
Abstract
Background
The multidrug resistance (MDR) 1 gene encodes a 170-kDa membrane transporter called P-glycoprotein, which plays an important role in protecting cells against lipophilic xenobiotics by the way of an ATP-dependent cellular efflux mechanism. Three polymorphisms of MDR1, 3435C > T located in exon 26, 1236C > T in exon 12 and 2677G > T/A in exon 21 were the most extensively studied and were identified functionally important and ethnically diverse mapping to the gene region. Considering the potential influence of altering MDR1 activity, it is plausible that MDR1 polymorphisms might play a role in the development of cancer. Although the effects of MDR1 polymorphisms on susceptibility to human cancer have been investigated in many studies, the results still remain conflicting.
Methods
To resolve these conflicts, we performed a quantitative synthesis of the association between these three polymorphisms and cancer risk, including 52 studies (15789 cases and 20274 controls) for 3435C > T polymorphism, 10 studies (2101 cases and 2842 controls) for 1236C > T polymorphism and 18 studies (3585 cases and 4351 controls) for 2677G > T/A polymorphism.
Results
The stratified analyses for 3435C > T polymorphism, individuals with T-allele in 3435C > T had significantly higher ALL risks (TT versus CC: OR =1.286, 95% CI =1.123-1.474); significantly elevated risks were observed among Caucasian populations (TT versus CC: OR =1.276, 95% CI =1.112-1.464). When restricting the analysis to the source of controls, we found that HB (hospital-based) genetic models had higher risks (TT versus CC: OR =1.307, 95% CI =1.046-1.632), as well as in PB (population-based) genetic models (TT versus CC: OR =1.294, 95% CI =1.079-1.55).
The T/A-allele frequency of 2677G > T/A polymorphism was associated with higher risk of cancer (TT + TA + AA vs. GG: OR =1.348, 95% CI =1.031-1.762), significantly elevated risks were observed among Asian populations (TT + TA + AA vs. GG: OR =1.642, 95% CI =1.340-2.012), and elevated risks could be associated with PB models (TT + TA + AA vs. GG: OR =1.641, 95% CI =1.018-2.646).
Conclusions
Our meta-analysis suggested that 3435C > T polymorphism and 2677G > T/A polymorphism were associated with cancer risk when all studies were pooled together, while 1236C > T polymorphism not.
Similar content being viewed by others

Background
The human multidrug resistance 1 (MDR1 or ABCB1) gene encodes a 170-kDa membrane transport protein called P-glycoprotein. For minimizing the exposure of potential toxic compounds to the cellular homeostasis, P-glycoprotein is expressed primarily in regions that act as epithelial barriers or perform excretory functions, including blood-tissue barrier, the gastrointestinal tract, liver and kidney. Therefore, P-glycoprotein can play the role of sweeper by extruding several exogenous and endogenous substances, using ATP-dependent efflux pump [1–4]. The alteration of the cellular defense mechanism mediated by P-gp has been speculated to be closely associated with the development of various cancers including hepatocarcinoma, colorectal carcinoma, acute lymphoblastic leukemia and gall bladder tumors [5–8]. These suggest that MDR1 may play an important role in the elimination of carcinogens, and the mutation of MDR1 may lead to human malignancies [5]. Several studies try to prove the causal function of P-gp in tumorigenesis by animal experiments. One study by Mochida Y et al. suggested that the absence of the P-gp role suppressed the development of intestinal neoplasia in Apc (Min/+) mice, and a P-gp inhibitor was found to suppress tumorigenesis in rats subsequently [9, 10]. While Schinkel et al. conducted a study comparing normal MDR1 a (+/+) mice (MDR1 a is the mouse equivalent to the human MDR1 gene) to constructed MDR1 a (−/−) disrupted mice to find that the MDR1 a (−/−)mice resulted in cumulative toxicity of the pesticide, ivermectin due to decreased extrusion of these compounds [11]. Considering these data, we can infer that genetical absence of Pg-p expression may result in more exposure to environmental xenobiotics so that increased opportunity linked to the risk of malignancy was obtained. However, the causal relationship between MDR1 and the tumorigenesis has not been fully elucidated yet.
Recently, at least 50 single-nucleotide polymorphisms have been reported within MDR1 gene locus [12, 13]. Among the systematic screens of this gene, Hoffmeyer et al. reported significant role that a synonymous SNP played at position 3435 located in exon 26 in the P-glycoprotein function [14]. Recent studies have found that C3435T was in linkage disequilibrium with two other common SNPs, the synonymous C1236T (exon 12) and nonsynonymous triallelic G2677T/A (exon 21) [15–17].
Considering the potential influence of these SNPs, many molecular epidemiological studies were conducted to investigate the association between these SNPs and cancer risk in humans. However, the results from different studies are to some extent divergent, but nevertheless intriguing, which may be owing to limitations in individual studies. To clarify this issue, we performed a meta-analysis with subgroup analysis from all eligible studies focusing on 3435C > T, 1236C > T and 2677G > T/A, to obtain a more precise estimation of the relationship between polymorphisms and cancer risk.
Material and methods
Identification and eligibility of relevant studies
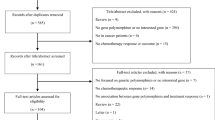
All case–control studies on the association between MDR1 polymorphisms and cancer risk published up to November 30, 2012 were identified through comprehensive searches using the PubMed and Medline database, ScienceDirect database, Springerlink database, Wiley Online Library, BioMed Central, Nature Series, Science Online, Cell Press Journals, CNKI, WanFang database with the following terms and keywords: “MDR1,” “ABCB1,” “single-nucleotide polymorphism,” and in combination with “leukemia,” “cancer,” “tumor” and “carcinoma.” The search was limited to human studies. In addition, we have especially reviewed the references cited in checked articles and identified some additional articles missed by the searching.
Inclusion criteria
The following criteria were used for the study selection: (1) a case–control study evaluating at least one of these three polymorphisms (3435C > T, 1236C > T and 2677G > T/A) and cancer risk; (2) studies with full-text articles; (3) no overlapping data. and (4) sufficient data for estimating an odds ratio (OR) with 95% confidence interval (95% CI).
