
Abstract
Background
The variant surface antigen family Plasmodium falciparum erythrocyte membrane protein-1 (PfEMP1) is an important target for protective immunity and is implicated in the pathology of malaria through its ability to adhere to host endothelial receptors. The sequence diversity and organization of the 3D7 PfEMP1 repertoire was investigated on the basis of the complete genome sequence.
Methods
Using two tree-building methods we analysed the coding and non-coding sequences of 3D7 var and rif genes as well as var genes of other parasite strains.
Results
var genes can be sub-grouped into three major groups (group A, B and C) and two intermediate groups B/A and B/C representing transitions between the three major groups. The best defined var group, group A, comprises telomeric genes transcribed towards the telomere encoding PfEMP1s with complex domain structures different from the 4-domain type dominant of groups B and C. Two sequences belonging to the var1 and var2 subfamilies formed independent groups. A rif subgroup transcribed towards the centromere was found neighbouring var genes of group A such that the rif and var 5' regions merged. This organization appeared to be unique for the group A var genes
Conclusion
The grouping of var genes implies that var gene recombination preferentially occurs within var gene groups and it is speculated that the groups reflect a functional diversification evolved to cope with the varying conditions of transmission and host immune response met by the parasite.
Similar content being viewed by others

Background
Plasmodium falciparum is the most virulent of the four species causing malaria and responsible for most malarial deaths. The particular virulence of P. falciparum is partly due to the ability of infected erythrocytes to adhere to a variety of host receptors and avoid splenic clearance[1, 2]. Unchecked growth and the accumulation of sequestered parasites in vital organs such as the brain[3] or placenta[4] are crucial elements in the pathogenesis of severe malaria[5]. CD36 is considered to be the major endothelial receptor for infected erythrocytes[6], but several other ligands have been identified, in particular ICAM-1[7], which has been associated with cerebral malaria and chondroitin sulfate A (CSA) associated with binding in the placenta and pregnancy-associated malaria (PAM) [8].
Plasmodium falciparum erythrocyte membrane protein-1 (PfEMP1) is a polymorphic family of high molecular weight adhesion antigens expressed on the surface of infected erythrocytes[9]. The accumulation of antibodies against a broad repertoire of PfEMP1s is probably the functional basis for the natural acquisition of immunity to malaria [10–13].
PfEMP1 antigens are encoded by the var gene family in two exon units[9, 14, 15]. Exon I codes for the extracellular and variable part of the protein as well as a transmembrane region and Exon II encodes the intracellular and relatively conserved acidic terminal segment (ATS). The most variable part of the protein contains a N-terminal segment followed by segments composed of three domain types; Duffy binding-like domains (DBL-domains): Cysteine-rich inter-domain regions (CIDRs) and C2 [16]. Besides the 59 full-length var genes found in the newly sequenced genome of P. falciparum clone 3D7 [17], the complete domain structures of PfEMP1s are only available in the databases for a handful of var genes from other P. falciparum isolates. The extent to which we can extrapolate from the organisation of 3D7 var genes, to the total diversity of PfEMP1 in the diverse global population of P. falciparum therefore remains somewhat uncertain.
P. falciparum genomes are estimated to contain 50 to 60 var genes. In the case of 3D7 these have been grouped into three major types based on sequence analysis of the intron and 5' and 3' un-translated regions (UTR) [17–19]. In a recent functional study, it was shown that the ability of CIDR domains to bind CD36 could be predicted on the basis of sequence analysis and that binding and non-binding domains fell into two separate CIDR clusters [20]. The rif genes constitute another multigene family which has 149 members in the 3D7 genome. They encode proteins (RIFINs) exposed on the surface of infected erythrocytes [21, 22]. The functions of these proteins are not known and they have not been shown to mediate binding.
With the completion of the 3D7 genome[17], it has become possible to study a complete PfEMP1 and RIFIN repertoire of a single genome. We have analysed both coding and non-coding regions of 3D7 var and rif genes and assigned the var genes into different groups. These groups appear evolutionarily conserved, possibly because selection favours gene segments 'shuffling' within particular groups, but not exchanges between different groups. We speculate that these PfEMP1 groups have arisen as a result of diversifying selection for antigenic divergence being superimposed on strong selective constraints maintaining a particular ligand-receptor binding interaction.
Methods
Sequences
Nucleotide and deduced amino acid sequences as well as location and transcriptional directions of 3D7 var and rif genes were obtained from the Plasmodium Genome Resource – http://www.plasmodb.org[23]. RT-PCR, cloning and sequencing of 5' flanking regions of var2 genes was performed as previously described[24].
Alignments – defining homologous sequence boundaries
Alignments were performed using the ClustalW multiple alignment method, European Molecular Biology Laboratory, Heidelberg, Germany at default parameters (Gap Open: 10.00; Gap Extention: 0.20, Gap Separation Distance: 4, Protein weight matrix: Gonnet, DNA weight matrix: identity (IUB)). Alignments were corrected by hand using Bioedit[25] to assure homologous sequences for sequence analysis and tree-building. For distance tree-building, the var gene 5' flanking regions were defined as the 600 bp, 1.4 kb or 2.0 kb upstream of the translation initiation codon and the 3' flanking region as the 500 bp downstream of the translation stop codon. The available var flanking sequences from other strains than 3D7 varied between 250 and 2100 bp in length. For the most part 3' sequences were those retrieved by Mercereau-Puijalon et al [19]. Var gene domain structures were defined using definitions described in Smith et al. [16]. DBLαCIDR1 domains were aligned from Pro-Cys (PC) of DBLα homology block A to the conserved Glu-Trp (EW) motif of CIDR M2 area, resulting in sequence lengths of 550 to 650 aa. The exon II encoded acidic terminal segments (ATS) were aligned over their entire sequences. For analysis of DBL relationships sequences covering Pro-X-Arg-Arg (PXRR) of DBL homology block B to Glu-Trp (EW) of homology block H were aligned. RIFINs were aligned over the full-length protein sequences.
