
Abstract
Bortezomib is a highly selective, reversible inhibitor of the 26S proteasome that is indicated for single-agent use in the treatment of patients with multiple myeloma who have received at least 2 prior therapies and are progressing on their most recent therapy. Clinical investigations have been completed or are under way to evaluate the safety and efficacy of bortezomib alone or in combination with chemotherapy in multiple myeloma, both at relapse and presentation, as well as in other cancer types. The antiproliferative, proapoptotic, antiangiogenic, and antitumor activities of bortezomib result from proteasome inhibition and depend on the altered degradation of a host of regulatory proteins. Exposure to bortezomib has been shown to stabilize p21, p27, and p53, as well as the proapoptotic Bid and Bax proteins, caveolin-1, and inhibitor κB-α, which prevents activation of nuclear factor κB-induced cell survival pathways. Bortezomib also promoted the activation of the proapoptotic c-Jun-NH2 terminal kinase, as well as the endoplasmic reticulum stress response. The anticancer effects of bortezomib as a single agent have been demonstrated in xenograft models of multiple myeloma, adult T-cell leukemia, lung, breast, prostate, pancreatic, head and neck, and colon cancer, and in melanoma. In these preclinical in vivo studies, bortezomib treatment resulted in decreased tumor growth, angiogenesis, and metastasis, as well as increased survival and tumor apoptosis. In several in vitro and/or in vivo cancer models, bortezomib has also been shown to enhance the antitumor properties of several antineoplastic treatments. Importantly, bortezomib was generally well tolerated and did not appear to produce additive toxicities when combined with other therapies in the dosing regimens used in these preclinical in vivo investigations. These findings provide a rationale for further clinical trials using bortezomib alone or in combination regimens with chemotherapy, radiation therapy, immunotherapy, or novel agents in patients with hematologic malignancies or solid tumors.
Similar content being viewed by others


Introduction
Bortezomib (VELCADE®, formerly PS-341) was approved for the treatment of patients with relapsed or refractory multiple myeloma in May 2003 by the US Food and Drug Administration [1] and in April 2004 by the Committee for Proprietary Medicinal Products of the European Union. A number of clinical studies evaluating the activity and safety of bortezomib in multiple myeloma, as well as in other types of cancer, have been conducted [2–10] or are ongoing [11]. Therefore, a review of key preclinical studies that have explored the mechanisms of action and provided the rationale for clinical investigation of this novel agent in multiple myeloma and other cancer types is warranted.
Mechanism of action
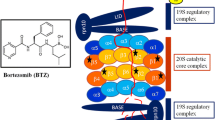
Bortezomib, a boronic acid dipeptide (Figure 1A) [12], is a highly selective, reversible inhibitor of the 26S proteasome that was first shown to exhibit antitumor properties in a panel of 60 cancer cell lines from the US National Cancer Institute [13]. The proteasome is an enzyme complex that primarily functions in the degradation of misfolded proteins and is essential for the regulation of the cell cycle. Proteasomes are localized in the nucleus and cytosol, where they are largely associated with centrosomes, the cytoskeleton, and the outer endoplasmic reticulum [14]. Damaged intracellular proteins are targeted for elimination by the proteasome through ubiquitination (Figure 1B). Many of the substrates that have been identified are proteins that function in the regulation of transcriptional activation, signal transduction, cell cycle proliferation, and apoptosis (Figure 2, Table 1) [15–29].

Chemical structure of the proteasome inhibitor bortezomib: pyrazylcarbonyl-Phe-Leu-boronate (A). Schematic illustration of the ubiquitin-proteasome pathway. Misfolded proteins (e.g., p53) are targeted for degradation by the 26S proteasome via phosporylation and ubiquitination (B). Following substrate degradation, the ubiquitin tags and peptides are recycled for future use.

Inhibition of the proteasome by bortezomib results in activation of JNK, stabilization of p53, Bid, Bax, p21, p27, caveolin-1, and IκBα, resulting in inhibition of NF-κB. Alteration of the levels of these cellular proteins leads to inhibition of proliferation, migration, and angiogenesis and promotion of apoptosis of cancer cells.
Several independent investigators have found that bortezomib inhibits activation of the transcription factor nuclear factor κB (NF-κB) [15–20, 30]. NF-κB is important for cell survival and is activated in response to cell stress, including that induced by cytotoxic agents, radiation, or DNA damage. NF-κB is overexpressed in several tumors and regulates the expression of genes involved in apoptosis (including Bcl-2 and Bcl-xL), cell cycle progression, inflammation, and angiogenesis (including interleukin [IL]-6, IL-8, and vascular endothelial growth factor [VEGF]) [31–33]. NF-κB is normally bound in the cytosol to inhibitor κB-α (IκBα). Phosphorylation, ubiquitination, and degradation of IκBα are required for NF-κB to translocate to the nucleus and activate the transcription of target genes. Bortezomib blocks the activation of NF-κB by preventing proteasomal degradation of IκBα. Through inhibition of NF-κB, bortezomib not only promotes apoptosis of cancer cells but also sensitizes these cells to chemotherapy [15, 20, 30], radiation [16], or immunotherapy [19]. However, because specific NF-κB inhibition alone via PS-1145 only partially inhibits proliferation of tumor cells [18], the cytotoxic activity of bortezomib must also depend on altered regulation of other signal transduction pathway targets [18].