Data extraction
Information was carefully extracted from all the eligible publications. The following data were collected from each study: first author’s name, publication date, country, ethnicity, cancer type, source of controls (population-based [PB] or hospital-based [HB] controls), genotyping method, total numbers of cases and controls and number of cases and controls for each MDR1 polymorphism. Meanwhile, different case–control groups in one study were considered as independent studies. For each study, we did not define a minimum number of patients for inclusion in our meta-analysis.
Statistical methods
The strength of association between MDR1 polymorphisms and cancer risk was measured by ORs with 95% CIs. The risks (ORs) of cancer associated with the three
polymorphisms were estimated for each study. In our study, the C- allele, C-allele and G-allele were considered the reference genotypes, respectively in 3435C > T, 1236C > T and 2677G > T/A. The pooled ORs were performed for co-dominant model (TT vs. CC and TC vs. CC, TT + TA + AA vs. GG and GT + GA vs. GG), dominant model (TC + TT vs. CC, TT + TA + AA + GT + GA vs. GG) and recessive model (TT vs. TC + CC, TT + TA + AA vs. GT + GA + AA), respectively. Heterogeneity assumption was checked by a χ 2-based Q-test. A p-value of >0.05 for the Q-test indicated a lack of heterogeneity among studies, so that the pooled OR estimate of each study was calculated by the fixed-effects model (the Mantel–Haenszel method). Otherwise, the random-effects model (the DerSimonian and Laird method) was used. Heterogeneity was quantified with the I2 metric, which is independent of the number of studies in the meta-analysis (I2 < 25%: no heterogeneity; I2 = 25–50%: moderate heterogeneity; I2 = 50–75%: large heterogeneity, I2 > 75%: extreme heterogeneity). Subgroup analyses were performed by cancer type (if one cancer type contained fewer than three individual studies, it was combined into an “other cancers” group), ethnicity and source of controls. Before analysis for each study, we examined whether the genotype distribution of controls was consistent with Hardy– Weinberg equilibrium using the χ 2 test. We performed One-way sensitivity analysis by deleting a single study in the meta-analysis each time to reflect the influence of the individual data set to the pooled OR to assess the stability of the results. An estimate of potential publication bias was carried out by the funnel plot, in which the standard error of log (OR) of each study was plotted against its log (OR). An asymmetric plot suggests a possible publication bias. Funnel plot asymmetry was assessed by the method of Egger’s linear regression test, a linear regression approach to measure funnel plot asymmetry on the natural logarithm scale of the OR. The significance of the intercept was determined by the t-test suggested by Egger (p < 0.05 was considered a significant publication bias). All of the statistical tests used in our meta-analysis were performed by SPSS version 13.0 and STATA version 11.0 (Stata, College Station, TX).
Results
Eligible studies and meta-analysis databases
A total of 48 publications with 52 case–control studies exploring the association between MDR1 3435C > T, 1236C > T and 2677G > T/A polymorphisms and cancer risk were found [2, 5, 18–62]. Hence, as summarized in Table 1, 52 studies (15789 cases and 20274 controls) for 3435C > T polymorphism, 10 studies (2101 cases and 2842 controls) for 1236C > T polymorphism and 18 studies (3585 cases and 4351 controls) for 2677G > T/A polymorphism were selected in the meta-analysis, of which one publications had three independent studies and were considered separately. As summarized in Table 1, there were 25 hospital-based studies and 26 population-based studies. There were 8 acute lymphoblastic leukemia (ALL) studies, 14 colorectal cancer studies, 9 breast cancer studies, 4 gastric cancer studies, 3 renal cell cancer studies, 2 acute myelocytic leukemia (AML) studies, 2 lung cancer study, 2 B-cell chronic lymphocytic leukemia (CLL) studies, one endometrial cancer study, one esophageal squamous cell carcinoma study, one glioma study, one upper aerodigestive tract (UADT) cancers study, one multiple myeloma study, one leukemia study, one plasma cell myeloma study and one study with Hodgkin’s lymphoma (HL). Thirty-three studies were conducted in Europeans, seventeen studies were conducted in Asians. Two of the remained studies were conducted in Americans, and the other was in Mexicans. These studies indicate that the distribution of genotypes in controls was consistent with Hardy–Weinberg equilibrium. And the subjects of controls were matched for age and gender. Most of the cases were confirmed histologically or pathologically.
Quantitative synthesis
There was a wide variation in the T-allele and T/A-allele frequency of 3435C > T, 1236C > T and 2677G > T/A polymorphism between the two major ethnicities. For Asians, the T- allele frequency of 3435C > T was 25.52% (95% CI =23.84–27.21%), which was significantly higher than that in Caucasians (24.28%, 95% CI = 20.54–28.03%, p < 0.001). There was no statistical difference for the T- allele frequency of 1236C > T between Asians (24.28%, 95% CI = 11.37–26.56%) and Caucasians (36.11%, 95% CI = 27.74–44.47%, p =0.633). And for Asians (23.47%, 95% CI = 19.78–25.96%) whose T/A-allele frequency of 2677G > T/A polymorphism was not equivalent as Caucasians (22.87%, 95% CI = 11.12–35.82%, p =0.633).
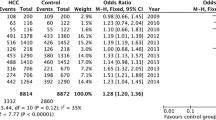
Tables 2 summarizes the main results of the meta-analysis for MDR1 3435C > T polymorphisms. Overall, we found that individuals with T-allele in 3435C > T had a higher risk of cancer (co-dominant model TT versus CC: OR =1.286, 95% CI =1.123–1.474; CT versus CC: OR = 1.126, 95% CI = 1.020–1.244; dominant model TT + CT versus CC: OR = 1.176, 95% CI = 1.068–1.295; recessive model TT versus CT + CC: OR =1.191, 95% CI =1.065–1.333). In the subgroup analysis by cancer type, the results indicated that individuals with T-allele in 3435C > T had significantly higher ALL risks (TT versus CC: OR =1.890, 95% CI =1.177–3.037), otherwise no significant association was found between higher CRC risks and T-allele in 3435C > T, neither was between breast cancer and T-allele in 3435C > T. When stratified by ethnicity, significantly elevated risks were observed among Caucasian populations(co-dominant model TT versus CC: OR =1.276, 95% CI =1.112–1.464; CT versus CC: OR = 1.172, 95% CI = 1.047–1.313; dominant model TT + CT versus CC: OR = 1.212, 95% CI = 1.083–1.357), whereas significantly elevated risks were not observed among Asian populations (co-dominant model TT versus CC: OR =1.314, 95% CI =0.894–1.933). When restricting the analysis to the source of controls, we found that HB genetic models had higher risks (TT versus CC: OR =1.307, 95% CI =1.046–1.632; dominant model: OR =1.170, 95% CI =1.009–1.357), and association was detected in PB genetic models also (TT versus CC: OR =1.294, 95% CI =1.079–1.552; CT versus CC: OR = 1.150, 95% CI = 1.019–1.299; dominant model TT + CT versus CC: OR = 1.459, 95% CI = 1.246–1.709; recessive model TT versus CT + CC: OR =1.183, 95% CI =1.033–1.355) (Figure 1).