Distance tree analysis
Distance trees were constructed the by p-distance/neighbour-joining (NJ) method as well as maximum parsimony (MP) using MEGA version 2.1[26]. Trees were bootstrapped 1000 times and compared between NJ and MP tree-building methods to assure confidence in topology. Observed clusters from each tree were confirmed visually on alignments.
Results
Sequence analysis of var 5' regions
The 1.5 kb 5' region of 3D7 var genes has previously been described to group into three major sequence groups, upsA, upsB and upsC [17]. To further investigate sequence similarities in this region, we analysed the 2 kb upstream sequences of all 3D7 var genes and the pseudo var gene PFE1640w. In agreement with Gardner et al. [17] the alignments revealed three major sequence groups with high similarity between sequences of each group. However, two sequences did not align well with any of the groups and within the groups, subgroups could be identified. Thus, each of the groups were analysed separately.
UpsA
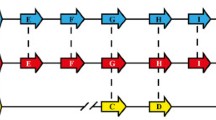
Ten var genes had 5' regions belonging to this group and all but one were positioned head-to-head with a rif gene, the exception PF08_0141 was head-to-head with another var gene (figure 1). The distance from the start codons of the var to the rif gene was either 3 or 4 kb. From the var gene translation initiation codon to about 1.2 kb upstream the ten sequences were almost identical up to a stretch of TA repeats, which we propose identifies the 5' end of the upsA. This conclusion is based on analysis of the sequences further upstream from the TA repeats, which probably identifies 5' regions of the neighbouring genes. Thus, six sequences were almost identical until the translation initiation codon of the flanking rif gene. Another group of three sequences also shared this similarity but had an insertion of around 1 kb at the proposed upsA end. Characteristic of this group is that the PfEMP1s consist of only two DBL domains. The last sequence flanking PF08_0141 constitutes the upstream region of another var gene.

Schematic representation of head-to-head genomic organisation of rif and upsA flanked var genes. Nine genes are flanked by a rif gene, which has its initiation codon approximately 3 or 4 kb upstream from the var initiation codon, and one var gene by another var gene at -2 kb. Punctured lines represent upsA, dotted lines upsA-rif and full line upsBsh. The diamond marks the putative termination site of upsA characterised by a stretch of TA repeats. Sizes of genes are not in scale.
Sequences with high similarity to the 3D7 upsB and upsC regions have been found in other parasite isolates[18]. Regions with similarity to upsA have yet to be found in other parasites than 3D7. Using a primer set targeting upsA 3D7 sequences around -900 bp from the translation initiation codon, we could PCR amplify products of the expected sizes in 3D7 genomic DNA, as well as in five of five field-isolates tested (data not shown). Although sequence data are not available this data suggests that upsA regions not are unique to 3D7.
UpsB
35 var genes of which 22 genes are telomeric had 5' regions belonging to the upsB group. 33 upsB sequences were highly similar over approximately 650 bp from the var gene translation initiation site until a polyT15–20 stretch. The upsB region of the telomeric genes was very similar over the entire region between the coding var sequence and the repetitive elements of the telomere. Eleven sequences were highly similar over varying shorter stretches downstream to the 650 bp. In most of these cases similarity was abruptly lost at polyT15–20 stretches. These upsB regions were defined as upsBsh. In the case of PF08_140 and MAL6P1.316 the upstream region shifts from upsB to upsA and enabling the identification of a minimal observed upsB length of 650 bp. In the case of MAL6P1.316 the flanking upsA related gene is the var pseudogene MAL6P1.317. The similarity of MAL6P1.316 and PFL1950w upstream sequences to other upsB sequences were atypically low and fragmented.
UpsC
13 var genes had 5' regions belonging to this group. The 10 closest related sequences of upsC aligned until a stretch of TA repeats around -4 kb from the translation initiation codon. The remaining three upsC sequences showed high upsC sequence similarity through the first 400 bp, but relatively low similarity over short stretches upstream. These were marked upsC^ in figure 7.
Ups D
PFE1640w did not group with any of the other 5' sequences and was classified as upsD. PFE1640w is a pseudogene with high similarity to the var1 sub-family of var genes present in many parasite genomes [27, 28]. The 5' region of the var1 sub-family is also conserved across parasite isolates [29]. DBL3γ regions of the var1 family can bind CSA [30] and antibodies against these can prevent CSA mediated parasite binding to brain and lung endothelial cells[31], However other studies have failed to show any up-regulation of this gene after CSA selection[24, 32].
Ups E
The 5' region of PFL0030c was unique among the 5' sequences analysed. It was therefore classified as upsE. It has recently been demonstrated that PFL0030c belongs to another family of conserved var genes, the var2 sub-family[24, 32]. var2 is the dominant var gene transcribed in CSA binding parasites and is also transcribed at high levels in parasites isolated from placentas[24, 32]. The PFL0030c 5' region was also found upstream of the pseudogene MAL13P1.354, which apart from single frameshift in exonI is almost identical to PFL0030c. Interestingly, unlike any other var gene PFL0030c appears to have an upstream open reading frame (uORF), which encodes a 119 amino acid sequence, which ends 274 bp before the start codon. To examine whether this gene segment was an exon of PFL0030c, a single transcript or a real uORF, RT-PCR was performed with primers targeting sequences on each side of the segment. A product of the expected size was amplified indicating that the uORF and PFL0030c were situated on the same mRNA molecules and that splicing had not occurred during the transcription process. This conclusion was supported by real time quantitative RT-PCR experiments showing that transcription of the segment with the uORF was up-regulated 77 times in NF54 parasites selected for CSA binding compared to the transcription in the parental non selected line. This up regulation corresponded to that observed when using primers targeting other parts of the PFL0030c gene (data not shown). To investigate whether upsE is present in other parasite genomes, the region spanning the uORF sequence to the transcription initiation site of PFL0030c from five parasites was PCR amplified, cloned and sequenced. All five parasites had both the uORF and the var2 gene, and a near 100% bp similarity was seen throughout the sequenced region.