The intracellular levels of a number of other proteins that regulate gene transcription, apoptosis, and proliferation are significantly affected by bortezomib (Table 1). c-Jun-NH2 terminal kinase (JNK) is a protein that promotes cell death in response to stress and increased levels of misfolded proteins [34]. Bortezomib treatment leads to activation of JNK in multiple myeloma [21, 22] and non-small cell lung cancer cells [23]. These studies further showed that specific inhibition of JNK activation, either genetic or pharmacologic, prevented mitochondrial release of cytochrome c and Smac, activation of caspase-8, -9, and -3, and apoptosis.
Proteasome inhibition has also been shown to stabilize the cyclin-dependent kinase inhibitors p21 and p27, the tumor suppressor p53, and the proapoptotic proteins Bid and Bax [15, 21, 23–27]. The increased levels of activated p21, p27, p53, Bid, and Bax result in inhibition of cell cycle progression and/or promotion of apoptosis in response to bortezomib. Interestingly, sensitivity to proteasome inhibition was partially dependent on the p53 status of breast [35] and lung cancer in vitro [36], but bortezomib-induced apoptosis and/or chemosensitization were p53 independent in prostate [13], multiple myeloma [15], and colon cancer cells [30]. Therefore, the degree of variability in the sensitivity to bortezomib with respect to p53 status appears cell-type dependent.
A recently published study found that bortezomib prevented activation of caveolin-1 in multiple myeloma cells [28]. Activation of caveolin-1, a protein that functions in cell motility or migration in a number of tissues, requires phosphorylation. In this report, bortezomib was shown to prevent phosphorylation of caveolin-1 by VEGF, a proangiogenic cytokine and transcriptional target of NF-κB [37]. Bortezomib also inhibited VEGF secretion by the bone marrow. Together, these findings demonstrate important mechanisms by which bortezomib may inhibit migration of cancer cells as well as tumor angiogenesis.
The specific proteins mentioned have all been shown to be at least partially responsible in various models for the antiproliferative, proapoptotic, antiangiogenic, and antitumor effects of bortezomib. However, recent studies have found that bortezomib results in cytotoxic activity through activation of the endoplasmic reticulum stress response [38–41]. The mechanism appears to involve blockade of retrograde transportation and degradation of damaged endoplasmic reticulum proteins by proteasome inhibition [42]. Further studies are necessary to link these new findings with the specific intracellular signals that have been previously implicated in the anticancer activities of bortezomib.
Bortezomib alone
Bortezomib has shown promising antitumor activity in a number of preclinical cancer murine models in vivo (Table 2) [13, 17, 19, 24, 30, 43–49]. In a xenograft model of multiple myeloma, bortezomib treatment resulted in significant inhibition of tumor growth, an increase in overall survival, and a decrease in tumor angiogenesis [43]. The proteasome inhibitor was well tolerated up to 0.5 mg/kg intravenously (IV) twice weekly for 4 weeks, with dose-limiting toxicities, including weight loss, at 1 mg/kg. Two recent reports evaluating the efficacy of bortezomib in murine xenograft models of adult T-cell leukemia have reached contradictory conclusions. Tan and Waldman found that bortezomib treatment alone, 0.06 mg/kg intraperitoneally (IP), daily for 3 weeks, did not produce significant antitumor effects [19]. However, a second group reported that bortezomib 1.0 mg/kg IP twice weekly for 2 weeks resulted in antitumor activity [44]. Whereas Tan and Waldman reported lethality at a more aggressive dosing regimen of bortezomib 0.1 mg/kg IP twice daily for 2 weeks, slight, temporary weight loss was the only adverse effect described by the second group of investigators [44]. It seems plausible that the discrepancy in activity and toxicity may be due to the differences in the doses and dosing regimens. Based on the finding that proteasome inhibition by bortezomib lasts for up to 72 hours [8], more recent preclinical studies have used twice-weekly, rather than daily or twice-daily dosing, and have shown overall greater activity with less toxicity.
An evaluation of the effects of bortezomib in murine xenograft models of both lung and breast cancer was also conducted [45]. Treatment with oral bortezomib 1.0 mg/kg daily for 18 days caused tumor growth delays, as well as a decrease in the number of metastases in the Lewis lung cancer model. Furthermore, in a murine model, bortezomib at a single IP dose of up to 5 mg/kg significantly decreased the surviving fraction of breast tumor cells. A decrease in the level of colony-forming-unit granulocyte macrophages was the only toxicity noted in these experiments.
Two groups of investigators have evaluated the efficacy of bortezomib in murine xenograft models of prostate cancer. The first study concluded that bortezomib 1.0 mg/kg IV weekly for 4 weeks reduced tumor growth by 60% [13]. The second study, in which bortezomib 1.0 mg/kg IV every 72 hours for 15 days was administered, produced similar results, with 50% and 80% inhibition in tumor growth in two xenograft models [46]. This report further showed that bortezomib significantly inhibited tumor angiogenesis in one of the models, as measured by a decrease in the number of CD31+ vessels in tumor sections. No toxicities were detected in either of these studies.