Forest plot of cancer risk associated with the MDR1 3435C > T polymorphism (TT vs. CC, random effects). The squares and horizontal lines correspond to the study-specific OR and 95% CI. The area of the squares reflects the study-specific weight (inverse of the variance). The diamonds represent the pooled OR and 95% CI.
We also found that T/A-allele frequency of 2677G > T/A polymorphism was associated with higher risk of bearing cancer (2677G > T/A: TT + TA + AA vs. GG: OR =1.348, 95% CI =1.031–1.762; dominant model: OR = 1.161, 95% CI = 1.051–1.281; recessive model: OR = 1.278, 95% CI =1.022–1.597). In the subgroup analysis by ethnicity, significantly elevated risks were observed among Asian populations (co-dominant model TT + TA + AA vs. GG: OR =1.642, 95% CI =1.340–2.012; dominant model: OR = 1.273, 95% CI = 1.101–1.471; recessive model: OR = 1.481, 95% CI = 1.244–1.763), however no significantly elevated risks were observed among Caucasian populations. When stratified by the source of controls, we couldn’t find association between HB genetic models and higher cancer risks, but elevated risks could be associated with PB models (TT + TA + AA vs. GG: OR =1.641, 95% CI =1.018–2.646; GA + GT vs. GG: OR =1.164, 95% CI =1.008–1.345; dominant model: OR = 1.284, 95% CI =1.122–1.470).
The combined result based on all studies showed that there was no statistically significant link between cancer risk and 1236C > T (1236C > T: TT vs. CC: OR =1.325, 95% CI =0.824–2.133; CT vs. CC: OR = 1.133, 95% CI = 0.817–1.573; dominant model: OR = 1.173, 95% CI = 0.825–1.688; recessive model: OR = 1.191, 95% CI =0.840–1.690) (Table 2).
Heterogeneity and sensitivity analyses
For 3435C > T polymorphism, there was substantial heterogeneity among these studies for homozygote comparison (TT vs. CC: p heterogeneity = 0.000), and recessive model comparison (TT vs. CT + CC: p heterogeneity < 0.001). Then, we assessed the source of heterogeneity for homozygote comparison (TT vs. CC) by ethnicity, cancer type and source of controls. We found that cancer type (χ 2 = 18.51, df = 2 and p < 0.001), ethnicity (χ 2 = 9.58, df = 1 and p = 0.002) and the source of controls (χ 2 = 4.42, df = 1 and p = 0.036) all contributed to substantial heterogeneity. Although the sample size for cases and controls in 52 studies ranged from 38 to 5,486, the corresponding pooled ORs were not qualitatively altered with or without the study of small sample. Similarly, no other single study influenced the pooled OR materially as indicated by sensitivity analysis.
And for 2677G > T/A polymorphism, there was also heterogeneity for homozygote comparison (TT + TA + AA vs. GG: p heterogeneity = 0.000), recessive model comparison (p heterogeneity < 0.001). The heterogeneity we decided to analyze was homozygote comparison (TT + TA + AA vs. GG). Due to cancer type (χ 2 = 20.14, df = 7 and p = 0.005) and the source of controls (χ 2 = 14.53, df = 1 and p < 0.001), but not the ethnicity (χ 2 = 1.28, df = 1 and p = 0.258).
Publication bias
We performed Begg’s funnel plot and Egger’s test to assess the publication bias of literatures. As shown in Figure 2, the shape of the funnel plots did not reveal any evidence of obvious asymmetry. The results of Egger’s test still did not suggest any evidence of publication bias for 3435C > T polymorphism (p =0.085 for TT vs. CC, p =0.273 for CT vs. CC, p = 0.102 for dominant model, respectively and p = 0.176 for recessive model). There are neither Publication bias for 1236C > T polymorphism (p =0.247 for TT vs. CC, p =0.208 for CT vs. CC, p = 0.215 for dominant model, respectively and p = 0.332 for recessive model), nor for 2677G > T/A polymorphism (p =0.716 for TT + TA + AA vs. GG and p = 0.656 for recessive model, p =0.841 for GT + GA vs. GG, p = 0.971 for dominant model, respectively).

Begg’s funnel plots for publication bias test (3435C > T TT vs. CC). Each point represents a separate study for the indicated association. Log (OR), natural logarithm of OR. Horizontal line, means effect size.
Discussion
ATP-binding cassette (ABC) is one of the largest families of active importers and exporters that are located mainly in tissues acting as a barrier or having an excretory function. Most of the ABC transporters play a role in cell defense against environmental attacks generated by xenobiotics and intraceullar metabolic waste. The multidrug resistance 1 (MDR1/ABCB1) gene codes for P-glycoprotein, a membrane-bound transporter. Various cytokines, such as interleukin-1beta, benzo [a]pyrene and chemokines involved in inflammation seem to be the substrates of P-glycoprotein, which leading P-glycoprotein to be a potential cause of inflammation-induced carcinogenesis. MDR1 also seems to play a role in preventing early apoptosis in tumor cells.