Voss et al.[18] identified a 30 bp degenerated motif common to upsB and upsC sequences at -637 and -1227 from var translation initiation site in upsB and upsC, respectively. A similar but not identical motif was also found in the upsA at -675 to -850. In upsE the motif was found at -1672 from var ATG and -1058 from the uORF ATG. The consensus motif common to upsA, most upsB, upsC and upsE was A(T/G)A (C/A/T)AT AT(T/G) (G/A)TA GAT A(A/G)T A(A/G/C)(A/G) GAT AGA (A/G)A(A/G). This motif was not found in upsD or the upsBsh of PF08_140, MAL6P1.316 and PF08_103. In another paper, Voss et al. identifies three conserved var promoter motifs interacting with distinct DNA-binding proteins[33]. Subtelomeric var gene promoter element 1 and 2 (SPE1 and SPE2) are associated with upsB sequences and the chromosome-central var gene promoter element (CPE) with upsC. We identified the SPE1 and SPE2 elements in all upsB but not upsBsh or upsB^ sequences. The CPE was found in all upsC sequences except the three upsC^ sequences with relatively low upsC similarity. None of the elements were found in upsA, upsD or upsE.
Sequence analysis of var 3' regions
Alignments and tree-building (figure 2) of the 500 bp var 3' regions divided most sequences into four clusters (A-D). 13 sequences fell outside these clusters, and the relationship between these sequences could not be confirmed by bootstrapping or comparison the two tree-building methods used.

Distance tree of 3D7 var gene 500 bp 3' region generated using the p-distance/NJ method. The four dense clusters A through D were supported by both bootstrapping and maximum parsimony (MP) tree (not shown). The relationship of the remaining sequences could not be verified by the MP tree making method. Numbers at the nodes represent bootstrap proportions (BP) on 1000 replicates. The scale bar represents the proportion of different nucleotide compared. PlasmoDB accession numbers are shown. Genes with assigned cluster are collected in figure 7.
var gene introns
Calderwood et al. [34]have recently shown that var gene introns can be divided into 3 regions on basis of nucleotide composition. Selecting a representative subset of var genes we analysed 30 intron sequences manually and identified the three-region structure in all introns except the three smallest var genes with a two-DBL-domain structure. These have unusual short introns, which appears to mainly consist of region 2. Though alignments showed high similarity between four intron sequences belonging to var genes with a upsA 5' region, it was not possible to group all 3D7 var gene intron sequences on the basis of tree building methods.
Sequence analysis of coding var gene regions
Because most PfEMP1 molecules contain a semi-conserved head structure comprising of DBL1α and CIDR1, and all contain the acidic terminal segment (ATS) encoded by exon II, we restricted the analysis of coding sequences to these domains. In 3D7 all but one var gene encode a DBLlα as the first domain and in all but four genes DBL1 is followed by a CIDR1. Since alignment and tree constructions of DBL1 and CIDR1 domains individually yielded almost identical clusters, we decided to analyse the head structure sequences from the N terminal region of DBL1 to a conserved motif in the C-terminal region of CIDR1 (figure 3). Fifty-two sequences, including that of pseudogene PFE1640w, could be grouped into five clusters, and four sequences could not be assigned any of these. When all CIDR sequences are aligned most CIDR1s fall into separate clusters of CIDRα or CIDRα1 domains[20]. The exceptions are three sequences (PF08_0141, PF11_0008, PF13_0003), which fall into a CIDRγ cluster. In figure 3, the head structures of these genes fall into group A. Robinson et al. [20] found that most CIDR domains bind CD36 but identified nine, which did not. These constitute cluster A. In the analysis of DBLα domains three small almost identical PfEMP1s (PFA0015c, MAL6P1.314, PFI1820w) clustered with the DBL1α of group A (data not shown). Thus, we named their DBL1 "A*" (Figure 7). Alignments and tree construction of all var ATS sequences (figure 4) identified four clusters. Though less well defined by bootstrap proportions the clusters were supported by both NJ and MP tree making methods. Four sequences could not be assigned to any of these clusters.

Distance tree of DBLαCIDR1 domains of 3D7 PfEMP1 and pseudogene PFE1640w generated using the p-distance/NJ method. The clusters A through E were supported by both bootstrapping and maximum parsimony tree (not shown). Numbers at the nodes represent bootstrap proportions (BP) on 1000 replicates. The scale bar represents the proportion of different amino acids compared. PlasmoDB accession numbers are shown. Genes with assigned cluster are collected in figure 7.

Distance tree of ATS domains of 3D7 PfEMP1 generated using the p-distance/NJ method. Though A, B and C clusters were vaguely supported by the bootstrapping, they were supported by maximum parsimony tree making. Numbers at the nodes represent bootstrap proportions (BP) on 1000 replicates. The scale bar represents the proportion of different amino acids compared. PlasmoDB accession numbers are shown. Genes with assigned cluster are collected in figure 7.