In a study of pancreatic cancer murine xenografts, treatment with bortezomib 1.0 mg/kg IV or IP weekly for 4 weeks resulted in a 72% or 84% reduction in tumor growth, as well as an increase in tumor cell apoptosis, with no evidence of toxicity [24]. Another group found that bortezomib 1.0 mg/kg IV biweekly for 2 to 3 weeks significantly inhibited tumor growth and angiogenesis and promoted apoptosis in 1 of 2 pancreatic cancer xenograft murine models [47]. These investigators also reported adverse events, including decreased body weight, diarrhea, and gastrointestinal inflammation at doses above 1.0 mg/kg and lethality at doses above 1.5 mg/kg. Finally, bortezomib produced significant antitumor, proapoptotic, and/or antiangiogenic effects in murine xenograft models of head and neck [17] and colon cancer [30], as well as melanoma [49]. Adverse events, including dehydration, lethargy, weight loss, and death, were noted at doses of 2.0 mg/kg IP 3 times weekly for 3 weeks in one of these studies [17], whereas the other studies did not report any toxicities at lower doses (Table 2).
These preclinical investigations have collectively demonstrated the antitumor activity of bortezomib as a single agent at tolerable levels in a variety of murine cancer models. However, because the standard approach to antineoplastic therapy generally involves the administration of more than one agent or modality in an effort to prevent the development of chemoresistance and increase tumor cell kill, the effects of bortezomib in combination with chemotherapy, radiation, immunotherapy, or novel agents have been investigated in vitro and/or in vivo.
Combination therapy
A number of preclinical studies have evaluated the activity of bortezomib in combination with other therapies (Table 3) [15, 16, 19, 20, 24, 30, 39, 40, 45, 48–61]. The finding that several cytotoxic agents as well as radiation [62, 63] activate NF-κB is a major rationale for combining proteasome inhibitors with these therapies in the treatment of cancer. Activated NF-κB is free to translocate to the nucleus and induce the expression of proinflammatory and antiapoptotic genes, such as Bcl-2 and Bcl-xL, which promote tumor cell survival [31–33]. Furthermore, inhibition of NF-κB has been implicated as an important mechanism by which bortezomib sensitizes tumor cells to various drugs or radiation [15, 16, 20, 24, 30, 48–50, 55, 57, 59, 60].
Bortezomib with chemotherapy
In an investigation of lung cancer, bortezomib in combination with chemotherapeutic agents, including 5-fluorouracil, cisplatin, paclitaxel, or doxorubicin, produced additive antitumor and antimetastatic effects [45]. In this same study, bortezomib also increased tumor-cell killing by cyclophosphamide and cisplatin, as well as tumor-cell killing by radiation, in an in vitro-in vivo model of breast cancer. Although toxicity is difficult to assess in preclinical models, no added toxicities were observed when bortezomib was added to the other therapies, and bortezomib dose modifications were not required.
Several studies evaluating the effects of bortezomib in combination with other therapies have been conducted in multiple myeloma. These in vitro experiments collectively confirmed that bortezomib enhanced the antiproliferative and proapoptotic effects of conventional antimyeloma agents, including melphalan, doxorubicin, and dexamethasone [15, 20, 50]. These investigators also reported that multiple myeloma cell lines that were previously resistant to melphalan, doxorubicin, dexamethasone, or mitoxantrone were sensitized up to 1,000,000-fold by prior exposure to subtoxic concentrations of bortezomib. Finally, the researchers showed that bortezomib was not only directly cytotoxic to the multiple myeloma cells but that it also altered the microenvironment through inhibition of IL-6 to prevent the growth of tumor cells in proximity to the bone marrow [15].
Two groups of researchers examined the effects of bortezomib in combination with the topoisomerase inhibitor irinotecan in murine xenograft models of colon [30] and pancreatic [24] cancer. Both these studies concluded that combined inhibition of the proteasome and topoisomerase resulted in enhanced antiproliferative and proapoptotic effects. The combination of bortezomib and irinotecan therapy further resulted in a 94% [30] or 89% [24] decrease in tumor size compared with controls. These tumors were also significantly smaller than those of the mice receiving either bortezomib (26% or 65% decrease in tumor size relative to controls) or irinotecan (48% or 43% decrease in tumor size relative to controls) as single agents.
Xenograft models of pancreatic cancer were also used to evaluate the activity of bortezomib in combination with other standard chemotherapies [48, 53]. Inhibition of the proteasome increased the sensitivity of tumors to both gemcitabine [48] and docetaxel [53], because bortezomib in combination with these agents resulted in significant enhancement of antiproliferative, proapoptotic, antitumor, and/or antiangiogenic activities.
The combination of bortezomib with temozolomide was studied in models of malignant melanoma [49]. Bortezomib enhanced the antiproliferative and cytotoxic effects of temozolomide in melanoma cells in vitro, and this combined therapy produced complete remission of tumors lasting more than 200 days in murine xenografts in vivo. In contrast, tumors eventually progressed in mice receiving either drug alone. Although specific toxicity evaluations were not performed, no toxicities were observed in any of these investigations.