Significant MDR1 gene heterogeneity, namely multiple mutations in the ABCB1/MDR1 gene has been demonstrated in previous studies. Analysis of all 28 exons of the ABCB1/MDR1 gene demonstrated at least 48 single-nucleotide polymorphisms (SNPs) to date, including promoter and the intron–exon region. The most frequent SNP MDR1 2677G > T/A in exon 21 (RefSNP ID: rs2032582), leads to amino acid exchange from Ala to Ser or Thr. The silent mutation in exon 26 MDR1 3435C > T (RefSNP ID: rs1045642) is associated with altered protein function. The third common polymorphism of ABCB1/MDR1 gene is a silent mutation in exon 12 MDR1 1236C > T (RefSNP ID: rs1128503). These three polymorphisms are closely related to linkage disequilibrium (LD). It was suggested that SNP is connected with susceptibility to many cancer types, such as renal epithelial tumors and acute lymphoblastic leukemia and CRC.
Our results showed that MDR1 3435C > T polymorphism is associated with cancer risk when all studies were pooled together (TT versus CC: OR =1.286, 95% CI =1.123–1.474; CT versus CC: OR = 1.126, 95% CI = 1.020–1.244; dominant model TT + CT versus CC: OR = 1.176, 95% CI = 1.068–1.295; recessive model TT versus CT + CC: OR =1.191, 95% CI =1.065–1.333). In the stratified analysis by cancer type, the results indicated that individuals with T-allele in 3435C > T had significantly higher ALL risks (TT versus CC: OR =1.890, 95% CI =1.177–3.037; TT + CT versus CC: OR = 1.240, 95% CI = 1.046–1.470; recessive model TT versus CT + CC: OR = 1.829, 95% CI =1.095–3.054), otherwise no significant association was found between higher CRC risks and T-allele in 3435C > T, neither was between breast cancer and T-allele in 3435C > T. When stratified by ethnicity, significantly elevated risks were observed among Caucasian populations(co-dominant model TT versus CC: OR =1.276, 95% CI =1.112–1.464; CT versus CC: OR = 1.172, 95% CI = 1.047–1.313; dominant model TT + CT versus CC: OR = 1.212, 95% CI = 1.083–1.357), whereas significantly elevated risks were not observed among Asian populations (co-dominant model TT versus CC: OR =1.314, 95% CI =0.894–1.933). When restricting the analysis to the source of controls, we found that HB genetic models had higher risks (TT versus CC: OR =1.307, 95% CI =1.046–1.632; dominant model: OR =1.170, 95% CI =1.009–1.357), and association was detected in PB genetic models also (TT versus CC: OR =1.294, 95% CI =1.079–1.552; CT versus CC: OR = 1.150, 95% CI = 1.019–1.299; dominant model TT + CT versus CC: OR = 1.459, 95% CI = 1.246–1.709; recessive model TT versus CT + CC: OR =1.183, 95% CI =1.033–1.355).
Inconsistent results might be attributed to the different roles MDR1 played in different cell types or tissues. We’ve found the association between MDR1 3435C > T polymorphism and ALL risk in subgroup analyses, as well as subgroup based on HB and PB genetic models. And in the stratified analysis by ethnicity, individuals carrying the T-allele in 3435C > T were significantly associated with elevated cancer risk in Asian as well as Caucasian populations compared with C-allele carriers.
We also found that T/A-allele frequency of 2677G > T/A polymorphism was associated with higher risk of cancer (2677G > T/A: TT + TA + AA vs. GG: OR =1.348, 95% CI =1.031–1.762). In the subgroup analysis by ethnicity, significantly elevated risks were observed among Asian populations (co-dominant model TT + TA + AA vs. GG: OR =1.642, 95% CI =1.340–2.012), however no significantly elevated risks were observed among Caucasian populations. When stratified by the source of controls, we couldn’t find association between HB genetic models and higher cancer risks, but elevated risks could be associated with PB models (TT + TA + AA vs. GG: OR =1.641, 95% CI =1.018–2.646; GA + GT vs. GG: OR =1.164, 95% CI =1.008–1.345; dominant model: OR = 1.284, 95% CI =1.122–1.470).
The combined result based on all studies showed that there was no statistically significant link between cancer risk and 1236C > T (1236C > T: TT vs. CC: OR =1.325, 95% CI =0.824–2.133; CT vs. CC: OR = 1.133, 95% CI = 0.817–1.573; dominant model: OR = 1.173, 95% CI = 0.825–1.688; recessive model: OR = 1.191, 95% CI =0.840–1.690).
It should be considered that the apparent inconsistency of these results may be caused by differences in disease prevalence, lifestyle, as well as possible limitations due to the relatively small sample size. The current knowledge of carcinogenesis indicates it is a process developed step by step, as well as influenced by multiple factors that involve various genetic alterations and several signaling pathways. Thus, it is unlikely that risk factors of cancer work in isolation from each other. Besides, even the same polymorphisms may act distinct roles in each cancer type, for different genetic backgrounds may contribute to the cancer discrepancy. And more importantly, the appearance determined by polymorphisms may largely depend on synthetically interaction with each polymorphism or a particular environmental exposure. Thus, it is possible that the effects of DNA repair function on cancer risk may be modified by multiple genetic polymorphisms. Also should we consider the chance findings as another plausible reason for the inconsistency of the results.
Although we have put considerable resources and efforts into discovering the association between MDR1 polymorphism and cancer risk as possible as we can, there still exists some limitations. First, when stratified by the source of controls, our results indicated that studies using hospital-based controls rather than population-based controls had a significantly increased risk. The reason may be that the hospital-based studies have some biases because such controls may contain certain other diverse diseases which can cause different risks of developing into cancer of various organs and may not be so representative as the general population. Therefore, using proper and more representative cancer-free control subjects are crucial for reducing biases in such case–control studies. Second, our results were based on single-factor estimates without adjustment for other risk factors such as age, smoking and drinking status, environmental factors and other variables, which might have caused serious confounding bias. Third, some inevitable publication bias might exist. Finally, the number of the published studies was not sufficiently large for a comprehensive analysis, particularly for the 1236C > T polymorphism. Hence we had to give up subgroup analysis for the polymorphism. For these limitations, our results should be interpreted with caution.
Our meta-analysis also has several strengths. First, statistically, a systematic review of the association of MDR1 polymorphism between cancer risks is more powerful than any single study. Second, the quality of eligible studies included in current meta-analysis was satisfactory and met our inclusion criterion. Third, we did not detect sufficient publication bias indicating that the whole results might be unbiased.