Sequence analysis of DBL domains
Among the var genes of 3D7 PFL0030c (var2) differs markedly with respect to domain structure and 5' region. The gene does not encode a CIDR domain and of the six encoded DBL domains, three have previously been classified as DBLx [17], indicating that they along with ten other DBL domains did not fit into the existing classification by Smith et al. [16]. In attempt to classify these domains we randomly choose 50 DBL domains to represent DBLα-ε and figure 5 shows a tree of these together with the DBLx domains. Although the analysis was based on 3D7 sequences only and cannot be considered definitive some patterns emerged. The DBL1x domain was seen as a side branch to DBLα cluster. The var2 DBL2x and DBL3x domains did not fall into any of the clusters, but was most closely related to the DBLε sequences. Of the ten other DBLx domains, three formed a separate cluster, which in this study were named DBLζ. These DBLζ domains were part of PfEMP1s with identical domain structure. Six DBLx domains fell within or clustered closely to DBLδs. Like other DBLδ they were flanked by a CIDR domain, and were classified as DBLδ in figure 7. One DBLx domain clustered with DBLγ sequences and was classified as such in figure 7.

Distance tree of all 3D7 DBLx and randomly chosen 50 DBL domains representing all DBL subtypes generated using the p-distance/NJ method. All clusters were supported by their bootstrap proportions and by maximum parsimony tree making method (data not shown) Numbers at the nodes represent bootstrap proportions (BP) on 1000 replicates. The scale bar represents the proportion of different amino acids compared. PlasmoDB accession numbers are shown.
3D7 var and rif gene groups
As described, all but one of the upsA sequences were flanked by a rif gene transcribed in the opposite direction and all of these rif genes appeared to share a 5' region, here named upsA-rif. rif genes exhibit a chromosomal organisation similar to var genes, i.e. a small subset genes including the upsA-rif flanked genes were found to be transcribed in direction opposite to the majority. Thus, the rif genes located near the telomere and transcribed towards the centromere were organised as one to three successive genes with the most 5' gene flanked by a sequence with high similarity to upsA-rif. Sequence analysis of 3D7 RIFINs revealed that 12 of 16 RIFINs with a upsA-rif grouped into two separate clusters (figure 6). BLAST search of randomly chosen 1.7 kb RIFIN 5' regions showed that other RIFIN clusters shared distinct upstream sequences (not shown).

Distance tree of 3D7 RIFINs generated using the p-distance/NJ method. MAL6P1.251 was left out as it aligned closer to STEVORs in preliminary alignments (data not shown). Red dots mark RIFINs flanked by upsA-rif. Though, the tree topology could not be confirmed by bootstrapping, the clusters containing the marked RIFINs were verified by the maximum parsimony tree (not shown). The scale bar represents the proportion of different amino acids compared. No accession numbers are given to simplify the graphics.

A) Schematic presentation of all 3D7 var gene sequence analyses. Gene names, chromosomal location, transcriptional direction and domain structure are shown along with the cluster to which each gene was assigned by the sequences analyses. Sequences that could not be assigned to any cluster were named X. Three major var gene groups (group A-C), two intermediate groups group B/A and group B/C and two unique genes representing var1 and var2 var gene families were defined (framed). B) Sequence analyses of var genes from other P. falciparum strains than 3D7. Protein accession numbers, originating strain, domain structure and the closest related 3D7 var 5' sequence are shown along with sequence group allocations as defined in 3D7. *) The genes were assigned to group A, as their DBL1α sequences clustered together with other group A sequences in analysis of DBLα sequences. **) Pseudogene, belongs to the var1 family ^) Upstream sequences with atypically low similarity to upsB or upsC sequences.
Grouping of var genes
Figure 7 schematically sums up the findings of all the var gene sequence analyses. The combination of clusters and chromosomal organization of the var genes indicate that var genes can be grouped into three major subgroups, var group A, B and C and two intermediate groups group B/A and group B/C, which appear to represent transitions between these three groups. The two genes previously shown to belong to conserved var families, var1 and var2, fell outside these groups. Group A var genes were most easily defined, whereas the borders of the proposed group B and C were less clear (figure 7A). The grouping was supported by analyses of both coding and non-coding sequences. However, the best predictors for the groups were the upstream region and chromosomal organization. Thus, genes placed near the telomere and with a transcriptional direction towards the telomere all had upsA sequences and formed group A. Group B were dominated by telomeric located but centromeric transcribed genes flanked by upsB and finally group C harboured all centromeric located genes with a upsC 5'region.
Group A comprise most large PfEMP1s with a domain structure different from the most common 4-domain type, which is the predominant domain structure of Group B and C. Two genes PF08_0140 and MAL6P1.316 were classified as B/A because they had upsBsh 5' regions and chromosomal characteristics in common with group B genes, but had DBLα-CIDR1 sequences and domain structure characteristic for group A genes. Interestingly, these two genes are adjacent to a group A var or a pseudo var gene with an upsA region both transcribed in the opposite direction, thereby merging their 5'regions. Adding the DBL1α-CIDR1 of the flanking pseudogene MAL6P1.317 to the alignments placed this pseudogene within DBLα-CIDR1 cluster A.