Bortezomib with radiation, immunotherapy, or novel agents
A number of studies have reported on the radiosensitizing properties of bortezomib. In addition to the previously mentioned study in breast cancer [45], other laboratories have demonstrated that bortezomib sensitizes colon [16] and prostate [54] cancers to radiation-induced cytotoxicity. In both clonogenic cell survival assays and murine xenograft tumor models, pretreatment with bortezomib enhanced the anticancer effects of irradiation with no observed toxicity.
Another murine xenograft model was used to investigate the effects of bortezomib in combination with daclizumab, a humanized anti-IL-2Rα antibody in adult T-cell leukemia [19]. Although either agent alone resulted in partial or no responses, bortezomib plus daclizumab resulted in prolongation of the survival of tumor-bearing mice. The only adverse effect of the combined therapy was a slight temporary weight loss during the treatment regimen.
Bortezomib has also demonstrated enhanced in vitro and/or in vivo anticancer effects when combined with novel agents such as TRAIL/Apo2L, a cell death-inducing ligand [55, 56], 17-N-allylamino-17-demethoxygeldanamycin (17-AAG), an inhibitor of heat shock protein 90 [39], and suberoylanilide hydroxamic acid (SAHA) or sodium butyrate, both histone deactylase inhibitors [40, 57, 59, 60]. Pretreatment with bortezomib sensitized multiple myeloma, myeloid leukemia, and renal cancer cells but not normal B lymphocytes to TRAIL/Apo2L-induced apoptosis [55, 56]. In an in vivo experiment, bone marrow and renal cancer cell mixtures, with or without bortezomib and/or TRAIL/Apo2L, were transplanted into the bone marrow of mice. Whereas all the mice receiving cells treated with TRAIL/Apo2L died of leukemia within 35 days, 50% of those receiving cells treated with bortezomib and 90% of those receiving cells treated with both TRAIL/Apo2L and bortezomib survived more than 100 days [56]. A mild transient thrombocytopenia was the only toxicity observed in this study. Finally, while the combination of bortezomib and 17-AAG, SAHA, or sodium butyrate resulted in synergistic antiproliferative and proapoptotic effects in vitro [39, 40, 57–60], these combinations have yet to be evaluated in tumor xenograft models in vivo.
Sequence of administration
One of the areas of controversy has been the appropriate timing of therapy with bortezomib in relation to other antineoplastic agents. A brief discussion of some of the conflicting results is warranted as these preclinical studies may ultimately provide the rationale for clinical trials. In vitro experiments conducted by Mitsiades and colleagues revealed that optimal antimyeloma activity was achieved when bortezomib was administered 24 hours after doxorubicin [50]. The sequence of chemotherapy (gemcitabine) followed by bortezomib was also found to be the most effective in a model of pancreatic cancer [64]. In contrast, Pei et al showed that bortezomib followed by a second antineoplastic agent (SAHA) yielded the highest level of apoptosis in a model of myeloma [60], and Mimnaugh and colleagues found that maximal antiproliferative effects were observed upon simultaneous administration of bortezomib and a heat shock protein 90 inhibitor in a model of breast cancer [39]. It is important to note that these studies utilized bortezomib in combination with different antineoplastic agents in unique cancer models. Further studies may elucidate the reasons for the inconsistent findings.
Conclusion
The proteasome inhibitor bortezomib exhibits antiproliferative, proapoptotic, antiangiogenic, and antitumor activities in several cancer models. The mechanism of action of bortezomib involves stabilization of NF-κB, p21, p27, p53, Bid, and Bax, inhibition of caveolin-1 activation, and activation of JNK as well as the endoplasmic reticulum stress response. These preclinical evaluations have found that bortezomib is well tolerated at doses that demonstrated antitumor activity in xenograft models of multiple myeloma, adult T-cell leukemia, cancer of the lung, breast, prostate, pancreas, head and neck, and colon, as well as melanoma. Bortezomib also enhances the anticancer effects of chemotherapy, radiation therapy, immunotherapy, or novel agents, without added toxicities requiring dose modifications. The studies provide a rationale for clinical trials of bortezomib alone or in combination with other therapies in patients with solid tumors or hematologic malignancies.
Abbreviations
- NF-κB:
-
nuclear factor-κB
- IκBα:
-
inhibitor κB-α
- VEGF:
-
vascular endothelial growth factor
- ER:
-
endoplasmic reticulum.
References
Kane RC, Bross PF, Farrell AT, Pazdur R: Velcade: U.S. FDA approval for the treatment of multiple myeloma progressing on prior therapy. Oncologist. 2003, 8: 508-513. 10.1634/theoncologist.8-6-508.
Aghajanian C, Soignet S, Dizon DS, Pien CS, Adams J, Elliott PJ, Sabbatini P, Miller V, Hensley ML, Pezzulli S, Canales C, Daud A, Spriggs DR: A phase I trial of the novel proteasome inhibitor PS341 in advanced solid tumor malignancies. Clin Cancer Res. 2002, 8: 2505-2511.