In conclusion, our meta-analysis suggests that the MDR1 3435C > T and 2677G > T/A polymorphism may contribute to genetic susceptibility of cancers. And the results support that the minor T-allele of the MDR1 3435C > T polymorphism is associated with a higher risk of acute lymphocytic leukemia, and significantly elevated risks were observed among Asian and Caucasian populations as well as HB and PB subgroups. And in the 2677G > T/A polymorphism, those who carry the T-allele and A-allele were associated with higher cancer risks among Asians and PB subgroup. However, it is necessary to conduct large sample studies using standardized unbiased genotyping methods, homogeneous cancer patients and well-matched controls. Moreover, further studies estimating the effect of gene–gene and gene–environment interactions may eventually lead to our better, comprehensive understanding of the association between the MDR1 polymorphism and cancer risk.
Abbreviations
- 95% CI:
-
95% confidence interval
- OR:
-
Odds ratio
- ALL:
-
Acute lymphoblastic leukemia
- HL:
-
Hodgkin lymphoma
- HB:
-
Hospital-based
- PB:
-
Population-based
- PCR-RFLP:
-
Polymerase chain reaction-restriction fragment length polymorphism
- MAF:
-
Minor allele frequency
- APEX:
-
Arrayed primer extensions.
References
Laura M, Hodges SMM, Chinn LW, Gow JM, Kroetz DL, Klein TE, Altman RB: Very important pharmacogene summary: ABCB1 (MDR1, Pglycoprotein). Pharmacogenet Genomics. 2011, 21 (3): 152-161. 10.1097/FPC.0b013e3283385a1c.
Urayama KY, Wiencke JK, Buffler PA, Chokkalingam AP, Metayer C, Wiemels JL: MDR1 Gene variants, indoor insecticide exposure, and the risk of childhood acute lymphoblastic leukemia. Cancer Epidemiol Biomarkers Prev. 2007, 16: 1172-1177. 10.1158/1055-9965.EPI-07-0007.
Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM: Biochemical, cellular, and pharmacological aspects of the multidrug transporter. Annu Rev Pharmacol Toxicol. 1999, 39: 361-398. 10.1146/annurev.pharmtox.39.1.361.
Schinkel AH: The physiological function of drug-transporting P-glycoproteins. Semin Cancer Biol. 1997, 8: 161-170. 10.1006/scbi.1997.0068.
Hattori H, Suminoe A, Morimasa W, Yuhki K, Kimitoshi K, Okamura J, Toshiro H, Akinobu M: Regulatory polymorphisms of multidrug resistance 1 (MDR1) gene are associated with the development of childhood acute lymphoblastic leukemia. Leuk Res. 2007, 31: 1633-1640. 10.1016/j.leukres.2007.04.009.
Wang BL, Zhai HY, Chen BY, Zhai SP, Yang HY, Chen XP: Clinical relationship between MDR1 gene and gallbladder cancer. Hepatobiliary Pancreat Dis Int. 2004, 3: 296-299.
Weinstein RS, Jakate SM, Dominguez JM, Lebovitz MD, Koukoulis GK, Kuszak JR: Relationship of the expression of the multidrug resistance gene product (P-glycoprotein) in human colon carcinoma to local tumor aggressiveness and lymph node metastasis. Cancer Res. 1991, 51: 2720-2726.
Thorgeirsson SS, Huber BE, Sorrell S, Fojo A, Pastan I, Gottesman MM: Expression of the multidrug-resistant gene in hepatocarcinogenesis and regenerating rat liver. Science. 1987, 236: 1120-1122. 10.1126/science.3576227.
Mochida Y, Taguchi K, Taniguchi S, Tsuneyoshi M, Kuwano H, Tsuzuki T, Michihiko K, Morimasa W: The role of P-glycoprotein in intestinal tumorigenesis: disruption of mdr1a suppresses polyp formation in Apc(Min/+)mice. Carcinogenesis. 2003, 24: 1219-1224. 10.1093/carcin/bgg073.
Yamada T, Mori Y, Hayashi R, Takada M, Ino Y, Naishiro Y, Tadashi K, Setsuo H: Suppression of intestinal polyposis in Mdr1-deficient ApcMin/+ mice. Cancer Res. 2003, 63: 895-901.
Schinkel AH, Smit JJM, van Tellingen O, Beijnen JH, Wagenaar E, van Deemter L, Mol CAAM, van der Valk MA, Robanus-Maandag EC, te Riele HPJ, Berns AJM, Borst P: Disruption of the mouse mdr1a P-glycoprotein gene leads to a deficiency in the blood–brain barrier and to increased sensitivity to drugs. Cell. 1994, 77: 491-502. 10.1016/0092-8674(94)90212-7.
Cascorbi I, Gerloff T, Johne A, Meisel C, Hoffmeyer S, Schwab M, Schaeffeler E, Eichelbaum M, Brinkmann U, Roots I: Frequency of single nucleotide polymorphisms in the P-glycoprotein drug transporter MDR1 gene in white subjects. Clin Pharmacol Ther. 2001, 69: 169-174. 10.1067/mcp.2001.114164.
Kroetz DL, Pauli-Magnus C, Hodges LM, Huang CC, Kawamoto M, Johns SJ, Stryke D, Ferrin TE, DeYoung J, Taylor T, Carlson EJ, Herskowitz I, Giacomini KM, Clark AG, Pharmacogenetics of Membrane Transporters Investigators: Sequence diversity and haplotype structure in the human ABCB1 (MDR1, multidrug resistance transporter) gene. Pharmacogenetics. 2003, 13: 481-494. 10.1097/00008571-200308000-00006.
Hoffmeyer S, Burk O, von Richter O, Arnold HP, Brockmöller J, Johne A, Cascorbi I, Gerloff T, Roots I, Eichelbaum M, Brinkmann U: Functional polymorphisms of the human multidrug-resistance gene: multiple sequence variations and correlation of one allele with P- glycoprotein expression and activity in vivo. Proc Natl Acad Sci U S A. 2000, 97: 3473-3478. 10.1073/pnas.97.7.3473.