Sequence analysis DBLαCIDR1 domains of other parasite strains than 3D7
To investigate whether the suggested grouping of var genes also could be used to group var genes from other parasites than 3D7, eleven var genes available in Genbank were analysed. Figure 7B shows that eight of the nine 5' sequences were classified as upsB or upsBsh, and the remaining sequence was classified as upsC. Most 3D7 genes with an upsB 5'region encode two DBL and two CIDR domains. This was only the case for two of the eleven genes. Thus, the non 3D7 var genes in the database do not seem to represent the 3D7 var gene repertoire and genes with upsA 5' region were absent. Although the picture was not entirely clear, more detailed analysis of the individual non 3D7 var genes revealed interesting similarities to individual 3D7 var genes. AAA75397 from FCR3 has a upsBsh 5' region followed by a group A DBLα-CIDR1 sequence. The 5' region of this gene was 96% identical to the upsBsh of MAL6P1.316, which in itself showed rather low similarity to the other upsBsh regions in 3D7. As AAA75397, MAL6P1.316 carry a group A DBL1-CIDR1. AAC47438, AAB06961, and AF193424 carried DBLα-CIDR1 classified as X and had complex domain structures. When the DBLα sequences of these genes were analysed in with 3D7 DBLαs they clustered with PFL0020w, and PF07_0050 (data not shown), which also have complex domain structures. Attempts were made to group the large number of DBLα sequences[35] generated using the degenerate primer set designed by Taylor et al.[36]. However, grouping could not be maintained by these short sequences.
Discussion
The publication of the P. falciparum genome divided the var genes in different types according to the domain structure of the encoded proteins [17]. Other groups have described semi-conserved regions upstream from the translation initiation sites, and grouped var genes on this basis [18, 19, 29]. We have synthesised the available information and suggest a somewhat different division of the var genes into three major groups (A-C) and two intermediate groups (B/A and B/C), which represent transitions between A, B, and C. The genes were grouped according to chromosomal location and transcription direction, domain structure of the encoded proteins, and sequence similarities in coding and non-coding regions.
Group A consists of ten genes consistently identified as a distinct group by sequence analysis. Interestingly, recombinant CIDR domains based on the group A sequences do not bind CD36, by contrast to CIDR domains produced on the basis of groups B and C [20]. Group A var genes mainly encode large PfEMP1s with complex multi-domain structure. Nine of the Group A var genes are flanked by a rif gene, which is transcribed in the opposite direction. Thus, the 5' regions of the rif and var genes merge. The fact that this organisation has been maintained in the 3D7 genome indicates that the DNA between the coding regions constitutes a functional unit, possibly regulating either recombination or transcription. If the latter is the case the genes could be co-regulated and there might be a functional relationship between the encoded PfEMP1s and RIFINs.
The largest var group in 3D7, group B, comprise 22 genes sharing 5' upsB region. All genes but one are located in the telomeric region. The encoded proteins typically have the characteristic four-domain structure, DBLα-CIDRα-DBLδ-CIDR2. The 13 genes of group C are centromeric. The genes all share 5' upsC region and 12 of them encode proteins with the common four-domain structure. Genes of the B/A and B/C groups have characteristics indicating that they constitute intermediate forms between groups A and B, and groups B and C, respectively. Two genes, which have previously been shown to be present in most P. falciparum genomes, did not fit into any of the groups. Compared to other var genes they appear to be unusually conserved [28, 24, 37]and it has been suggested that they belong to var gene subfamilies named var1 and var2, respectively [24, 28].
To investigate whether the proposed groupings of 3D7 var genes could be used as a general classification of var genes, the available database sequences from other parasite isolates were analysed. Sufficient sequence data was only available for 11 genes, and with regard to domain structure of the predicted proteins, they were not particularly representative of the PfEMP1 repertoire in 3D7. Analysis of the 5' regions allocated ten of the genes to the upsB 3D7 cluster, and they could therefore be classified as group B or group B/A var genes. Further analysis of sequence and predicted domain structure showed that all the genes shared characteristics with at least one group B 3D7 var gene, and none of them shared characteristics with the 3D7 var genes belonging to group A. The upstream region identified one gene as belonging to group C. This encoded a protein with a domain structure typical of 3D7 group C PfEMP1s. Thus, although the data are limited, analysis of non 3D7 var genes suggested that the proposed nomenclature could be used in a general classification of var genes.
The suggested grouping of var genes is operational and based on best judgement. It is likely that future work will change the classification and move genes between groups, nevertheless we believe that this grouping is helpful as starting point for understanding the evolution of the var gene repertoire and developing hypotheses about their function.
The fact that 5' regions predict var gene chromosomal organisation and domain structure, and sequence similarities in coding and non-coding regions several thousand bases downstream from the translation initiation site implies that recombination, or other mechanisms of homogenizing exchange is much more likely to occur between var genes within a group than between var genes of different groupings. It can be proposed that an original ancestral var gene has been duplicated and diverged in the three main types, and each of these have then diverged into the genes of each group. In this process information may also have been exchanged between genes of different groupings. The data suggests that some exchange have taken place between groups B and C and some characteristics of group A have leaked into these groups, but that characteristics from groups B and C have not gained access to group A. It is tempting to speculate that distinct chromosomal organisation patterns restrict recombination and that the conserved flanking regions serve to align genes of similar group for recombination. The fact that a putative boundary of the upstream sequence could be determined for most var genes may suggest that these sites also serve as splicing sites for insertion of larger gene fragments or whole genes.
Why then are var genes structured into different groups? By mediating parasite binding to endothelium, PfEMP1 enables the parasite to sequester and avoid filtering through the spleen. Thus, parasites expressing PfEMP1, which are most effective in sequestering infected erythrocytes, will obtain the highest growth rates. How effective a given PfEMP1 is in binding in a particular host will depend on the binding characteristics of the PfEMP1, on the ligands that are available in the host [38], and the anti-PfEMP1 antibody repertoire in the infected individual [11, 39, 40]. Parasites causing severe malaria express phenotypes that are more often recognised by antibodies in children's plasma than the phenotypes expressed by parasites causing uncomplicated disease [41];[40]. The phenotypes associated with severe disease also tend to be serologically cross-reactive (Nielsen et al., in preparation). Given that immunity to severe malaria is developed relatively early in life, it is possible to speculate that the most severe forms of malaria are caused by fast growing parasites expressing PfEMP1s optimized to mediate a very effective binding in non immune hosts. To maintain effective binding these PfEMP1 types are probably functionally constrained, and consequently have tight limits to the degree to which they can vary. The fact that recombination within var genes of group A appear to be the most constrained, suggests that the PfEMP1s associated with severe malaria will be encoded by group A var genes. This hypothesis is in agreement with findings from China indicating that parasites from individuals suffering from cerebral malaria compared with cases of non-severe malaria expressed high molecular weight PfEMP1s [42] and a study from Brazil where expression of DBLα domains lacking 1–2 cysteine residues in DBLα homology block G were mainly found among severe malaria cases [43]. In 3D7 this is a feature of all genes of group DBLα-CIDR1 group A (var gene group A).