Orlowski RZ, Stinchcombe TE, Mitchell BS, Shea TC, Baldwin AS, Stahl S, Adams J, Esseltine DL, Elliott PJ, Pien CS, Guerciolini R, Anderson JK, Depcik-Smith ND, Bhagat R, Lehman MJ, Novick SC, O'Connor OA, Soignet SL: Phase I trial of the proteasome inhibitor PS-341 in patients with refractory hematologic malignancies. J Clin Oncol. 2002, 20: 4420-4427. 10.1200/JCO.2002.01.133.
Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, Srkalovic G, Alsina M, Alexanian R, Siegel D, Orlowski RZ, Kuter D, Limentani SA, Lee S, Hideshima T, Esseltine DL, Kauffman M, Adams J, Schenkein DP, Anderson KC: A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med. 2003, 348: 2609-2617. 10.1056/NEJMoa030288.
Davis NB, Taber DA, Ansari RH, Ryan CW, George C, Vokes EE, Vogelzang NJ, Stadler WM: Phase II trial of PS-341 in patients with renal cell cancer: a University of Chicago phase II consortium study. J Clin Oncol. 2004, 22: 115-119. 10.1200/JCO.2004.07.165.
Papandreou CN, Daliani DD, Nix D, Yang H, Madden T, Wang X, Pien CS, Millikan RE, Tu SM, Pagliaro L, Kim J, Adams J, Elliott P, Esseltine D, Petrusich A, Dieringer P, Perez C, Logothetis CJ: Phase I trial of the proteasome inhibitor bortezomib in patients with advanced solid tumors with observations in androgen-independent prostate cancer. J Clin Oncol. 2004, 22: 2108-2121. 10.1200/JCO.2004.02.106.
Kondagunta GV, Drucker B, Schwartz L, Bacik J, Marion S, Russo P, Mazumdar M, Motzer RJ: Phase II trial of bortezomib for patients with advanced renal cell carcinoma. J Clin Oncol. 2004, 22: 3720-3725. 10.1200/JCO.2004.10.155.
Cortes J, Thomas D, Koller C, Giles F, Estey E, Faderl S, Garcia-Manero G, McConkey D, Patel G, Guerciolini R, Wright J, Kantarjian H: Phase I study of bortezomib in refractory or relapsed acute leukemias. Clin Cancer Res. 2004, 10: 3371-3376.
Jagannath S, Barlogie B, Berenson J, Siegel D, Irwin D, Richardson PG, Niesvizky R, Alexanian R, Limentani SA, Alsina M, Adams J, Kauffman M, Esseltine DL, Schenkein DP, Anderson KC: A phase 2 study of two doses of bortezomib in relapsed or refractory myeloma. Br J Haematol. 2004, 127: 165-172. 10.1111/j.1365-2141.2004.05188.x.
Shah MH, Young D, Kindler HL, Webb I, Kleiber B, Wright J, Grever M: Phase II study of the proteasome inhibitor bortezomib (PS-341) in patients with metastatic neuroendocrine tumors. Clin Cancer Res. 2004, 10: 6111-6118.
Bruno B, Rotta M, Giaccone L, Massaia M, Bertola A, Palumbo A, Boccadoro M: New drugs for treatment of multiple myeloma. Lancet Oncol. 2004, 5: 430-442. 10.1016/S1470-2045(04)01511-6.
Adams J: The development of proteasome inhibitors as anticancer drugs. Cancer Cell. 2004, 5: 417-421. 10.1016/S1535-6108(04)00120-5.
Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ: Proteasome inhibitors: a novel class of potent and effective antitumor agents. Cancer Res. 1999, 59: 2615-2622.
Wojcik C, DeMartino GN: Intracellular localization of proteasomes. Int J Biochem Cell Biol. 2003, 35: 579-589. 10.1016/S1357-2725(02)00380-1.
Hideshima T, Richardson P, Chauhan D, Palombella VJ, Elliott PJ, Adams J, Anderson KC: The proteasome inhibitor PS-341 inhibits growth, induces apoptosis, and overcomes drug resistance in human multiple myeloma cells. Cancer Res. 2001, 61: 3071-3076.
Russo SM, Tepper JE, Baldwin ASJ, Liu R, Adams J, Elliott P, Cusack JCJ: Enhancement of radiosensitivity by proteasome inhibition: implications for a role of NF-kappaB. Int J Radiat Oncol Biol Phys. 2001, 50: 183-193. 10.1016/S0360-3016(01)01446-8.
Sunwoo JB, Chen Z, Dong G, Yeh N, Crowl BC, Sausville E, Adams J, Elliott P, Van Waes C: Novel proteasome inhibitor PS-341 inhibits activation of nuclear factor-kappa B, cell survival, tumor growth, and angiogenesis in squamous cell carcinoma. Clin Cancer Res. 2001, 7: 1419-1428.
Hideshima T, Chauhan D, Richardson P, Mitsiades C, Mitsiades N, Hayashi T, Munshi N, Dang L, Castro A, Palombella V, Adams J, Anderson KC: NF-kappa B as a therapeutic target in multiple myeloma. J Biol Chem. 2002, 277: 16639-16647. 10.1074/jbc.M200360200.