Ji-Ye Y, Qiong H, Youyun Y, Jian-Ting Z, Mei-Zuo Z, Hong-Hao Z, Zhao-Qian L: Characterization and analyses of multidrug resistance-associated protein 1 (MRP1/ABCC1) polymorphisms in Chinese population. Pharmacogenet Genomics. 2009 March, 19 (3): 206-216. 10.1097/FPC.0b013e328323f680.
Sai K, Kaniwa N, Itoda M, Saito Y, Hasegawa R, Komamura K, Ueno K, Kamakura S, Kitakaze M, Shirao K, Minami H, Ohtsu A, Yoshida T, Saijo N, Kitamura Y, Kamatani N, Ozawa S, Sawada J: Haplotype analysis of ABCB1/MDR1 blocks in a Japanese population reveals genotype-dependent renal clearance of irinotecan. Pharmacogenetics. 2003, 13: 741-757. 10.1097/00008571-200312000-00005.
Tang K, Ngoi SM, Gwee PC, Chua JM, Lee EJ, Chong SS, Lee CG: Distinct haplotype profiles and strong linkage disequilibriumat the MDR1 multidrug transporter gene locus in three ethnic Asian populations. Pharmacogenetics. 2002, 12: 437-450. 10.1097/00008571-200208000-00004.
Rongrong L: The role of multidrug resistance gene 1 polymorphism to prognosis of adult acute lymphoblastic leukemia patients. Central South University. 2008, 5: 1-48.
Zhi-zhuo D: The singnificance of the expression of the CYP3A5 and MDRl genetic polymorphism in childhood acute lymphoblastic leukemia. Journal Of Clinical Pediatrics. 2011, 29 (3): 283-289.
Jamroziak KM, Lłynarski W, Balcerc Zak E, Mistygacz M, Trelin Ska J, Mirowski M, Bodalski J, Robak T: Functional C3435T polymorphism of MDR1 gene: an impact on genetic susceptibility and clinical outcome of childhood acute lymphoblastic leukemia. Eur J Haematol Eur J Haematol. 2004, 72: 314-321.
Evelia L-U, Melva G-A, Macías-Gómez NM, Valeria P-L, Jorge D-G, María De La Luz A-M, Miriam P-P, Patricio B-N, Dinorah R-D, Moreno-Ortiz JM, Jorge P-S, Juan Pablo M-E: MDR1 C3435T Polymorphism in Mexican children with acute lymphoblastic leukemia and in healthy individuals. Human Biol. 2008, 80 (4): 449-455. 10.3378/1534-6617-80.4.449.
Agnes F, Semsei Daniel J, Erdelyi I, Ungvari E, Kamory Be la C, Hajnalka A, Attila T, Edit C, Andras F, Kovacs GT, Csaba S: Association of some rare haplotypes and genotype combinations in the MDR1 gene with childhood acute lymphoblastic leukaemia. Leukemia Res. 2008, 32: 1214-1220. 10.1016/j.leukres.2007.12.009.
Vibeke A, Lene A, Dorte J, Østergaard M, Sæbø M, Hamfjord J, Kure E, Vogel U: Polymorphisms in the xenobiotic transporter multidrug resistance 1 (MDR1) and interaction with meat intake in relation to risk of colorectal cancer in a Danish prospective case-cohort study. BMC Cancer. 2009, 10: 10-18.
Campa D, Sainz J, Pardini B, Vodickova L, Naccarati A, Rudolph A, Novotny J, Forsti A, Buch S, von Witigo S, Schafmayer C, Volzke H, Hoffmeister M, Frank B, Barale R, Hemminki K, Hampe J, Chang-Claude J, Brenner H, Vodicka P, Canzian F: A comprehensive investigation on common polymorphisms in the MDR1/ABCB1 transporter gene and susceptibility to colorectal cancer. PLoS ONE. 2012, 7 (3): e32784-10.1371/journal.pone.0032784.
Potocnik U, Glava D, Dean M: Common germline MDR1/ABCB1 functional polymorphisms and haplotypes modify susceptibility to colorectal cancers with high microsatellite instability. Cancergencyto. 2008, 183: 28-34.
Mhaidat NM, Alshogran OY, Khabour OF, Alzoubi KH, Matalka II, Haddadin WJ, Mahasneh IO, Aldaher AN: Multi-drug resistance 1 genetic polymorphism and prediction of chemotherapy response in Hodgkin’s Lymphoma. J Exper Clin Cancer Res. 2011, 30 (68): 1-8.
Bae S-Y, Sun-Keun C, Kyung-Rae K, Chang-Shin P, Sung-Keun L, Hyung-Keun R, Dong-Woon S, Jae-Eun P, Ze-Hong W, Ju-Hee K: Effects of genetic polymorphisms of MDR1, FMO3 and CYP1A2 on susceptibility to colorectal cancer in Koreans. Cancer Sci. 2006, 97: 774-779. 10.1111/j.1349-7006.2006.00241.x.
Khedri A, Nejat-Shokouhi A, Salek R, Esmaeili H, Mokhtarifar A, Heravi RE, Tatari F, Behravan J, Miladpour B, Shahireh Omidvar SO: Association of the colorectal cancer and MDR1 gene polymorphism in an Iranian population. Mol Biol Rep. 2011, 38: 2939-2943. 10.1007/s11033-010-9957-9.
Kurzawski M, Drozdzik M, Suchy J, Kurzawski G, Białecka M, Gornik W, Lubinski J: Polymorphism in the P-glycoprotein drug transporter MDR1 gene in colon cancer patients. Eur J Clin Pharmacol. 2005, 61: 389-394. 10.1007/s00228-005-0926-5.
Komoto C, Nakamura T, Sakaeda T, Deanna I: MDR1 Haplotype frequencies in Japanese and Caucasian, and in Japanese patients with colorectal cancer and esophageal cancer. Drug Metab Pharmacokinet. 2006, 21 (2): 126-132. 10.2133/dmpk.21.126.
