opinion formed after the first section. Up to seven sections per brain were examined in those cases with no staining detectable, and these comprised sections from two to four blocks, which provided an element of depth within individual blocks as well as a spread of location across the brain. With few exceptions, staining was either absent, moderate, or strong, and if strong was sometimes remarkably intense and even, despite the high dilution of primary antibody. Mid-brain sections from three adults who had died of coronary artery disease, and from another three who died of non-infectious, non-cerebral conditions (Brain Bank for Sydney Central Area Health Science Approval X980216), were included. Single blocks only were available for lung and liver. Two examiners (IC and CH), blinded to the diagnosis, examined the sections independently.
Results
Cerebral malaria
Brain of the 32 cases clinically diagnosed as cerebral malaria were examined after staining with anti-HO-1 antibody as described. Differences in degree of staining between the various parts of the brain in any single individual were slight to negligible, with the picture observed in the first section examined holding true throughout the series for that case. Staining was restricted to monocytes, and where microhaemorrhages were present HO-1 was detected in the macrophages that migrated into the immediate area. In contrast to iNOS staining, a characteristic of HO-1 detection was its either strong presence or absence, with little between. Only single blocks of lung and liver were available, and these stained uniformly across the section. Staining in lung was restricted to monocytes and alveolar macrophages, and in liver to Kupffer cells, except for a very pale even stain across the parenchyma of a few livers.
Category A cerebral malaria
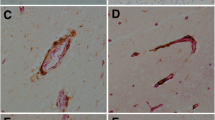
In the 11 cases that comprised category A (no discernible histological changes, only the occasional rare malarial pigment or parasites within vessels, and in most cases an additional explanation for death identified at general autopsy) eight had no detectable HO-1 staining. in the brain (Table 1). One showed isolated moderately stained monocytes, and the other two showed a light scattering of strongly stained monocytes across the brain parenchyma. Examples are shown in Fig. 1. The identity of these cells was confirmed with CD68 staining (not shown). Of the 11 lung sections, six exhibited strongly stained monocytes and alveolar macrophages (Fig. 1), two were pale, and one was negative. In the other two the section was largely obscured by a heavy influx of neutrophils, a cell type we have not seen stain for HO-1. Nine of the 11 liver sections showed strongly staining Kupffer cells (Fig. 1), and in the other two these cells were moderately stained only.

CM (A). HO-1 staining of tissues from cerebral malaria cases showing apparently absent physical brain pathology. A and B brain, C and D lung, and E and F liver, the left hand column (A, C and E) being from case 22, an example of the more common (8 of 11) combination, in which cerebral vasculature monocytes were quiescent. B, D and F are from case 38. As in both examples, monocytes in lung, and Kupffer cells, were commonly (6 of 11 and 9 of 11 respectively) strongly stained. Scale bar, 100 μm.
Category B cerebral malaria
In the seven category B cases (no discernible pathological changes in the examined sections, but sequestered parasitised erythrocytes present) 2 had rare strongly staining monocytes in cerebral blood vessels, and another four showed a scattering of strongly stained monocytes (Table 1). The identity of these cells was confirmed with CD68 staining (not shown) An example of each type of density is shown in Fig. 2. Five out of seven lung sections showed strong staining of monocytes and alveolar macrophages, one was moderately stained, and the other section was largely obscured by a heavy influx of neutrophils, with no HO-1 staining discernible. All six of the available seven liver sections showed strong staining of Kupffer cells, again as illustrated in Fig. 2.

CM (B). HO-1 staining of tissues from cerebral malaria cases showing apparently absent physical brain pathology, but sequestered parasites common. A and B brain, C and D lung, and E and F liver. The left hand column (A, C and E) is from an example (case 25) of the more common (4 of 7) combination, in which cerebral vasculature monocytes were not common, though parasite sequestration sometimes intense. B, D and F are from case 21. As in both examples, monocytes in lung, and Kupffer cells, were commonly strongly stained. Scale bar, 100 μm.