In most endemic settings transmission does not occur continuously, but is highly seasonal and in some areas restricted to a few months of every year [44]. In such a situation the ability to establish chronic infections is important for parasite survival and transmission. Chronic human malaria infections are associated with 'shift' in PfEMP1 expression [45] and it has been proposed that such shifts are driven by antibody forcing parasites to express PfEMP-1 molecules which are less optimal for adhesion, but not recognised by cross reactive antibodies. It is possible to speculate that PfEMP1s of groups B and C could serve this function.
In areas of high malaria endemicity, women who have acquired malaria immunity during childhood become susceptible to malaria during their first pregnancies [46] and are infected by parasites expressing antigens that mediate binding to CSA in placenta [8]. Parasites of this phenotype can apparently only expand and establish infection in individuals carrying a placenta and these parasites do not cross-react serologically with non-placental parasites [38]. It has been recently reported that PFL0030c is the dominant var gene transcribed in parasites selected for CSA binding and that most parasite genomes carry very similar genes, the var2 family [28], Interestingly, the var2 upstream region (upsE) is markedly different from the 5' regions of the other var genes and appears to be conserved. The upstream upsE region of var2 is also the only such region containing an ORF. Upstream ORFs are uncommon in known genomes, and primarily described in association with genes that are under tight translational control, such as oncogenes and genes involved in cellular differentiation (reviewed by Kozak, 2002). The function of the uORF 5' of var2 remains unclear.
Conclusion
The analysis of the var gene repertoire in 3D7 indicates that the var genes can be assigned to different groups and that exchange of genetic information is more likely to take place between the genes in a group than between genes of different grouping. The groups are probably maintained by inhibition of recombination due to differences in chromosomal locations and direction of transcription. It is likely that the proteins encoded within the groups have evolved to serve different functions enabling the parasite to survive under a range of transmission patterns in non-pregnant and pregnant hosts with varying degree of acquired immunity.
References
David PH, Hommel M, Miller LH, Udeinya IJ, Oligino LD: Parasite sequestration in Plasmodium falciparum malaria: spleen and antibody modulation of cytoadherence of infected erythrocytes. Proc Natl Acad Sci U S A. 1983, 80: 5075-5079.
Howard RJ, Barnwell JW: Roles of surface antigens on malaria-infected red blood cells in evasion of immunity. Contemp Top Immunobiol. 1984, 12: 127-191.
Turner GDH, Morrison H, Jones M, Davis TME, Looareesuwan S, Buley ID, Gatter KC, Newbold CI, Pukritayakamee S, Nagachinta B, White NJ, Berendt AR: An immunohistochemical study of the pathology of fatal malaria: evidence for widespread endothelial activation and a potential role for intercellular adhesion molecule-1 in cerebral sequestration. Am J Pathol. 1994, 145: 1057-1069.
Fried M, Duffy PE: Maternal malaria and parasite adhesion. J Mol Med. 1998, 76: 162-171. 10.1007/s001090050205.
Newbold CI, Craig AG, Kyes S, Berendt AR, Snow RW, Peshu N, Marsh K: PfEMP1, polymorphism and pathogenesis. Ann Trop Med Parasitol. 1997, 91: 551-557. 10.1080/00034989760923.
Barnwell JW, Asch AS, Nachman RL, Yamaya M, Aikawa M, Ingravallo P: A human 88-kD membrane glycoprotein (CD36) functions in vitro as a receptor for a cytoadherence ligand on Plasmodium falciparum-infected erythrocytes. J Clin Invest. 1989, 84: 765-772.
Berendt AR, Simmons DL, Tansey J, Newbold CI, Marsh K: Intercellular adhesion molecule-1 is an endothelial cell adhesion receptor for Plasmodium falciparum. Nature. 1989, 341: 57-59. 10.1038/341057a0.
Fried M, Duffy PE: Adherence of Plasmodium falciparum to chondroitin sulphate A in the human placenta. Science. 1996, 272: 1502-1504.
Smith JD, Chitnis CE, Craig AG, Roberts DJ, Hudson-Taylor DE, Peterson DS, Pinches R, Newbold CI, Miller LH: Switches in expression of Plasmodium falciparum var genes correlate with changes in antigenic and cytoadherent phenotypes of infected erythrocytes. Cell. 1995, 82: 101-110.
Marsh K, Otoo L, Hayes RJ, Carson DC, Greenwood BM: Antibodies to blood stage antigens of Plasmodium falciparum in rural Gambians and their relation to protection against infection. Trans R Soc Trop Med Hyg. 1989, 83: 293-303.
Bull PC, Lowe BS, Kortok M, Molyneux CS, Newbold CI, Marsh K: Parasite antigens on the infected red cell are targets for naturally acquired immunity to malaria. Nature Med. 1998, 4: 358-360.