Lee B-I, Kyu-Yong C, Kang-Moon L, Woo-Chul C, Byung-Wook K, Hwang C, Se-Hyun C, Hyong-Ju K, Jin-Sun L, Myung-Seok K, Hiun-Suk C, In-Sik C: Is C3435T polymorphism of MDR1 related to inflammatory bowel disease or colorectal cancer in Korean?. 2006, 47: 22-29.
Osswald E, Andreas J, Laschinski G, Arjomand-Nahad F, Uwe M, Kirchheiner J, Gerloff T, Meisel C, Mrozikiewicz PM, Chernov J, Roots I, Köpke K: Association of MDR1 genotypes with susceptibility to colorectal cancer in older non-smokers. Eur J Clin Pharmacol. 2007, 63: 9-16.
Panczyk M, Ewa B, Sylwester P, Krzysztof J, Grażyna P-W, Mirowski M: ABCB1 gene polymorphisms and haplotype analysis in colorectal cancer. Int J Colorectal Dis. 2009, 24: 895-905. 10.1007/s00384-009-0724-0.
Petrova DT, Nedeva P, Maslyankov S, Toshev S, Yaramov N, Atanasova S, Toncheva D, Oellerich M, von Ahsen N: No association between MDR1 (ABCB1) 2677G > T and 3435C > T polymorphism and sporadic colorectal cancer among Bulgarian patients. J Cancer Res Clin Oncol. 2008, 134: 317-322. 10.1007/s00432-007-0279-9.
Sainz J, Rudolph A, Hein R, Hoffmeister M, Buch S, von Schonfels W, Hampe J, Schafmayer C, Volzke H, Frank B, Brenner H, Forsti A, Hemminki K, Chang-Claude J: Association of genetic polymorphisms in ESR2, HSD17B1, ABCB1, and SHBG genes with colorectal cancer risk. Endocrine-Related Cancer. 2011, 18: 265-276. 10.1530/ERC-10-0264.
Cizmarikova M, Wagnerova M, Schonova L, Habalova V, Kohut A, Linkova A, Sarissky M, Mojzis J, Mirossay L, Mirossay A: MDR1 (C3435T) polymorphism: relation to the risk of breast cancer and therapeutic outcome. Pharmacogenomics J. 2010, 10: 62-69. 10.1038/tpj.2009.41.
Taheri M, Mahjoubi1 F, Omranipour R: Effect of MDR1 polymorphism on multidrug resistance expression in breast cancer patients. Genet Mol Res. 2010, 9 (1): 34-40. 10.4238/vol9-1gmr669.
Tatari F, Roham S, Fatemeh M, Azam K, Behravan J: Association of C3435T single-nucleotide polymorphism of MDR1 gene with breast cancer in an Iranian population. DNA and Cell Biol. 2009, 28 (5): 259-263. 10.1089/dna.2008.0826.
George J, Kadambari D, Srinivasan K, Adithan C, Soya Sisy S, Elangovan S: A single-nucleotide polymorphism in the MDR1 gene as a predictor of response to neoadjuvant chemotherapy in breast cancer. Clin Breast Cancer. 2009, 9 (3): 161-165. 10.3816/CBC.2009.n.026.
Henriquez-Hernandez LA, Murias Rosales A, Hernandez Gonzalez A, Cabrera De Leon A, Diaz-Chico BN, Mori De Santiago M, Fernandez Perez L: Gene polymorphisms in TYMS, MTHFR, p53 and MDR1 as risk factors for breast cancer: a case–control study. Oncol Reports. 2009, 22: 1425-1433.
Nordgard SH, Ritchie MD, Jensrud SD, Motsinger AA, Alnæs GIG, Lemmon G, Marianne B, Geisler S, Moore JH, Lønning PE, Børresen-Dale AL, Kristensen VN: ABCB1 and GST polymorphisms associated with TP53 status in breast cancer. Pharmacogenetics Genomics. 2007, 17: 127-136. 10.1097/FPC.0b013e328011abaa.
Turgut S, Arzu Y, Raziye K, Gunfer T: MDR1 C3435T Polymorphism in patients with breast cancer. Archives Med Res. 2007, 38: 539-544. 10.1016/j.arcmed.2007.02.005.
Wu H, Kang H, Liu Y, Tong W, Duo L, Xiuli Y, Minqiong L, Weifan Y, Haishan Z, Desheng H, Xianzheng S, Enhua W, Minjie W: Roles of ABCB1 gene polymorphisms and haplotype in susceptibility to breast carcinoma risk and clinical outcomes. J Cancer Res Clin Oncol. 2012, 138: 1449-1462. 10.1007/s00432-012-1209-z.
Sabahi Z, Salek R, Heravi RE, Mosaffa F, Avanaki ZJ, Behravan J: Association of gastric cancer incidence with MDR1 gene polymorphism in an ethnic Iranian population. 2010, 47 (3): 317-321.
Sugimoto M, Takahisa F, Naohito S, Chise K, Masafumi N, Mihoko Y, Mutsuhiro I, Haruhiko S, Takashi I, Akira H: MDR1 C3435T polymorphism has no influence on developing Helicobacter pylori infection-related gastric cancer and peptic ulcer in Japanese. Life Sci. 2008, 83: 301-304. 10.1016/j.lfs.2008.06.022.
Tahara T, Tomiyasu A, Tomoyuki S, Ichiro H, Hiroshi N: Multi-drug resistance 1 polymorphism is associated with reduced risk of gastric cancer in the Japanese population. J Gastroenterol Hepatol. 2007, 22: 1678-1682. 10.1111/j.1440-1746.2007.04848.x.
Mrozikiewicz PM, Seremak-mrozikiewicz A, Semczuk A, Landt O, Breborowicz GH, Drews K: The significance of C3435T point mutation of the MDR1 gene in endometrial cancer. Int J Gynecol Cancer. 2007, 17: 705-734. 10.1111/j.1525-1438.2007.00843.x.
Gemignani F, Landi S, Neonilia S-D, Zaridze D, Jolanta L, Rudnai P, Eleonora F, Dana M, Lenka F, Vladimir J, Vladimir B, Valerie G, Gioia-Patricola L, Ilaria B, Barale R, Canzian F, Hall J, Boffetta P, Hung RJ, Brennan P: Development of lung cancer before the age of 50: the role of xenobiotic metabolizing genes. Carcinogenesis. 2007, 28 (6): 1287-1293. 10.1093/carcin/bgm021.