Category C cerebral malaria
The brains of 14 category C cases (microhaemorrhages, intravascular mononuclear cell accumulations, and also sequestered parasites) all exhibited strong monocyte HO-1 staining in mononuclear leucocytes forming accumulations, and also in those scattered singly, within blood vessels (Table 1). The two cases illustrated in Fig. 3 had neither a low haematocrit, nor hypoglycaemia. Monocyte accumulations were not seen in sections of two of the brains, and thus the individual cells are referred to in the Table as "scattered, strong". In addition the macrophages that had aggregated at microhaemorrhages were stained strongly (Fig. 3). The identity of these cells was confirmed with CD68 staining (not shown). Eleven of the 13 available lung sections showed strongly staining monocytes and alveolar macrophages (Fig. 3), with the monocytes often in accumulations similar to those observed in the brain. One section was moderately stained, and the other section was largely obscured by a heavy influx of neutrophils. As shown in the examples in Fig. 3, all of the 14 livers had strongly stained Kupffer cells.

CM (C). HO-1 staining of tissues from cerebral malaria cases showing physically apparent brain pathology, plus sequestered parasites present. A and B brain, C and D lung, and E and F liver. Both case 9 (A, C and E) and case 26 (B, D and F) show the brain monocyte, lung monocyte and macrophage, and Kupffer cell staining that dominate this group. Scale bar, 100 μm.
Coma associated with bacterial infections
Another five patients were comatose on admission and before death, had very low or no peripheral malaria parasitaemia, and were considered on clinical grounds to be suffering from a disease other than malaria [20]. Two had septic meningitis (one each culture positive for Haemophilus influenzae and S. pneumoniae), two had a positive blood culture (one Salmonella enteritidis, one Escherichia coli) and one, with a lymphocytic infiltrate in the cerebrospinal fluid, was diagnosed histologically, including the presence of acid-fast organisms, as tuberculous meningitis. The E. coli and H. influenzae cases had no detectable HO-1 staining. in the brain, and the other three showed a variable scattering of stained monocytes (Table 1). In the tuberculosis case staining was limited to the areas adjacent to large blood vessels. Two of the five lung sections (S. enteritidis and E. coli cases) showed strongly staining monocytes and alveolar macrophages, while none were evident in the H. influenzae and S. pneumonia cases, and they were restricted to the region near chronic lesions in the tuberculosis case. All five livers showed HO-1 staining of the Kupffer cells, although in one case this was moderate rather than strong (Table 1). As examples, the changes in brain, lung and liver of the E. coli and H. influenzae cases are shown in Fig. 4.

HO-1 staining of tissues from bacterial sepsis cases. A and B brain, C and D lung, and E and F liver. Case14 (A, C and E), an E. coli infection, demonstrates no brain monocyte activity (A), but pulmonary monocytes and macrophages (C), and Kupffer cells (E) stained strongly. While case 18 (B, D and F), a H. influenzae infection, had no HO-1 staining in the brain or lung, Kupffer cells stained strongly. Scale bar, 100 μm.
Control Tissues
Brain sections from all three African controls, and lung sections from two of them, and the liver of one, showed no staining for HO-1 (Table 1, Fig. 5). The lung of the one, and the liver of two, showed few, palely stained cells only. Likewise, the 6 Australian-origin adult brains showed no staining, whether the patients had died from coronary artery disease or non-infectious, non-cerebral conditions (Fig. 5G). The subcutaneous tissues from Australian adult controls were also negative (not shown). In contrast, macrophages in the section of inflamed pilonidal sinus included in every staining run invariably were strongly positive (Fig. 5H), as were the macrophages in the splenic red pulp, a location where free haem can be expected to induce HO-1, in all African cases (not shown).

HO-1 staining of tissues from controls. A and B brain, C and D lung, and E and F liver of African controls, cases 41 (A, C and E) and 43 (B, D and F). Australian origin coronary artery disease brain (G) and chronically infected pilonidal sinus (H) are also shown. Pattern shown was the same in all cases illustrated. Scale bar, 100 μm.
CD 68 staining
CD36 staining confirmed the identity of the mononuclear phagocytic lineage of stained cells, including the cells attracted to ring haemorrhages, that were positive for HO-1. Both single and double staining was undertaken (not shown). This confirmed that negative HO-1 staining of tissues could not be attributed to an absolute absence of monocytes and macrophages in various locations, but to them being present and not staining.