Tan C, Waldmann TA: Proteasome inhibitor PS-341, a potential therapeutic agent for adult T-cell leukemia. Cancer Res. 2002, 62: 1083-1086.
Ma MH, Yang HH, Parker K, Manyak S, Friedman JM, Altamirano C, Wu ZQ, Borad MJ, Frantzen M, Roussos E, Neeser J, Mikail A, Adams J, Sjak-Shie N, Vescio RA, Berenson JR: The proteasome inhibitor PS-341 markedly enhances sensitivity of multiple myeloma tumor cells to chemotherapeutic agents. Clin Cancer Res. 2003, 9: 1136-1144.
Hideshima T, Mitsiades C, Akiyama M, Hayashi T, Chauhan D, Richardson P, Schlossman R, Podar K, Munshi NC, Mitsiades N, Anderson KC: Molecular mechanisms mediating antimyeloma activity of proteasome inhibitor PS-341. Blood. 2003, 101: 1530-1534. 10.1182/blood-2002-08-2543.
Chauhan D, Li G, Podar K, Hideshima T, Mitsiades C, Schlossman R, Munshi N, Richardson P, Cotter FE, Anderson KC: Targeting mitochondria to overcome conventional and bortezomib/proteasome inhibitor PS-341 resistance in multiple myeloma (MM) cells. Blood. 2004, 104: 2458-2466. 10.1182/blood-2004-02-0547.
Yang Y, Ikezoe T, Saito T, Kobayashi M, Koeffler HP, Taguchi H: Proteasome inhibitor PS-341 induces growth arrest and apoptosis of non-small cell lung cancer cells via the JNK/c-Jun/AP-1 signaling. Cancer Sci. 2004, 95: 176-180.
Shah SA, Potter MW, McDade TP, Ricciardi R, Perugini RA, Elliott PJ, Adams J, Callery MP: 26S proteasome inhibition induces apoptosis and limits growth of human pancreatic cancer. J Cell Biochem. 2001, 82: 110-122. 10.1002/jcb.1150.
Williams SA, McConkey DJ: The proteasome inhibitor bortezomib stabilizes a novel active form of p53 in human LNCaP-Pro5 prostate cancer cells. Cancer Res. 2003, 63: 7338-7344.
Breitschopf K, Zeiher AM, Dimmeler S: Ubiquitin-mediated degradation of the proapoptotic active form of bid. A functional consequence on apoptosis induction. J Biol Chem. 2000, 275: 21648-21652. 10.1074/jbc.M001083200.
Li B, Dou QP: Bax degradation by the ubiquitin/proteasome-dependent pathway: involvement in tumor survival and progression. Proc Natl Acad Sci U S A. 2000, 97: 3850-3855. 10.1073/pnas.070047997.
Podar K, Shringarpure R, Tai YT, Simoncini M, Sattler M, Ishitsuka K, Richardson PG, Hideshima T, Chauhan D, Anderson KC: Caveolin-1 is required for vascular endothelial growth factor-triggered multiple myeloma cell migration and is targeted by bortezomib. Cancer Res. 2004, 64: 7500-7506.
Hideshima T, Chauhan D, Hayashi T, Akiyama M, Mitsiades N, Mitsiades C, Podar K, Munshi NC, Richardson PG, Anderson KC: Proteasome inhibitor PS-341 abrogates IL-6 triggered signaling cascades via caspase-dependent downregulation of gp130 in multiple myeloma. Oncogene. 2003, 22: 8386-8393. 10.1038/sj.onc.1207170.
Cusack JCJ, Liu R, Houston M, Abendroth K, Elliott PJ, Adams J, Baldwin ASJ: Enhanced chemosensitivity to CPT-11 with proteasome inhibitor PS-341: implications for systemic nuclear factor-kappaB inhibition. Cancer Res. 2001, 61: 3535-3540.
Zong WX, Edelstein LC, Chen C, Bash J, Gelinas C: The prosurvival Bcl-2 homolog Bfl-1/A1 is a direct transcriptional target of NF-kappaB that blocks TNFalpha-induced apoptosis. Genes Dev. 1999, 13: 382-387.
Dong QG, Sclabas GM, Fujioka S, Schmidt C, Peng B, Wu T, Tsao MS, Evans DB, Abbruzzese JL, McDonnell TJ, Chiao PJ: The function of multiple IkappaB : NF-kappaB complexes in the resistance of cancer cells to Taxol-induced apoptosis. Oncogene. 2002, 21: 6510-6519. 10.1038/sj.onc.1205848.
Loercher A, Lee TL, Ricker JL, Howard A, Geoghegen J, Chen Z, Sunwoo JB, Sitcheran R, Chuang EY, Mitchell JB, Baldwin ASJ, Van Waes C: Nuclear factor-kappaB is an important modulator of the altered gene expression profile and malignant phenotype in squamous cell carcinoma. Cancer Res. 2004, 64: 6511-6523.
Gabai VL, Meriin AB, Yaglom JA, Volloch VZ, Sherman MY: Role of Hsp70 in regulation of stress-kinase JNK: implications in apoptosis and aging. FEBS Lett. 1998, 438: 1-4. 10.1016/S0014-5793(98)01242-3.