Dodoo D, Staalsoe T, Giha H, Kurtzhals JAL, Akanmori BD, Koram K, Dunyo S, Nkrumah FK, Hviid L, Theander TG: Antibodies to variant antigens on the surfaces of infected erythrocytes are associated with protection from malaria in Ghanaian children. Infect Immun. 2001, 69: 3713-3718. 10.1128/IAI.69.6.3713-3718.2001.
Giha HA, Staalsoe T, Dodoo D, Roper C, Satti GM, Arnot DE, Hviid L, Theander TG: Antibodies to variable Plasmodium falciparum-infected erythrocyte surface antigens are associated with protection from novel malaria infections. Immunol Lett. 2000, 71: 117-126. 10.1016/S0165-2478(99)00173-X.
Su X, Heatwole VM, Wertheimer SP, Guinet F, Herrfeldt JA, Peterson DS, Ravetch JA, Wellems TE: The large diverse gene family var encodes proteins involved in cytoadherence and antigenic variation of Plasmodium falciparum-infected erythrocytes. Cell. 1995, 82: 89-100.
Baruch DI, Pasloske BL, Singh HB, Bi X, Ma XC, Feldman M, Tarachi TF, Howard RJ: Cloning the Plasmodium falciparum gene encoding PfEMP1, a malarial variant antigen and adherence receptor on the surface of parasitized human erythrocytes. Cell. 1995, 82: 77-87.
Smith JD, Subramanian G, Gamain B, Baruch DI, Miller LH: Classification of adhesive domains in the Plasmodium falciparum erythrocyte membrane protein 1 family. Mol Biochem Parasitol. 2000, 110: 293-310. 10.1016/S0166-6851(00)00279-6.
Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DMA, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, Mcfadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B: Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002, 419: 498-511. 10.1038/nature01097.
Voss TS, Thompson JK, Waterkeyn J, Felger I, Weiss N, Cowman AF, Beck HP: Genomic distribution and functional characterisation of two distinct and conserved Plasmodium falciparum var gene 5' flanking sequences. Mol Biochem Parasitol. 2000, 107: 103-115. 10.1016/S0166-6851(00)00176-6.
Mercereau-Puijalon O, Barale JC, Bischoff E: Three multigene families in Plasmodium parasites: facts and questions. Int J Parasitol. 2002, 32: 1323-1344. 10.1016/S0020-7519(02)00111-X.
Robinson BA, Welch TL, Smith JD: Widespread functional specialization of Plasmodium falciparum erythrocyte membrane protein 1 family members to bind CD36 analysed across a parasite genome. Mol Microbiol. 2003, 47: 1265-1278. 10.1046/j.1365-2958.2003.03378.x.
Fernandez V, Hommel M, Chen QJ, Hagblom P, Wahlgren M: Small, clonally variant antigens expressed on the surface of the Plasmodium falciparum-infected erythrocyte are encoded by the rif gene family and are the target of human immune responses. J Exp Med. 1999, 190: 1393-1403. 10.1084/jem.190.10.1393.
Kyes SA, Rowe JA, Kriek N, Newbold CI: Rifins: a second family of clonally variant proteins expressed on the surface of red cells infected with Plasmodium falciparum. Proc Natl Acad Sci U S A. 1999, 96: 9333-9338. 10.1073/pnas.96.16.9333.
Bahl A, Brunk B, Coppel RL, Crabtree J, Diskin SJ, Fraunholz MJ, Grant GR, Gupta D, Huestis RL, Kissinger JC, Labo P, Li L, McWeeney SK, Milgram AJ, Roos DS, Schug J, Stoeckert CJ: PlasmoDB: the Plasmodium genome resource. An integrated database providing tools for accessing, analyzing and mapping expression and sequence data (both finished and unfinished). Nucleic Acids Res. 2002, 30: 87-90. 10.1093/nar/30.1.87.
Salanti A, Staalsoe T, Lavstsen T, Jensen A, Sowa M, Arnot D, Hviid L, Theander TG: Selective up-regulation of a single distinctly structured var gene in CSA-adhering Plasmodium falciparum involved in pregnancy-associated malaria. Mol Microbiol. 2003, 49: 179-191. 10.1046/j.1365-2958.2003.03570.x.
Hall TA: BioEdit:a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucl Acids Symp Ser. 1999, 41: 95-98.
Kumar S, Tamura K, Jakobsen I, Nei M: MEGA2 : Molecular Evolutionary Genetics Analysis. Ver. 2.1. Bioinformatics. 2001, 17: 1244-1245. 10.1093/bioinformatics/17.12.1244.
Rowe JA, Kyes SA, Rogerson SJ, Babiker HA, Raza A: Identification of a conserved Plasmodium falciparum var gene implicated in malaria in pregnancy. J Infect Dis. 2002, 185: 1207-1211. 10.1086/339684.
Salanti A, Jensen AT, Zornig HD, Staalsoe T, Joergensen L, Nielsen MA, Khattab A, Arnot DE, Klinkert MQ, Hviid L, Theander TG: A sub-family of common and highly conserved Plasmodium falciparum var genes. Mol Biochem Parasitol. 2002, 122: 111-115. 10.1016/S0166-6851(02)00080-4.
Vazquez-Macias A, Martinez-Cruz P, Castaneda-Patlan MC, Scheidig C, Gysin J, Scherf A, Hernandez-Rivas R: A distinct 5' flanking var gene region regulates Plasmodium falciparum variant erythrocyte surface antigen expression in placental malaria. Mol Microbiol. 2002, 45: 155-167. 10.1046/j.1365-2958.2002.02999.x.