Discussion
Here is described the cellular distribution of HO-1 in several key organs in African children who died of clinically defined cerebral malaria or coma accompanying a bacterial infection. Endothelial cells and vascular smooth muscle, and skeletal muscle, so often MIF and iNOS positive [20], were devoid of HO-1. Previous reports of HO-1 staining of human tissues includes the cytotrophoblast cells within the placental bed of the normal human placenta [23], the alveolar macrophages of normal and inflamed lung [24], endothelium and macrophages of atherosclerosis lesions [25], Kupffer cells and hepatocytes in portal hypertension [26], tubular epithelial cells in kidney diseases [27], and microglia and macrophages in focal cerebral infarcts and brain trauma [28]. Malarial brains, but no other organ, from Asian [22] and European adults [21] have been stained for HO-1, and, consistent with the literature of the time, HO-1 was discussed only in terms of being a stress protein [22] and a generator of CO that could contribute to cerebral malaria by influencing neurons directly [21].
What induced HO-1 in these tissues remains unclear. It is well accepted that a high concentration of haem induces HO-1 [29], and the invariable staining observed in spleen red pulp in all cases (not shown), and at cerebral haemorrhage sites where red cells have been phagocytosed (Fig. 3B) in category C brains is consistent with this. Haem from local haemorrhage cannot, however, account for the presence of HO-1 in mononuclear phagocytes in CM(A) or CM(B) brains, and other organs In addition, cellular deprivation of glucose has been shown to induce the HO-gene 1 in vitro [30], raising the possibility of the hypoglycaemia sometimes seen in severe childhood falciparum malaria being instrumental in generating the observed HO-1. No difference was seen in the intensity of HO-1 staining between cases with normal peripheral blood glucose concentration and those with systemic hypoglycaemia, but this observation does not exclude local tissue hypoglycaemia as a possible stimulus to HO-1 induction. IL-10, a major modulator of inflammation increased in malaria [10], is now known to induce HO-1 [14]. Plasmas for IL-10 assay were not available from these cases, but the precedence exists of high levels being associated with severe malarial illness [10, 11].
In view of the evidence that the inflammatory cascade is activated in this Malawian [20, 31] and other [32–34] populations with severe malaria infection, the observed HO-1 is additional evidence that in fatal malaria a widespread host inflammatory response occurs, similar to that seen in other acute infections (reviewed in [35]). This is consistent with the HO-1 staining seen in the sepsis cases in this series (Fig. 4), in that systemic bacterial infections are broadly accepted to be examples of systemic inflammation. This is not to suggest that some of the cerebral malaria cases in this series did not have additional or alternative disease mechanisms, arising from local cerebral lesions, such as vessel obstruction from large parasite loads in the apparent absence of other lesions (Fig. 2B). Nevertheless, strong systemic inflammation was present in all these cases. Local cerebral changes arising from post-schizogony secondary inflammatory events, as demonstrated by the presence of monocytes accumulations, high iNOS, haemorrhages and fibrin in other CM(C) cases (Fig. 3A and 3B) may well also contribute significantly.
Taken with the evidence that eight of the 11 category A brains had no evidence of HO-1 staining, these results, along with the apparent absence of sequestration or monocytic accumulation (now reinforced by examining this large series of new sections) and iNOS staining in these same brains [20], add to previous evidence [20] that these 11 sets of brains sections, one third of the total from clinically diagnosed cases of cerebral malaria, were largely devoid of significant intravascular change. These patients, together with those in the group of patients with non-malarial comatose illness, illustrate the fact that coma may accompany infections with extensive systemic inflammation but without evidence of intracerebral inflammatory responses. It is, therefore, possible that, in the CM(B) and CM(C) patients (with cerebral sequestration and cerebral intravascular HO-1 induction), the observed widespread systemic inflammation may have been similarly important in the pathogenesis of coma. This adds strength to the argument that systemic events, not only those in the brain, should be borne in mind when attempting to understand disease outcome in African children clinically diagnosed with cerebral malaria. The CM(A) group, one third of the cases, in which malaria was only one of several conditions that could have contributed to death, might have provided insight into the state the cerebral vasculature of the CM(B) and CM(C) cases after they had become comatose, but some time before death.