Haenisch S, Zimmermann U, Dazert E, Wruck CJ, Dazert P, Siegmund S, Kroemer HK, Warzok RW, Cascorbi I: Influence of polymorphisms of ABCB1 and ABCC2 on mRNA and protein expression in normal and cancerous kidney cortex. Pharmacogenomics J. 2007, 7: 56-65. 10.1038/sj.tpj.6500403.
Jamroziak K, Ewa B, Cebula B, Janus A, Mirowski M, Tadeusz R: No influence of 3435C > T ABCB1 (MDR1) gene polymorphism on risk of adult acute myeloid leukemia and P-glycoprotein expression in blast cells. Ther Drug Monit. 2006, 28 (28): 707-711.
Miller KL, Kelsey KT, Wiencke JK, Moghadassi M, Rei M, Mei L, Wrensch M: The C3435T Polymorphism of MDR1 and Susceptibility to Adult Glioma. Neuroepidemiol. 2005, 25: 85-90. 10.1159/000086288.
Jamroziak K, Ewa B, Smolewski P, Robey RW, Cebula B, Panczyk M, Kowalczyk M, Szmigielska-Kapon A, Mirowski M, Bates SE, Tadeusz R: MDR1 (ABCB1) gene polymorphism C3435T is associated with P-glycoprotein activity in B-cell chronic lymphocytic leukemia. Pharmacological Reports. 2006, 58: 720-728.
Siegsmund M, Ulrich B, Elke Scha F, Gregor W, Schwab M, Eichelbaum M, Fritz P, Burk O, Decker J, Alken P, Uwe R, Reinhold K, Sven H, Hiltrud B: Association of the P-glycoprotein transporter MDR1 C3435T polymorphism with the susceptibility to renal epithelial tumors. J Am Soc Nephrol. 2002, 13: 1847-1854. 10.1097/01.ASN.0000019412.87412.BC.
Gervasini G, Carrillo JA, Montserrat G, Carmen San J, Cabanillas A, Benitez J: Adenosine triphosphate-binding cassette B1 (ABCB1) (multidrug resistance 1) G2677T/a gene polymorphism is associated with high risk of lung cancer. Cancer. 2006, 107 (12): 2850-2857. 10.1002/cncr.22332.
Sam SS, Vinod T, Kumaran S: Kanipakapatanam Sathyanarayana Reddy, Gopalakrishnan Surianarayananc and Adithan Chandrasekaran. ABCB1 genetic polymorphism and risk of upper aerodigestive tract cancers among smokers, tobacco chewers and alcoholics in an Indian population. Pharmacogenetics Genomics. 2007, 17: 861-866. 10.1097/FPC.0b013e3282c4b18a.
Jamroziak K, Ewa B, Calka K, Piaskowski S, Urbanska-Rys H, Salagack A, Mirowski M, Robak T: Polymorphisms and haplotypes in the multidrug resistance 1 gene (MDR1/ABCB1) and risk of multiple myeloma. Leukemia Res. 2009, 33: 332-335. 10.1016/j.leukres.2008.06.008.
Rocha V, Porcher R, Fernandes JF, Filion A, Bittencourt H, Silva W, Vilela G, Zanette DL, Ferry C, Larghero J, Devergie A, Ribaud P, Skvortsova Y, Tamouza R, Gluckman E, Socie G, Zago MA: Association of drug metabolism gene polymorphisms with toxicities, graft-versus-host disease and survival after HLA-identical sibling hematopoietic stem cell transplantation for patients with leukemia. Leukemia. 2009, 23: 545-556. 10.1038/leu.2008.323.
Drain S, Mark A, Catherwood NO, Galligan LI, Maeve R, Hodkinson C, Drake MB, Kettle PJ, Curly Morris TCM, Alexander DH: ABCB1 (MDR1) rs1045642 is associated with increased overall survival in plasma cell myeloma. Leukemia Lymphoma. 2009, 50 (4): 566-570. 10.1080/10428190902853144.
Penna G, Allegra A, Alonci A, Aguennouz M, Garufi A, Cannavo A, Gerace D, Alibrandi A, Musolino C: MDR-1 polymorphisms (G2677T and C3435T) in B-chronic lymphocytic leukemia: an impact on susceptibility and prognosis. Med Oncol. 2011, 28: 1549-1554. 10.1007/s12032-010-9561-9.
The MARIE-GENICA Consortium on Genetic Susceptibility for Menopausal Hormone Therapy Related Breast Cancer Risk: Polymorphisms in the BRCA1 and ABCB1 genes modulate menopausal hormone therapy associated breast cancer risk in postmenopausal women. Breast Cancer Res Treat. 2010, 120: 727-736.
Chang H, Sun Young R, Hei-Cheul J, Chong Kun I, Sung Hoon N, Jin Ju K, Hyun Cheol C: Association of the ABCB1 3435C > T polymorphism and treatment outcomes in advanced gastric cancer patients treated with paclitaxel-based chemotherapy. Oncol Reports. 2010, 23: 271-278.
Rao DN, Anuradha C, Vishnupriya S, Sailaja K, Surekha D, Raghunadharao D, Senthil R: Association of an MDR1 gene (C3435T) polymorphism with acute leukemia in India. Asian Pacific J Cancer Prev. 2010, 11: 1063-1066.
Acknowledgements
The author would like to acknowledge the Guangdong Supporting Grant for Outstanding Talents (2001–12) that led to the writing of this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The author declares that there are no competing interests and that this work has not been published or submitted concurrently for publication elsewhere.
Authors’ contributions
LHW contributed solely to the writing and submission of this work. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
Open Access This article is published under license to BioMed Central Ltd. This is an Open Access article is distributed under the terms of the Creative Commons Attribution License ( https://creativecommons.org/licenses/by/2.0 ), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
About this article
Cite this article
Wang, LH., Song, YB., Zheng, WL. et al. The association between polymorphisms in the MDR1 gene and risk of cancer: a systematic review and pooled analysis of 52 case–control studies. Cancer Cell Int 13, 46 (2013). https://doi.org/10.1186/1475-2867-13-46
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/1475-2867-13-46