MacLaren AP, Chapman RS, Wyllie AH, Watson CJ: p53-dependent apoptosis induced by proteasome inhibition in mammary epithelial cells. Cell Death Differ. 2001, 8: 210-218. 10.1038/sj.cdd.4400801.
Ling YH, Liebes L, Jiang JD, Holland JF, Elliott PJ, Adams J, Muggia FM, Perez-Soler R: Mechanisms of proteasome inhibitor PS-341-induced G(2)-M-phase arrest and apoptosis in human non-small cell lung cancer cell lines. Clin Cancer Res. 2003, 9: 1145-1154.
Bancroft CC, Chen Z, Dong G, Sunwoo JB, Yeh N, Park C, Van Waes C: Coexpression of proangiogenic factors IL-8 and VEGF by human head and neck squamous cell carcinoma involves coactivation by MEK-MAPK and IKK-NF-kappaB signal pathways. Clin Cancer Res. 2001, 7: 435-442.
Lee AH, Iwakoshi NN, Anderson KC, Glimcher LH: Proteasome inhibitors disrupt the unfolded protein response in myeloma cells. Proc Natl Acad Sci U S A. 2003, 100: 9946-9951. 10.1073/pnas.1334037100.
Mimnaugh EG, Xu W, Vos M, Yuan X, Isaacs JS, Bisht KS, Gius D, Neckers L: Simultaneous inhibition of hsp 90 and the proteasome promotes protein ubiquitination, causes endoplasmic reticulum-derived cytosolic vacuolization, and enhances antitumor activity. Mol Cancer Ther. 2004, 3: 551-566.
Denlinger CE, Rundall BK, Jones DR: Proteasome inhibition sensitizes non-small cell lung cancer to histone deacetylase inhibitor-induced apoptosis through the generation of reactive oxygen species. J Thorac Cardiovasc Surg. 2004, 128: 740-748. 10.1016/j.jtcvs.2004.07.010.
Fribley A, Zeng Q, Wang CY: Proteasome inhibitor PS-341 induces apoptosis through induction of endoplasmic reticulum stress-reactive oxygen species in head and neck squamous cell carcinoma cells. Mol Cell Biol. 2004, 24: 9695-9704. 10.1128/MCB.24.22.9695-9704.2004.
Kostova Z, Wolf DH: For whom the bell tolls: protein quality control of the endoplasmic reticulum and the ubiquitin-proteasome connection. EMBO J. 2003, 22: 2309-2317. 10.1093/emboj/cdg227.
LeBlanc R, Catley LP, Hideshima T, Lentzsch S, Mitsiades CS, Mitsiades N, Neuberg D, Goloubeva O, Pien CS, Adams J, Gupta D, Richardson PG, Munshi NC, Anderson KC: Proteasome inhibitor PS-341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res. 2002, 62: 4996-5000.
Satou Y, Nosaka K, Koya Y, Yasunaga JI, Toyokuni S, Matsuoka M: Proteasome inhibitor, bortezomib, potently inhibits the growth of adult T-cell leukemia cells both in vivo and in vitro. Leukemia. 2004, 18: 1357-1363. 10.1038/sj.leu.2403400.
Teicher BA, Ara G, Herbst R, Palombella VJ, Adams J: The proteasome inhibitor PS-341 in cancer therapy. Clin Cancer Res. 1999, 5: 2638-2645.
Williams S, Pettaway C, Song R, Papandreou C, Logothetis C, McConkey DJ: Differential effects of the proteasome inhibitor bortezomib on apoptosis and angiogenesis in human prostate tumor xenografts. Mol Cancer Ther. 2003, 2: 835-843.
Nawrocki ST, Bruns CJ, Harbison MT, Bold RJ, Gotsch BS, Abbruzzese JL, Elliott P, Adams J, McConkey DJ: Effects of the proteasome inhibitor PS-341 on apoptosis and angiogenesis in orthotopic human pancreatic tumor xenografts. Mol Cancer Ther. 2002, 1: 1243-1253.
Bold RJ, Virudachalam S, McConkey DJ: Chemosensitization of pancreatic cancer by inhibition of the 26S proteasome. J Surg Res. 2001, 100: 11-17. 10.1006/jsre.2001.6194.
Amiri KI, Horton LW, LaFleur BJ, Sosman JA, Richmond A: Augmenting chemosensitivity of malignant melanoma tumors via proteasome inhibition: implication for bortezomib (VELCADE, PS-341) as a therapeutic agent for malignant melanoma. Cancer Res. 2004, 64: 4912-4918.
Mitsiades N, Mitsiades CS, Richardson PG, Poulaki V, Tai YT, Chauhan D, Fanourakis G, Gu X, Bailey C, Joseph M, Libermann TA, Schlossman R, Munshi NC, Hideshima T, Anderson KC: The proteasome inhibitor PS-341 potentiates sensitivity of multiple myeloma cells to conventional chemotherapeutic agents: therapeutic applications. Blood. 2003, 101: 2377-2380. 10.1182/blood-2002-06-1768.