Finally, the inhibitory activity of CO, the product of HO-1, against TNF [4] parallels that of NO, the product of iNOS [5]. Ideas on how iNOS polymorphisms might have been selected for in African populations [36, 37] through their interaction with malarial disease might also, from these results, apply to known HO-1 polymorphisms [38, 39] in these populations.
References
Boczkowski J, Lanone S, Ungureanulongrois D, Danialou G, Fournier T, Aubier M: Induction of diaphragmatic nitric oxide synthase after endotoxin administration in rats – role in diaphragmatic contractile dysfunction. J Clin Invest. 1996, 98: 1550-1559.
Lanone S, Mebazaa A, Heymes C, Henin D, Poderoso JJ, Panis Y, Zedda C, Billiar T, Payen D, Aubier M: Muscular contractile failure in septic patients – Role of the inducible nitric oxide synthase pathway. Am J Resp Crit Care Med. 2000, 162: 2308-2315.
Mitaka C, Hirata Y, Yokoyama K, Wakimoto H, Hirokawa M, Nosaka T, Imai T: Relationships of circulating nitrite/nitrate levels to severity and multiple organ dysfunction syndrome in systemic inflammatory response syndrome. Shock. 2003, 19: 305-309. 10.1097/00024382-200304000-00002.
Peng HB, Libby P, Liao JK: Induction and stabilization of I kappa B alpha by nitric oxide mediates inhibition of NF-kappa B. J Biol Chem. 1995, 270: 14214-14219. 10.1074/jbc.270.23.14214.
Otterbein LE, Bach FH, Alam J, Soares M, Tao Lu H, Wysk M, Davis RJ, Flavell RA, Choi AM: Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000, 6: 422-428. 10.1038/74680.
Sarady JK, Otterbein SL, Liu F, Otterbein LE, Choi AM: Carbon monoxide modulates endotoxin-induced production of granulocyte macrophage colony-stimulating factor in macrophages. Am J Respir Cell Mol Biol. 2002, 27: 739-745.
Hartsfield CL: Cross talk between carbon monoxide and nitric oxide. Antioxid Redox Signal. 2002, 4: 301-307. 10.1089/152308602753666352.
Schmidt HHHW: NO., CO and .OH – Endogenous soluble guanylyl cyclase-activating factors. FEBS Letters. 1992, 307: 102-107. 10.1016/0014-5793(92)80910-9.
Wang P, Wu P, Siegel MI, Egan RW, Billah MM: Interleukin (IL)-10 inhibits nuclear factor kappa B (NF kappa B) activation in human monocytes. IL-10 and IL-4 suppress cytokine synthesis by different mechanisms. J Biol Chem. 1995, 270: 9558-9563. 10.1074/jbc.270.16.9558.
Peyron F, Burdin N, Ringwald P, Vuillez JP, Rousset F, Banchereau J: High levels of circulating IL-10 in human malaria. Clin Exp Immunol. 1994, 95: 300-303.
Wenisch C, Parschalk B, Narzt E, Looareesuwan S, Graninger W: Elevated serum levels of IL-10 and IFN-gamma in patients with acute Plasmodium falciparum malaria. Clin Immunol Immunopathol. 1995, 74: 115-117. 10.1006/clin.1995.1017.
Lin RY, Astiz ME, Saxon JC, Saha DC, Rackow EC: Relationships between plasma cytokine concentrations and leukocyte functional antigen expression in patients with sepsis. Crit Care Med. 1994, 22: 1595-1602.
Ho M, Sexton MM, Tongtawe P, Looareesuwan S, Suntharasamai P, Webster HK: Interleukin-10 inhibits tumor necrosis factor production but not antigen-specific lymphoproliferation in acute Plasmodium falciparum malaria. J Infect Dis. 1995, 172: 838-844.
Lee TS, Chau LY: Heme oxygenase-1 mediates the anti-inflammatory effect of IL-10 in mice. Nat Med. 2002, 8: 240-246. 10.1038/nm0302-240.