Buffet PA, Gamain B, Scheidig C, Baruch D, Smith JD, Hernandez-Rivas R, Pouvelle B, Oishi S, Fujii N, Fusai T, Parzy D, Miller LH, Gysin J, Scherf A: Plasmodium falciparum domain mediating adhesion to chondroitin sulfate A: a receptor for human placental infection. Proc Natl Acad Sci U S A. 1999, 96: 12743-12748. 10.1073/pnas.96.22.12743.
Costa FT, Fusai T, Parzy D, Sterkers Y, Torrentino M, Douki JB, Traore B, Petres S, Scherf A, Gysin J: Immunization with recombinant duffy binding like gamma3 induce pan reactive and adhesion blocking antibodies against placental chondroitin sulfate A binding Plasmodium falciparum parasites. J Infect Dis. 2003, 188: 153-164. 10.1086/375800.
Kyes S, Christodoulou Z, Raza A, Horrocks P, Pinches R, Rowe A, Newbold CI: A well-conserved Plasmodium falciparum var gene shos an unusual stage-specific transcript pattern. Mol Microbiol. 2003, 48: 1339-1348. 10.1046/j.1365-2958.2003.03505.x.
Voss TS, Kaestli M, Vogel D, Bopp S, Beck HP: Identification of nuclear proteins that interact differentially with Plasmodium falciparum var gene promoters. Mol Microbiol. 2003, 48: 1593-1607. 10.1046/j.1365-2958.2003.03528.x.
Calderwood MS, Gannoun-Zaki L, Wellems TE, Deitsc : Plasmodium falciparum var genes are regulated by two regions with separate promoters, on upstream of the coding region and a second within the intron. J Biol Chem. 2003,
Ward CP, Clottey GT, Dorris M, Ji D-D, Arnot DE: Analysis of Plasmodium falciparum PfEMP-1/ var genes indicates that recombination rearranges constrained sequences. Mol Biochem Parasitol. 1999, 102: 167-177. 10.1016/S0166-6851(99)00106-1.
Taylor HM, Kyes SA, Harris D, Kriek N, Newbold CI: A study of var gene transcription in vitro using universal var gene primers. Mol Biochem Parasitol. 2000, 105: 13-23. 10.1016/S0166-6851(99)00159-0.
Rowe JA, Kyes SA, Rogerson SJ, Babiker HA, Raza A: Identification of a conserved Plasmodium falciparum var gene implicated in malaria in pregnancy.
Ricke CH, Staalsoe T, Koram K, Akanmori BD, Riley EM, Theander TG: Plasma antibodies from malaria-exposed pregnant women recognize variant surface antigens on Plasmodium falciparum-infected erythrocytes in a parity-dependent manner and block parasite adhesion to chondroitin sulphate A. J Immunol. 2000, 165: 3309-3316.
Bull PC, Lowe BS, Kortok M, Marsh K: Antibody recognition of Plasmodium falciparum erythrocyte surface antigens in Kenya: evidence for rare and prevalent variants. Infect Immun. 1999, 67: 733-739.
Nielsen MA, Staalsoe T, Kurtzhals JA, Goka BQ, Dodoo D, Alifrangis M, Theander TG, Akanmori BD, Hviid L: Plasmodium falciparum variant surface antigen expression varies between isolates causing severe and nonsevere malaria and is modified by acquired immunity. J Immunol. 2002, 168: 3444-3450.
Bull PC, Kortok M, Kai O, Ndungu F, Ross A, Lowe BS: Plasmodium falciparum-infected erythrocytes: agglutination by diverse Kenyan plasma is associated with severe disease and young host age. J Infect Dis. 2000, 182: 252-259. 10.1086/315652.
Bian Z, Wang G, Tian X, Fan J: Expression of Plasmodium falciparum-infected erythrocyte membrane protein from cerebral malaria patients. Zhongguo Ji Sheng Chong Xue Yu Ji Sheng Chong. 1999, 17: 359-362.
Kirchgatter K, Portillo HA: Association of severe noncerebral Plasmodium falciparum malaria in Brazil with expressed PfEMP1 DBL1 alpha sequences lacking cysteine residues. Mol Med. 2002, 8: 16-23.
Giha HA, Rosthoj S, Dodoo D, Hviid L, Satti GMH, Scheike T, Arnot DE, Theander TG: The epidemiology of febrile malaria episodes in an area of unstable and seasonal transmission. Trans R. 2000, 94: 645-651.
Staalsoe T, Hamad AA, Hviid L, Elhassan IM, Arnot DE, Theander TG: In vivo switching between variant surface antigens in human Plasmodium falciparum infection. J Infect Dis. 2002, 186: 719-722. 10.1086/342390.
Brabin BJ: The risks and severity of malaria in pregnant women. TDR/FIELDMAL/1. 1991
Acknowledgements
The PlasmoDB database http://www.plasmodb.org was the essential resource for this work. We are thus indebted to all those who generated the data available at PlasmoDB. Odile Mercereau-Puijalon is thanked for supplying sequence material. This work was supported by grants from the Danish Medical Research Council (ref. No. 22-02-0220) and the Commission of the European Communities (grants no. QLK2-CT-1999-01293 (EUROMALVAC)). AS is funded by a Ph.D. grant from the Gates Malaria Partnership.
Author information
Authors and Affiliations
Corresponding author
Additional information
Authors' contributions
TL collected the sequences and performed the cluster analysis. AS did the laboratory experiments on the var2 uORF. All authors participated in the analysis and interpretation of data. TL produced the first draft. All authors contributed to writing the manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article
Cite this article
Lavstsen, T., Salanti, A., Jensen, A.T. et al. Sub-grouping of Plasmodium falciparum 3D7 var genes based on sequence analysis of coding and non-coding regions. Malar J 2, 27 (2003). https://doi.org/10.1186/1475-2875-2-27
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2875-2-27