Kamat AM, Karashima T, Davis DW, Lashinger L, Bar-Eli M, Millikan R, Shen Y, Dinney CP, McConkey DJ: The proteasome inhibitor bortezomib synergizes with gemcitabine to block the growth of human 253JB-V bladder tumors in vivo. Mol Cancer Ther. 2004, 3: 279-290.
Small GW, Shi YY, Edmund NA, Somasundaram S, Moore DT, Orlowski RZ: Evidence that mitogen-activated protein kinase phosphatase-1 induction by proteasome inhibitors plays an antiapoptotic role. Mol Pharmacol. 2004, 66: 1478-1490. 10.1124/mol.104.003400.
Nawrocki ST, Sweeney-Gotsch B, Takamori R, McConkey DJ: The proteasome inhibitor bortezomib enhances the activity of docetaxel in orthotopic human pancreatic tumor xenografts. Mol Cancer Ther. 2004, 3: 59-70.
Pervan M, Pajonk F, Sun JR, Withers HR, McBride WH: Molecular pathways that modify tumor radiation response. Am J Clin Oncol. 2001, 24: 481-485. 10.1097/00000421-200110000-00013.
Mitsiades CS, Treon SP, Mitsiades N, Shima Y, Richardson P, Schlossman R, Hideshima T, Anderson KC: TRAIL/Apo2L ligand selectively induces apoptosis and overcomes drug resistance in multiple myeloma: therapeutic applications. Blood. 2001, 98: 795-804. 10.1182/blood.V98.3.795.
Sayers TJ, Brooks AD, Koh CY, Ma W, Seki N, Raziuddin A, Blazar BR, Zhang X, Elliott PJ, Murphy WJ: The proteasome inhibitor PS-341 sensitizes neoplastic cells to TRAIL-mediated apoptosis by reducing levels of c-FLIP. Blood. 2003, 102: 303-310. 10.1182/blood-2002-09-2975.
Yu C, Rahmani M, Conrad D, Subler M, Dent P, Grant S: The proteasome inhibitor bortezomib interacts synergistically with histone deacetylase inhibitors to induce apoptosis in Bcr/Abl+ cells sensitive and resistant to STI571. Blood. 2003, 102: 3765-3774. 10.1182/blood-2003-03-0737.
Mitsiades CS, Mitsiades NS, McMullan CJ, Poulaki V, Shringarpure R, Hideshima T, Akiyama M, Chauhan D, Munshi N, Gu X, Bailey C, Joseph M, Libermann TA, Richon VM, Marks PA, Anderson KC: Transcriptional signature of histone deacetylase inhibition in multiple myeloma: biological and clinical implications. Proc Natl Acad Sci U S A. 2004, 101: 540-545. 10.1073/pnas.2536759100.
Denlinger CE, Keller MD, Mayo MW, Broad RM, Jones DR: Combined proteasome and histone deacetylase inhibition in non-small cell lung cancer. J Thorac Cardiovasc Surg. 2004, 127: 1078-1086. 10.1016/S0022-5223(03)01321-7.
Pei XY, Dai Y, Grant S: Synergistic induction of oxidative injury and apoptosis in human multiple myeloma cells by the proteasome inhibitor bortezomib and histone deacetylase inhibitors. Clin Cancer Res. 2004, 10: 3839-3852.
Sun K, Welniak LA, Panoskaltsis-Mortari A, O'Shaughnessy MJ, Liu H, Barao I, Riordan W, Sitcheran R, Wysocki C, Serody JS, Blazar BR, Sayers TJ, Murphy WJ: Inhibition of acute graft-versus-host disease with retention of graft-versus-tumor effects by the proteasome inhibitor bortezomib. Proc Natl Acad Sci U S A. 2004, 101: 8120-8125. 10.1073/pnas.0401563101.
Wang CY, Cusack JCJ, Liu R, Baldwin ASJ: Control of inducible chemoresistance: enhanced anti-tumor therapy through increased apoptosis by inhibition of NF-kappaB. Nat Med. 1999, 5: 412-417. 10.1038/10577.
Mayo MW, Baldwin AS: The transcription factor NF-kappaB: control of oncogenesis and cancer therapy resistance. Biochim Biophys Acta. 2000, 1470: M55-M62.
Fahy BN, Schlieman MG, Virudachalam S, Bold RJ: Schedule-dependent molecular effects of the proteasome inhibitor bortezomib and gemcitabine in pancreatic cancer. J Surg Res. 2003, 113: 88-95. 10.1016/S0022-4804(03)00201-4.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
M.B. has received consulting and lecture fees from Millennium Pharmaceuticals, Inc. G.M. declares that he has no competing interests; he is supported by the Leukaemia Research Fund and the International Myeloma Foundation. J.C. has received advisory board and speakers' bureau fees from Millennium Pharmaceuticals, Inc. and Ortho Biotech.
Authors' contributions
M.B. reviewed the literature and drafted the manuscript. G.M. and J.C. reviewed and revised the manuscript. All authors read and approved the final version.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
{kind=link}
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Boccadoro, M., Morgan, G. & Cavenagh, J. Preclinical evaluation of the proteasome inhibitor bortezomib in cancer therapy. Cancer Cell Int 5, 18 (2005). https://doi.org/10.1186/1475-2867-5-18
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2867-5-18