Cantoni L, Rossi C, Rizzardini M, Gadina M, Gheezi P: Interleukin-1 and tumor necrosis factor induce hepatic haem oxygenase. Biochem J. 1991, 891-894.
Platzer C, Meisel C, Vogt K, Platzer M, Volk HD: Up-regulation of monocytic IL-10 by tumor necrosis factor-alpha and cAMP elevating drugs. Int Immunol. 1995, 7: 517-523.
Gilroy DW, Colville Nash PR, Willis D, Chivers J, Paul Clark MJ, Willoughby DA: Inducible cyclooxygenase may have anti-inflammatory properties. Nat Med. 1999, 5: 698-701. 10.1038/9550.
Lee TS, Tsai HL, Chau LY: Induction of heme oxygenase-1 expression in murine macrophages is essential for the anti-inflammatory effect of low dose 15-deoxy-Delta(12,14)-prostaglandin J(2). J Biol Chem. 2003, 278: 19325-19330. 10.1074/jbc.M300498200.
Deininger MH, Kremsner PG, Meyermann R, Schluesener HJ: Focal accumulation of cyclooxygenase-1 (COX-1) and COX-2 expressing cells in cerebral malaria. J Neuroimmunol. 2000, 106: 198-205. 10.1016/S0165-5728(00)00187-9.
Clark IA, Awburn MM, Whitten RO, Harper CG, Liomba NG, Molyneux ME, Taylor TE: Tissue distribution of migration inhibitory factor and inducible nitric oxide synthase in falciparum malaria and sepsis in African children. Malaria J. 2003, 2: 6-10.1186/1475-2875-2-6.
Schluesener HJ, Kremsner PG, Meyermann R: Heme oxygenase-1 in lesions of human cerebral malaria. Acta Neuropathol. 2001, 101: 65-68.
Medana IM, Mai NTH, Day NPJ, Hien TT, Bethell D, Phu NH, Farrar J, White NJ, Turner GDH: Cellular stress and injury responses in the brains of adult Vietnamese patients with fatal Plasmodium falciparum malaria. Neuropath Appl Neurobiol. 2001, 27: 421-433. 10.1046/j.0305-1846.2001.00360.x.
Lyall F, Barber A, Myatt L, Bulmer JN, Robson SC: Hemeoxygenase expression in human placenta and placental bed implies a role in regulation of trophoblast invasion and placental function. FASEB J. 2000, 14: 208-219.
Lakari E, Pylkas P, Pietarinen Runtti P, Paakko P, Soini Y, Kinnula VL: Expression and regulation of hemeoxygenase 1 in healthy human lung and interstitial lung disorders. Hum Pathol. 2001, 32: 1257-1263. 10.1053/hupa.2001.28937.
Wang LJ, Lee TS, Lee FY, Pai RC, Chau LY: Expression of heme oxygenase-1 in atherosclerotic lesions. Am J Pathol. 1998, 152: 711-720.
Makino N, Suematsu M, Sugiura Y, Morikawa H, Shiomi S, Goda N, Sano T, Nimura Y, Sugimachi K, Ishimura Y: Altered expression of heme oxygenase-1 in the livers of patients with portal hypertensive diseases. Hepatology. 2001, 33: 32-42. 10.1053/jhep.2001.21161.
Morimoto K, Ohta K, Yachie A, Yang Y, Shimizu M, Goto C, Toma T, Kasahara Y, Yokoyama H, Miyata T: Cytoprotective role of heme oxygenase (HO)-1 in human kidney with various renal diseases. Kidney Int. 2001, 60: 1858-1866. 10.1046/j.1523-1755.2001.01000.x.
Beschorner R, Adjodah D, Schwab JM, Mittelbronn M, Pedal I, Mattern R, Schluesener HJ, Meyermann R: Long-term expression of heme oxygenase-1 (HO-1, HSP-32) following focal cerebral infarctions and traumatic brain injury in humans. Acta Neuropathol. 2000, 100: 377-384. 10.1007/s004010000202.
Maines MD, Trakshel GM, Kutty RK: Characterization of two constitutive forms of rat liver microsomal heme oxygenase. Only one molecular species of the enzyme is inducible. J Biol Chem. 1986, 261: 411-419.
Chang SH, Garcia J, Melendez JA, Kilberg MS, Agarwal A: Haem oxygenase 1 gene induction by glucose deprivation is mediated by reactive oxygen species via the mitochondrial electron-transport chain. Biochem J. 2003, 371: 877-885. 10.1042/BJ20021731.
Grau GE, Taylor TE, Molyneux ME, Wirima JJ, Vassalli P, Hommel M, Lambert P-H: Tumor necrosis factor and disease severity in children with falciparum malaria. N Engl J Med. 1989, 320: 1586-1591.
Kern P, Hemmer CJ, Van Damme J, Gruss H-J, Dietrich M: Elevated tumour necrosis factor alpha and interleukin-6 serum levels as markers for complicated Plasmodium falciparum malaria. Am J Med. 1989, 87: 139-143.
Kwiatkowski D, Hill AVS, Sambou I, Twumasi P, Castracane J, Manogue KR, Cerami A, Brewster DR, Greenwood BM: TNF concentration in fatal cerebral, non-fatal cerebral, and uncomplicated Plasmodium falciparum malaria. Lancet. 1990, 336: 1201-1204. 10.1016/0140-6736(90)92827-5.
Butcher GA, Garland T, Adjukiewicz AB, Clark IA: Serum TNF associated with malaria in patients in the Solomon Islands. Trans R Soc Trop Med Hyg. 1990, 84: 658-661.
Clark IA, Cowden WB: The pathophysiology of falciparum malaria. Pharmacol Ther. 2003, 99: 221-260. 10.1016/S0163-7258(03)00060-3.
Burgner D, Xu WM, Rockett K, Gravenor M, Charles IG, Hill AV, Kwiatkowski D: Inducible nitric oxide synthase polymorphism and fatal cerebral malaria. Lancet. 1998, 352: 1193-1194. 10.1016/S0140-6736(98)09176-4.
Hobbs MR, Udhayakumar V, Levesque MC, Booth J, Roberts JM, Tkachuk AN, Pole A, Coon H, Kariuki S, Nahlen BL: A new NOS2 promoter polymorphism associated with increased nitric oxide production and protection from severe malaria in Tanzanian and Kenyan children. Lancet. 2002, 360: 1468-1475. 10.1016/S0140-6736(02)11474-7.
Yamada N, Yamaya M, Okinaga S, Nakayama K, Sekizawa K, Shibahara S, Sasaki H: Microsatellite polymorphism in the heme oxygenase-1 gene promoter is associated with susceptibility to emphysema. Am J Hum Genet. 2000, 66: 187-195. 10.1086/302729.
Schillinger M, Exner M, Mlekusch W, Domanovits H, Huber K, Mannhalter C, Wagner O, Minar E: Heme oxygenase-1 gene promoter polymorphism is associated with abdominal aortic aneurysm. Thromb Res. 2002, 106: 131-136. 10.1016/S0049-3848(02)00100-7.
Acknowledgments
This work was supported by the Australian National Health and Medical Research Council, Grant 148902, the National Institute of Allergy and Infectious Diseases, USA (Grant 1-RO1-AI 34969) and the Wellcome Trust. We thank Dr Biziwick Mwale, Director of the Queen Elizabeth Central Hospital, and Professor Robin Broadhead, Department of Paediatrics of the College of Medicine, University of Malawi, for their hospitality and for access to patients in the Malaria Ward, and we thank Professor Terrie Taylor, who played a major part in running the Severe Malaria Ward and in arranging and conducting autopsies. We appreciate the cooperation of parents and guardians of the children studied, and the devoted care provided by the nursing and laboratory staff within the Paediatric Department and the research programme. This study received funding from the Australian Health and Medical Research Council, the National Institutes of Health, USA, and The Wellcome Trust, UK.
Author information
Authors and Affiliations
Corresponding author
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Clark, I.A., Awburn, M.M., Harper, C.G. et al. Induction of HO-1 in tissue macrophages and monocytes in fatal falciparum malaria and sepsis. Malar J 2, 41 (2003). https://doi.org/10.1186/1475-2875-2-41
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-2-41